

Abstract
The generation of β-lactosyl iodide was carried out under non-in situ-anomerization, metal free conditions by reacting commercially available β-per-O-acetylated lactose with trimethylsilyl iodide (TMSI). The β-iodide was surprisingly stable as evidenced by NMR spectroscopy. Introduction of octanol or cholesterol under microwave conditions gave high yields of α-linked glycoconjugates. Careful analysis of the reaction products and mechanistic considerations suggest an acid catalyzed rearrangement that provides α-linked glycosylation products with a free C2-hydroxyl. Accessibility to these compounds may further advance glycolipidomic profiling of immune modulating bacterial derived-glycans.
Keywords: H. pylori; 1,2-cis; Glycosyl iodide glycosylation; Mechanism
Carbohydrate chemists are continually challenged to control the stereochemistry of glycosidic bond formation. Although methodologies exist to provide 1,2-trans linkages with great confidence,1–3 procedures for achieving 1,2-cis glycosylations are limited.4 As early as 1975, Lemieux and co-workers reported efficient routes to 1,2-cis glycosides using in situ anomerization of anomeric bromides, Figure 1.5,6 Over the years, we and others have expanded upon these findings increasing the repertoire of available α-linked-O-glycosides.7–10 More recently, our laboratory has simplified the in situ anomerization of glycosyl iodides to enable step economical one-pot syntheses of α-linked glycolipids.11–16 The preferred protocol relies upon electron-rich monosaccharide donors that are reacted with TMSI to first generate the α-glycosyl iodide; addition of tetrabutylammonium iodide leads to in situ anomerization, and subsequent trapping with a suitable nucleophile affords 1,2-cis glycosides. In this fashion, several biologically relevant glycolipids have been prepared including the immune modulating α-cholesteryl glucosides associated with Helicobacter pylori infection.16–18 The pathogenicity of H. pylori involves bacterial uptake of host cholesterol and stereospecific biosynthesis of α-cholesteryl glucosides that lead to silencing of the host immune response.19,20 As part of a campaign to search for other variants of naturally occurring steryl glycosides and to understand the molecular basis of steryl glycoside immune modulation, our lab is developing facile methods for the synthesis of various analogs including oligosaccharide-containing constructs. Herein, we report extension of glycosyl iodide glycosylation to the synthesis of α-cholesteryl lactosides.
Figure 1.
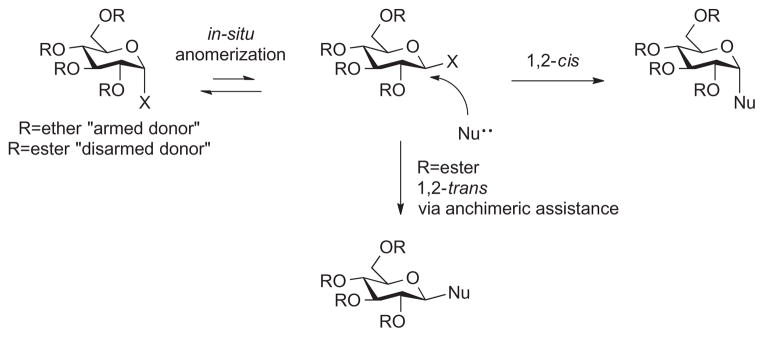
In situ anomerization to achieve 1,2-cis connectivity.
Establishing 1,2-cis connectivity is especially difficult when employing hindered acceptors such as cholesterol or oligosaccharide donors, which can undergo inter-residue glycosidic bond cleavage.21 The reactivity of glycosyl iodides is intricately linked to the protecting groups present on the donor. Ether protecting groups and especially silyl ether protecting groups are highly activating. Their electron releasing capacity has been said to ‘arm’ donors such as per-O-silyl glycosyl iodides toward nucleophilic attack. When attempting to react sterically hindered nucleophiles such as cholesterol, armed donors are preferred. However, unlike monosaccharide donors, per-O-silyl oligosaccharides cannot be used as donors in glycosyl iodide glycosylation because ether protecting groups also render the interglycosidic bond susceptible to cleavage into smaller units when treated with TMSI.2,3,22 In stark contrast, ester protecting groups are referred to as ‘disarming’ due to inductive effects; and while they stabilize interglycosidic linkages toward degradation, ester-protected glycosyl iodides are considerably less reactive than their ether protected counterparts. Moreover, if the ester is proximal to the anomeric center, for example, at the C-2 position, it typically guides glycosylation toward 1,2-trans linkages (Fig. 1). Considering these factors, the ideal oligosaccharide donor would have ester protecting groups on the hydroxyls remote from the anomeric center to protect against interglycosidic cleavage and an ether protecting group at the C-2 hydroxyl to activate the donor toward 1,2-cis attack. In fact, a monosaccharide corollary to this design was successfully employed by Demchenko and co-workers23 wherein 3,4,6-tri-Obenzoyl-2-O-benzyl-β-glucosyl bromide underwent reaction with carbohydrate acceptors giving α-stereoselectivity. Importantly, a relatively ‘disarmed’ β-glycosyl bromide was intercepted to achieve 1,2-cis connectivity. This work inspired us to explore the possibility of using ester-protected glycosyl iodides to synthesize cholesteryl-α-lactoside. Previous mechanistic studies in our lab had shown that β-glycosyl iodides could be generated from per-O-acetylated galactose and glucose encouraging us to consider per-O-acetylated lactose as a possible donor.24 We were hopeful that the increased reactivity of the iodide relative to the bromide might allow us to use per-acetylated donors and avoid multiple protecting group manipulations. Thus, the formation of the β-iodo-lactoside was explored with the intent of developing conditions to trap the reactive intermediate with cholesterol in order to achieve 1,2-cis glycosylation.
Initial studies were carried out on a 1:3 α/β-mixture of per-O-acetylated lactose and the progress of the reaction was studied by NMR (Fig. 2). Immediately upon introduction of TMSI into the NMR tube, the β-lactosyl acetate (2) began to react generating the β-anomeric iodide (3) due to anchimeric assistance from the C-2 acetate.24 However, the reaction of lactose was significantly slower than previously studied monosaccharides, which turned out to be fortuitous because the lifetime of the β-iodo-lactoside (3) was significantly extended. As shown in Figure 2, the α-acetate (1) was essentially unreactive for the first 15 min of the reaction during which time the β-acetate was nearly consumed. As the reaction proceeded over the course of 24 h, the equilibrium shifted to essentially quantitative formation of the α-iodide (4).
Figure 2.

NMR analysis of a 1:3 α/β-mixture of per-O-acetylated lactose reacting with TMSI.
The generation of 3 using pure per-O-acetylated-β-lactoside (2) continued by reacting 2 with TMSI in deuterated chloroform in order to monitor iodide formation via NMR spectroscopy (Fig. 3). As was observed with the α/β mixture, 2 was consumed within 10 min giving pure 3, which was stable enough at ambient temperature to persist for a few hours before completely anomerizing to the α-iodide (4). Having established conditions for generating a relatively long-lived β-lactosyl iodide, investigations continued with the introduction of acceptor alcohols.
Figure 3.

β-Per-O-acetylated 2 can cleanly generates 3.
Initial reactions of 3 with either octanol (2 equiv) or cholesterol (2 equiv) under microwave conditions afforded good glycoconjugate yields that favored 1,2-cis linkages and α-selectivity (Scheme 1). At the same time, these findings were surprising, as the major α-products 5 and 8 were missing the C-2 acetate. The reaction of 3 with octanol was completed in 30 min and afforded 87% overall yield of α-glycosides (44:1, α/β) in ~5:1 ratio of 5:6. Similarly, the reaction with cholesterol produced a 71% yield of α-glycosides (7:1, α/β) in approximately the same ratio ~5:1 ratio of 8:9, albeit the reaction required 2 h with the more hindered acceptor. In the latter reaction, significant amounts of acetylated cholesterol (13) and 3-β-iodo-5-cholestene (14) were also recovered.
Scheme 1.

Glycosylation with octanol and cholesterol.
Importantly, both α-linked compounds (8, 9) could be deacetylated using sodium methoxide to afford cholesteryl α-D-lactoside 15 (Scheme 2).
Scheme 2.

Deacetylation of cholesteryl α-lactosides 8 or 9.
A plausible mechanistic explanation for the observed products is presented in Scheme 3 and depends upon acid catalyzed rearrangement of the oxocarbenium resulting from anchimeric collapse onto the β-lactosyl iodide to give 16. Attack by the nucleophile at the acetate carbon rather than the anomeric center leads to formation of orthoacetates (17). Protonation of the C-2 oxygen to give 18 with concomitant introduction of iodide at the anomeric center would produce 19 or alternatively oxocarbenium formation to produce 20. Finally, nucleophilic attack on the highly reactive β-iodide would lead to the observed major products, 5 or 8. The other two major α-glycosides, 6 and 9 (Scheme 1), may result from direct nucleophilic attack on β-iodide 3. Similarly, the minor amounts of β-products, 7 and 10, may result from direct displacement of the α-iodide 4. It should be noted, that the reactivity of 19 is presumed to be much higher than 3 because it does not have an electron withdrawing acetate at the proximal C-2 position. Studies in our lab have repeatedly shown that the reactivity of glycosyl iodides is inversely correlated with the number of acetate groups on the pyranose.3,12,15,22
Scheme 3.

Proposed mechanism of HI catalyzed orthoester rearrangement to α-linked 2-hydroxy glycosylation products 5 and 8.
Achieving 1,2-cis glycosylation in the reaction of cholesterol addition to per-O-acetylated-β-lactosyl iodide contrasts earlier studies on monosaccharides. In 2008, Murakami, Sato, and Shibakami reported reacting α-iodo-2,3,4,6-O-tetraacetyl glucopyranoside with zinc halides and observed a 25–50% yield of β-linked product along with 15–20% yield of the 2-hydroxy product as an α/β-mixture.25 The same reaction when performed on α-iodo-2,3,4,6-O-tetrabenzoyl glucopyranoside gave mostly 1,2-cis products with the C-2 benzoate remaining intact, however the reaction did not work when employing cholesterol as an acceptor. The reactions reported herein do not require metal catalysis, result in 1,2-cis glycosylation and are high yielding even with hindered cholesterol acceptors. It is noteworthy that Murakami and co-workers also observed 30–40% of acetylated acceptor, further supporting the acid catalyzed rearrangement mechanism of the orthoester (Scheme 3), which depicts the departure of acetylated acceptor as iodide attacks. Indeed, a small amount of acetylated cholesterol (13) was isolated from the reaction mixture, but the 3-β-iodo-5-cholestene (14) was the major byproduct obtained. It is likely that compound 14 was generated by HI or TMSI catalyzed substitution of 13, explaining the small amounts of isolated acetylated cholesterol 13. To support this hypothesis, 13 was treated with TMSI under the same microwave conditions for 2 h and a 73% yield of 14 was obtained. Retention of stereochemistry at C-3 was confirmed from X-ray crystallography (Scheme 4). In related studies, formation of 3-β-iodo-5-cholestene (14) was observed in the reaction of cholesterol with HI,26 aluminum iodide,27 and treatment of 3-β-O-mesyl-5-cholestene with TMSI.28,29 Retention of stereochemistry at C-3 results from a non-classical cyclopropylcarbinyl cation generated from homoallylic stabilization of initial carbocation 22 (Scheme 4).30 Upon formation of cyclopropylcarbinyl cation (23) iodide attacks from the top face generating 14 (Scheme 4). The same reaction performed with dihydrocholesterol gave the inverted iodide as the major byproduct presumably due to SN2 displacement of the acetate.31
Scheme 4.

Reaction and mechanism of acetylated cholesterol (13) with TMSI to afford 3-β-iodo-5-cholestene (14).
The salient features of this research include the NMR characterization of the β-iodolactoside (3) carried out at ambient temperature, showing its unusual longevity and slow equilibration to the α-lactosyl iodide (4). Efficient trapping of the more reactive β-iodide anomer (3) via anchimeric assistance and then acid catalyzed rearrangement of the orthoester resulting from nucleophilic addition offers a new synthetic protocol for achieving 1,2-cis glycosylation of oligosaccharides. Importantly, the protocol accommodates sterically demanding nucleophiles such as cholesterol. Being able to carry out 1,2-cis glycosylations with per-O-acetylated sugars has the potential to rapidly access structurally diverse cholesteryl glycoside analogs because many per-O-acetylated oligosaccharides are commercially available and even more are readily accessible by a one-step protection. To date, the cholesteryl glycosides isolated from natural sources have consisted of primarily monosaccharides. Generation of cholesteryl α-lactoside and higher order oligosaccharide constructs will further the synthetic campaign toward providing cholesteryl glycoside standards to aid in the discovery of novel naturally occurring cholesteryl glycosides.
Supplementary Material
Acknowledgments
We would like to thank Dr. James C. Fettinger for collecting the X-ray diffraction pattern and refining the X-ray crystal structure of compound 14. We would also like to thank the following supporters for this work: NIH R01GM090262, NSF CHE-0196482, NSF CRIF Program (CHE-9808183), and NSF OSTI 97-24412. Grants for 600 and 800 MHz instruments include NSF DBIO 722538 and NIH PR1973.
Footnotes
Supplementary data (experimental date including NMR, MS, and X-ray diffraction data) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.tetlet.2015.05.012.
This work is dedicated to the memory and inspiration of Professor Harry Wasserman
References and notes
- 1.Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661. [Google Scholar]
- 2.Hsieh HW, Schombs MW, Gervay-Hague J. J Org Chem. 2014;79:1736. doi: 10.1021/jo402736g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh HW, Davis RA, Hoch JA, Gervay-Hague J. Chem Eur J. 2014;20:6444. doi: 10.1002/chem.201400024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demchenko AV. Curr Org Chem. 2003;7:35. [Google Scholar]
- 5.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056. [Google Scholar]
- 6.Lemieux RU, Driguez H. J Am Chem Soc. 1975;97:4069. doi: 10.1021/ja00847a034. [DOI] [PubMed] [Google Scholar]
- 7.Chernyak A, Oscarson S, Turek D. Carbohydr Res. 2000;329:309. doi: 10.1016/s0008-6215(00)00189-0. [DOI] [PubMed] [Google Scholar]
- 8.Baeschlin DK, Chaperon AR, Charbonneau V, Green LG, Ley SV, Lücking U, Walther E. Angew Chem, Int Ed. 1998;37:3423. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3423::AID-ANIE3423>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 9.Shingu Y, Nishida Y, Dohi H, Kobayashi K. Org Biomol Chem. 2003;1:2518. doi: 10.1039/b303984f. [DOI] [PubMed] [Google Scholar]
- 10.Shingu Y, Nishida Y, Dohi H, Matsuda K, Kobayashi K. J Carbohydr Chem. 2002;21:605. [Google Scholar]
- 11.Hadd MJ, Gervay J. Carbohydr Res. 1999;320:61. [Google Scholar]
- 12.Du W, Gervay-Hague J. Org Lett. 2005;7:2063. doi: 10.1021/ol050659f. [DOI] [PubMed] [Google Scholar]
- 13.Du W, Kulkarni SS, Gervay-Hague J. Chem Commun. 2007:2336. doi: 10.1039/b702551c. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni SS, Gervay-Hague J. Org Lett. 2008;10:4739. doi: 10.1021/ol801780c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schombs M, Park FE, Du W, Kulkarni SS, Gervay-Hague J. J Org Chem. 2010;75:4891. doi: 10.1021/jo100366v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis RA, Lin C-H, Gervay-Hague J. Chem Commun. 2012:9083. doi: 10.1039/c2cc33948j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis RA, Fettinger JC, Gervay-Hague J. J Org Chem. 2014;79:8447–8452. doi: 10.1021/jo501371h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen HQ, Davis RA, Gervay-Hague J. Angew Chem, Intl Ed. 2014;53:13400–13403. doi: 10.1002/anie.201406529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SJ, Lee BI, Suh SW. Proteins. 2011;79:2321. doi: 10.1002/prot.23038. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi M, Lee H, Nakayama J, Fukuda M. Curr Drug Metab. 2009;10:29. doi: 10.2174/138920009787048428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demchenko AV. Synlett. 2003:1225. [Google Scholar]
- 22.Hsieh HW, Schombs MW, Witschi MA, Gervay-Hague J. J Org Chem. 2013;78:9677. doi: 10.1021/jo4013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaeothip S, Yasomanee JP, Demchenko AV. J Org Chem. 2011;77:291. doi: 10.1021/jo2019174. [DOI] [PubMed] [Google Scholar]
- 24.Gervay J, Nguyen TN, Hadd MJ. Carbohydr Res. 1997;300:119. [Google Scholar]
- 25.Murakami T, Sato Y, Shibakami M. Carbohydr Res. 2008;343:1297. doi: 10.1016/j.carres.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Shoppee CW, Summers GHR. J Chem Soc. 1952:1786. [Google Scholar]
- 27.Broome J, Brown BR, Summers GHR. J Chem Soc. 1957:2071. [Google Scholar]
- 28.Sun Q, Cai S, Peterson BR. Org Lett. 2009;11:567. doi: 10.1021/ol802343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortega N, Feher-Voelger A, Brovetto M, Padron JI, Martin VS, Martin T. Adv Synth Catal. 2011;353:963. [Google Scholar]
- 30.Lambert JB, Ciro SM. J Org Chem. 1996;61:1940. [Google Scholar]
- 31.Davis RA. PhD Dissertation. University of California; Davis: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.