

Abstract
MiRNAs are small, nonprotein-coding RNA molecules involved in gene regulation. While bioinformatics help guide miRNA research, it is less clear how they perform when studying biological pathways. We used 13 criteria to evaluate effectiveness and usability of existing bioinformatics tools. We evaluated the performance of six bioinformatics tools with a cluster of 12 differentially expressed miRNAs in colorectal tumors and three additional sets of 12 miRNAs that are not part of a known cluster. MiRPath performed the best of all the tools in linking miRNAs, with 92% of all miRNAs linked as well as the highest based on our established criteria followed by Ingenuity (58% linked). Other tools, including Empirical Gene Ontology, miRó, miRMaid, and PhenomiR, were limited by their lack of available tutorials, lack of flexibility and interpretability, and/or difficulty using the tool. In summary, we observed a lack of standardization across bioinformatic tools and a general lack of specificity in terms of pathways identified between groups of miRNAs. Hopefully, this evaluation will help guide the development of new tools.
Keywords: miRNA, bioinformatics, miRPath, pathways
Introduction
MiRNAs are small, nonprotein-coding RNA molecules, ∼22 nucleotides in length,1–5 which are complementary to messenger RNA (mRNA) molecules. The mature miRNAs are generated from the cleavage of larger precursor RNA molecules by the enzymes Drosha and Dicer.6 The resulting mature miRNA then binds with an RNA-induced silencing complex, which has the ability to bind to mRNA molecules and alter gene expression by either inhibiting mRNA translation into protein or by promoting mRNA degradation,7 usually by mRNA cleavage or deadenylation.4 MiRNAs are thought to be involved in the carcinogenetic process given their ability to act as tumor suppressors and oncogenes.4 A single miRNA may regulate many different genes, and a single gene may be cooperatively regulated by different miR-NAs.7–9 Since the relationships between miRNAs and their products are numerous, and participate in several different biological processes, it is imperative to look at combinations of miRNAs and genes being expressed to better understand the role of miRNAs in the carcinogenic process. Coordinated gene expression involves the transcription of many genes that work together in specific cellular functions and biological pathways to create a required response.10 Figure 1 depicts this process for miRNAs. It has been suggested, “The recognition of coordinated expression profiles… enables inferences about biological pathways and genes functions to be made”.11 It has also been suggested that there are two classes of clustered miRNAs, those with similar sequences that work on a group of genes together, and those of a dissimilar sequence that produce a biological response by regulating different genes involved in that process at the same time.1 MiRNAs are seen as regulators of cell phenotypes6 and are being considered as potential biomarkers that can be used to help diagnosis, stage, and target cancer treatments.12
Figure 1.
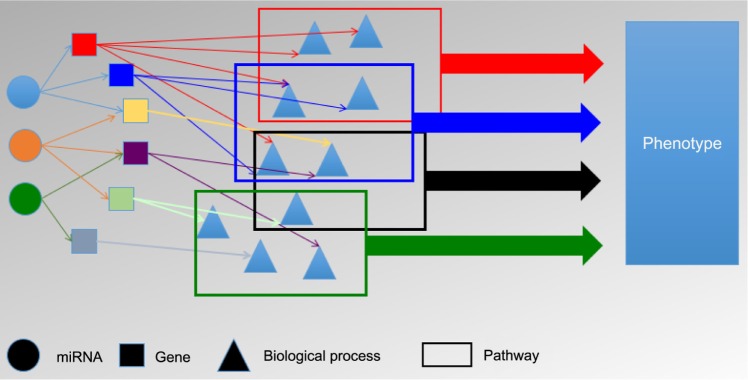
Coordinated expression with miRNAs.
Colorectal cancer (CRC) is a multifactorial disease,13 involving multiple biological pathways, and miRNA expression most likely plays a role in its progression.7,14 Studies have evaluated associations between expression of specific miRNAs and clusters of miRNAs and CRC. Because each miRNA may regulate many genes, the target gene list for even a relatively small miRNA cluster may be prohibitively large to curate manually (see Table 1 of one previously identified miRNA cluster associated with CRC). To investigate the nature of the miRNA clustering, a suitable tool for analyzing miRNAs in the context of functional biological pathways is needed. Currently, no usable standard exists for evaluating these bioinformatics tools and their attributes that pertain to miRNA pathway analysis. In this study, we evaluate six readily available miRNA bioinformatics tools focusing on their effectiveness and usability. We propose 13 criteria that can be used to measure the effectiveness and usability of bioinformatics tools in general and in the analysis of miRNAs within biological pathways.
Table 1.
MiRNA target gene regulation. This table shows the potential number of targets that need to be examined when analyzing a set of miRNA.
| miRNA | VALIDATED TARGETS1 | PREDICATED TARGETS2 | POTENTIAL TOTAL GENES |
|---|---|---|---|
| hsa-miR-106b-5p | 102 | 855 | 957 |
| hsa-miR-25-3p | 81 | 496 | 577 |
| hsa-miR-93-5p | 125 | 839 | 964 |
| hsa-miR-221-3p | 327 | 316 | 643 |
| hsa-miR-17-5p | 417 | 860 | 1277 |
| hsa-miR-20a-5p | 156 | 847 | 1003 |
| hsa-miR-92a-3p | 656 | 496 | 1152 |
| hsa-miR-20b-5p | 31 | 849 | 880 |
| hsa-miR-203a3 | 133 | 711 | 844 |
| hsa-miR-19a-3p | 108 | 788 | 896 |
| hsa-miR-7-5p | 300 | 434 | 734 |
| hsa-miR-224-5p | 118 | 441 | 559 |
| Total Genes | 2554 | 7932 | 10486 |
Notes:
Predicated targets from miRDB (http://mirdb.org/cgi-bin/search.cgi?searchType=miRNA&full=mirbase&searchBox=MIMAT0000081).
Validated targets are for hsa-miR-203.
We consider several methodological issues. First, it has been proposed that traditional gene set enrichment analysis (GSEA), which uses a hypergeometric distribution to identify a normal background for comparison, does not account for the bias in the gene list determined from a miRNA cluster15; therefore tools that incorporate GSEA must be evaluated for this bias. A key issue in assessing miRNA-related databases is the integration of disseminated information across multiple databases that is often in different formats,16 leading to a lack of standardization in miRNA nomenclature.17 While existing tools integrate this information differently, one component of our comparison is how effectively bioinformatics tools accomplish this integration. A third methodological challenge is the inconsistent versions of databases for each analysis tool. Tools that are not regularly updated potentially incorporate incomplete or obsolete data.
Methods
Twelve miRNAs have previously been reported as clustering in colon tumors; we replicated this miRNA cluster in a large miRNA colorectal study previously described using hierarchical clustering of miRNAs with similar expression profiles.18 These 12 miRNAs were evaluated in six bioinformatics tools as described below. For comparison in determination of the specificity of pathways generated by bioinformatics tools, we identified and used three additional sets of miRNAs that were not clustered by expression differences in tumor vs nontumor. The first set comprised 12 random miRNAs that were dysregulated in tumor compared to the nontumor in CRC cases but not clustered together. The second set of 12 miRNAs were only expressed in nontumor tissues from CRC cases but were not differentially expressed. The third set of 12 miR-NAs were identified from the literature, using PhenomiR19,20 as being upregulated in breast tumor cells.
Summary of Bioinformatics Databases and Tools
There are many open-source and freely available bioinfor-matics tools for investigating individual aspects of miR-NAs. Databases enabling validated gene target querying that are incorporated into the bioinformatics tools being evaluated include
miRTarBase (http://mirtarbase.mbc.nctu.edu.tw),21
TarBase (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index).22
Databases for predicting miRNA target genes include
TargetScanHuman (http://www.targetscan.org),8,9,23
DIANA TOOLS microT-CDS (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index),12
miRanda (http://www.microrna.org),20
miRDB (http://mirdb.org/miRDB/),24
miRGen (http://diana.cslab.ece.ntua.gr/mirgen/).25
Additionally, miRBase (http://www.mirbase.org)26 is a well-known repository containing published sequences and annotations. The bioinformatics tools investigated in this paper incorporate many of these listed repositories. The Gene Ontology (GO)27 is an ontology to enable structured querying of genes and annotation terms, it can be used to link miRNAs to molecular functions and biological processes via mRNAs annotated with these terms. The Kyoto Encyclopedia of Genes and Genomes (KEGG)28 is a large pathway repository, linked to participating genes, and is often utilized by bioinformatics tools in regard to pathway analysis.
The evaluated six bioinformatics tools that incorporate the aforementioned bioinformatics databases were Empirical GO,15 miRó,9 miRMaid,16 PhenomiR,19 miRPath,25 and Ingenuity Pathway Analysis (IPA).29 These tools did not require the upload of expression data or mRNA data and allowed the querying of only miRNA names. Descriptions of these tools and links to their sites can be seen in Table 2. These tools were chosen from a literature search that focused on miRNA analysis.
Table 2.
Description of miRNA analysis tools evaluated. This table describes the tools objectives and methods to the best of this study’s ability based on the information given in the tool’s paper and by the tool’s site.
| BIOINFORMATICS TOOL | DESCRIPTION |
|---|---|
| Empirical GO | One approach for looking at common biological pathways is through gene-annotation enrichment analysis. Since biological processes and pathways incorporate multiple genes, gene enrichment analysis considers the expression of all of the genes in a dataset as compared to the reference genome in question; if there are more genes in the dataset that correlate to a specific process than is expected by the background genome, this process is said to be enriched in the dataset.35 Recently, Bleazard et al.21 published an article reevaluating the approach to performing gene enrichment analysis on a list of genes generated as targets for a set of miRNAs; they state that due to “bias in target prediction algorithms, similarities among seed sequences, correlations between genes that are regulated together, and … preference for control of certain biological processes”, biases exist in the list of target genes generated by traditional methods and as such the reference background must be adjusted. The tool they have developed, empirical GO (http://sgjlab.org/empirical-go/), is written in Python and utilizes third-party, user-downloaded software and various text files containing sequence and annotation data as input to generate an unbiased enrichment analysis for miRNA target genes. |
| miRό | MiRό (http://ferrolab.dmi.unict.it/miro) is a tool that enables data-mining and querying over miRNA-phenotype associations in humans, including “identification of relationships among genes, processes, functions and diseases at the miRNA level”.36 This tool incorporates multiple data sources, including the aforementioned TargetScan, miRanda, miRBase and Gene Ontology. |
| miRMaid | MiRMaid (http://www.mirmaid.org/doku.php?do=search&id=sof)37 is a web service interface for miRBase data and can be queried by researchers as well as miRMaid plugins. It is written in Ruby and aims to eliminate the labor-intensive task of repetitively downloading and parsing data files from miRBase, as well as combining miRBase data into Models that represent concepts related to miRNA regulation.37 |
| PhenomiR | PhenomiR (http://mips.helmholtz-muenchen.de/phenomir/) is a manually annotated knowledgebase for miRNA studies that utilizes “well-established ontologies and resources” such as miRBase, GO and Medical Subject Headings (MeSH).38 Queries may be made on terms such as diseases, biological processes, genes, miRNAs, on specific qualities of studies, and many others using either a simple search or logical operators for more advanced searches; specific miRNA clusters expressed within diseases have been investigated using this tool as well.38 |
| miRPath v.2.0 | DIANA miRPath (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=mirpath/index)39 is a web-based tool designed specifically to link miRNAs to biological pathways, using a single or group of miRNAs as input. This tool also provides a number of other modules, such as enrichment analysis against KEGG and GO terms, hierarchical clustering, and heat maps. This tool claims to provide these functions for both predicted and validated gene targets.39 |
| IPA | Ingenuity Pathway Analysis (IPA) (http://www.ingenuity.com/products/ipa)40 is a commercially available option for the analysis of mRNA or miRNA lists in the context of biological pathways. IPA utilizes a large knowledgebase, the Ingenuity Knowledge Base (IKB), which is manually curated and contains almost 5 million findings.40 This software package contains many tools for the annotation of gene lists, and miRNA lists, including the discovery of up and downstream effects of genes, hypothesized mechanistic networks based on the functions of the genes, pathway involvement, and causal networks. |
Tool Evaluation Criteria Definitions
We utilized 13 criteria to compare miRNA database effectiveness and usability. The following were the criteria used.
Access
Many tools are offered as either web-based servers or locally installable applications. While web-based applications do not require large data files (>20 GB30), locally installed applications can be run when there is not sufficient Internet access.30
Appropriateness
According to Bottomley, the first step in tool evaluation is to ask the question “Does this software do the job it is supposed to do – is it fit for the purpose intended?”.31 This question is assessed by the criteria “appropriateness.”
Cost
The cost criterion was used here to help determine if a free tool performs comparably to a licensed tool in terms of the other criteria.
Documentation
An important feature of any bioinformatics tool is the availability of help or manual pages, examples of analysis, and/or peer-reviewed articles documenting the tool. These pages should be easy to access, and because of this, we measure this by the presence of these pages on the tool’s website.
Exportability
Exportability enables the user to calculate his/her own summary findings and statistics, save different analyses for comparison, and possibly utilize different tools for visualization.
Flexibility
One key criterion in a useful tool is the ability of the tool to accept various inputs, input formats, and different analysis criteria. In the case of analyzing miRNAs nomenclature, a number of miRNAs and target type are important variables that strongly influence the results. MiRBase is a repository that is widely used for miRNA information and has established certain nomenclature standards. According to miRBase, the spelling of “mir” typically refers to the miRNA gene and the precursor miRNA, while the spelling of “miR” refers to the mature miRNA.32 However, this is a small distinction that may not be caught by a user, and a tool should not ignore input because of this variation. If a database is using an outdated version of miRBase, does not link to miRBase for its information, or has incomplete information, differences between user input and the tool’s knowledge base will prevent a useful analysis. Therefore, it is important for the utilized tool to recognize alternate forms of miRNA names and/or suggest alternate entries. The second form of flexibility is the ability of the tool to allow various types of input, such as user-typed, user-uploaded text file, or a searchable list for the user to pick from. It is also important that the user need not format their data in an obscure, proprietary, or nonuniform format.31 Finally, the tool should enable miRNA target matching to both predicted and validated targets, to appeal to a wider range of analyses, and the user should be able to distinguish if he or she would like to include one or both of these options in their analysis.
Interpretability
We evaluated “interpretability” as the ability of the tool to provide summary calculations or statistics (such as a number of total genes and/or pathways linked to a miRNA, a functional enrichment analysis, and a multiple comparison analysis), to provide a visualization of the data (such as a graph, table, or heat map), and to link out to external databases containing additional information (such as Ensembl33 for gene information, KEGG for a pathway diagram, or miRBase for miRNA information).
Knowledge base
The tool reference data are important to both the quality and comprehensiveness of the output. There are two main methods of knowledge base generation utilized. The first is a manual curation of literature and third-party datasets, and the second is the integration of information from one or multiple databases and/or repositories. Tools in this study may incorporate one or both of these methods to generate their knowledge base. There has been some evidence that manual curation alone is not sufficient in providing complete and accurate curation.34
Methodologies
It is important that any methodology that the utilized tool incorporates in the analysis is up-to-date, whether this entails data from a repository (such as miRBase) or an updated analytic technique (such as GSEA).
Scalability
Scalability in a tool is necessary for versatile use. When studying miRNAs, it is important for the user to evaluate multiple miRNAs at a single time and pull together either the intersection or overlap of the miRNA’s pathway information. As the miRNA cluster evaluated in this study is 12 miRNAs large, the useful length for our purposes is 12; however, as a broader application this number should be much larger, or not restricted.
Standardization
The use of standardized terminology allows disparate databases to be compared for similar results, and this information can then be translated to reliability of the tools and validity of the results. The GO and KEGG repositories are reputable and standardized data sources for genes and pathways, respectively. Therefore, the criteria “standardization” will measure whether or not the investigated tools employ either or both of these repositories for the identification of functional terms and pathways, if the tool provides these services.
Sufficiency
In the context of measuring miRNAs in terms of biological pathways, sufficiency requires that one tool be utilized in the analysis and delivers functional information on biological pathways. Unlike appropriateness, “sufficiency” implies the presence or absence of an error incurred with the use of multiple tools and/or any manual curation.
Usability
“Usability” will be measured by the compatibility of the tool with multiple platforms, whether the user needs to acquire any additional files or information (not related to their input data) in order to run the tool, and whether the user must know any programming language. This criterion will not involve the use of logical operators (OR, AND, NOT), which are used in querying some databases. In this context, usability will not represent the ease of use of the user interface as is the case in computer science, but rather it will measure the general ease of use of the tool in the context of miRNA analysis.
Criteria Operationalization and Evaluation
The criteria were operationalized using “yes”/“no” questions, which can be seen in Table 4. The purpose of phrasing questions in yes/no format is to standardize the input for comparison without assigning an arbitrary numerical scale. While some of these criteria are more specific to the task of utilizing these tools for the purpose of evaluating miRNA clusters in terms of biological pathways, there are other criteria that are useful for evaluating the usefulness and effectiveness of these bioinformatics tools as in the context of miRNA analysis in general. If a criteria could not be answered, it received an “unknown.” In order to calculate overall tool effectiveness and usability, the categorical answers were coded with 1 for yes, −1 for no, and 0 for unknown. This enabled summary findings across tools to be determined; this would also enable the use of multiple evaluators’ scores to be combined. Bioinformatics tools were run on a PC running Windows 7 and a Mac running OS X Yosemite. Both computers were equipped with Java; the Windows computer was running Java 7 and the Mac was running Java 8. This was kept this way in order to investigate the usability across software versions, in the case that a tool required Java software to execute.
Table 4.
miRNA Input Number and Content. This table portrays the different evaluation criteria categories (‘Criteria’), the operationalized question (‘Definition’), and the different tools which were evaluated. Tools have a ‘Y’ if they fulfilled the criteria question, and ‘N’ if they did not, and a ‘U’ if this criteria question was not determined. At the bottom of the table there is a tally of the total criteria that the tool fulfilled (total number of ‘Y’s).
| CRITERIA | DEFINITION | EMPIRICAL GO | miRó | miRMaid | PhenomiR | miRPath | IPA |
|---|---|---|---|---|---|---|---|
| Access | Is the tool offered as a web-server? | N | Y | Y9 | Y | Y | Y |
| Is the tool downloadable? | Y | N | Y | N | N | N | |
| Appropriateness | Does the tool provide pathway output for a miRNA cluster input? | Y7 | N | N | N | Y | Y |
| Cost | Is the tool free? | Y | Y | Y | Y | Y | N |
| Documentation | Are there peer-reviewed articles provided on the tool’s web-site? | Y | Y | Y | Y | Y | Y |
| Are there examples or tutorials provided on the web-site? | N | Y | Y | N | Y | Y4 | |
| Are there manual pages provided on the tool’s web-site? | Y | N | Y | N | N | Y | |
| Efficiency | Is this the only tool needed to perform the desired analysis? | N | N | N | N | Y | Y |
| Exportability | Can the user download summary results? | Y | N | U8 | N | Y | Y |
| Does the tool allow data to be saved for future use? | Y | N | U | N | Y | Y | |
| Can the user download raw results? | U | N | U8 | Y | N | Y | |
| Flexibility | Does the tool find validated targets? | N | Y | U | Y2 | Y | Y |
| Does the tool find predicated targets? | Y | Y | U | N | Y | Y | |
| Does the tool allow predicted and validated target to be viewed together? | N | Y | U | N | Y | Y | |
| Does the tool allow predicted and validated target to be viewed separately? | N | N | U | N | Y | Y | |
| Does the tool discriminate between predicated and validated targets? | N | N11 | U | N | Y | Y | |
| Does the tool allow basic text file format? | Y | N | U | N | Y | Y | |
| Does the tool allow various nomenclature? | U | Y | Y1 | N | Y | Y | |
| Does the tool recognize equivalent names? | U | Y | Y1 | N | Y | Y | |
| Does the tool allow for species filtering? | Y | N | Y1 | Y | Y | N5 | |
| Does the tool suggest different miRNAs if the nomenclature does not match? | U | Y | Y1 | N | Y6 | U | |
| Does the tool alert the user when a miRNA does not link? | U | N | Y1 | Y | Y | Y | |
| Does the tool alert the user of how many miRNAs linked? | U | N | Y1 | Y | Y | Y | |
| Interpretability | Does the tool provide any summary statistics? | U | Y | U | Y | Y | Y |
| Does the tool provide any visualization? | U | Y | U | Y | Y | Y | |
| Does the tool link to external sites? | N | Y | Y | Y | Y | N | |
| Knowledgebase | Is the knowledgebase manually curated? | N | Y | Y | Y | Y | Y |
| Is the knowledgebase comprised of various repositories? | Y | Y | N | Y | Y | Y | |
| Methodologies | Are any utilized databases updated on a regular basis? | Y | N | N | N | N | Y |
| Are any statistical methodologies used up to date? | Y | U | U | U | U12 | U | |
| Scalability | Does the tool allow data to be saved for future use? | Y | N | U | N | Y | Y |
| Does the tool allow for different amounts of miRNAs input without limit? | Y | N | U | Y3 | Y | Y | |
| Standardization | Does the tool utilize KEGG pathways? | Y | N | N | N | Y | N |
| Does the tool utilize GO terms? | Y | Y | N | Y | N | N | |
| Usability | Is the tool compatible with the Windows platform? | Y | Y | N9 | Y | Y | Y |
| Is the tool compatible with the Mac platform? | Y | Y | Y | Y | Y | Y | |
| Is the tool compatible with the Linux platform?10 | U | U | Y | U | U | U | |
| Is the tool compatible with the UNIX platform? | U | U | U | U | U | U | |
| Can the user execute the tool without acquiring specific outside software?10 | N | Y | N | Y | Y | Y | |
| Can the user execute the tool using only their own input miRNA file(s), without acquiring additional files? | N | Y | Y | Y | Y | Y | |
| Can the user operate the tool without any programming language knowledge? | N | Y | N | Y | Y | Y | |
| Total summary score | 7 | 4 | 8 | 2 | 28 | 25 |
Notes:
Certain criteria were evaluated using miRMaid’s web-based server’s search tool only.
When the entry is entered with a validated target, this target is displayed in the results.
Twelve miRNA entries were allows in the query, however results were not obtained.
The examples are provided on the tool site, which is not neccesarily the web site.
Filtering is available, however only TargetScanHuman (predicted targets) will narrow the species.
When miRNAs are entered individually, the tool allows the user to pick a new name; if the miRNA is entered in a cluster it will not be added.
The authors re-ran the analysis using KEGG pathways in place of GO terms.
Raw results cannot be downloaded from the server-based tool; it is possible that this criterion doesn’t reflect full use of the tool.
The web-based tool is able to run on Windows; the Ruby-based claims to be incompatible with Windows, and compatible with Mac and Linux.
For the majority of tools, these criteria are supposed to be answerable as ‘Y’, however this was not verified by attempt by the user.
The tool does provide a link to miRBase, which provides links to validated and predicted targets, however this link is out of date.
MiRPath utilizes the hypergeometric distribution, which has been suggested to be inappropriate for miRNA GSEA; it is possible though that more research needs to be conducted on this before this criterion can conclusively be determined for this tool.
We ran each of the investigated tools with each of the miRNA inputs seen in Table 3. As can be seen, there were 41 unique input groups, derived from different versions and combinations of the chosen input cluster of miRNA as individuals and as clusters, used in the evaluation of each tool. These groups consisted of three cluster groups (the mature miRNA spelling group, the miRNA gene spelling group, and the alternative spelling group), as well as each individual miRNA by itself. The purpose of different groups was to determine whether the tool accepted individual and group-formatted inputs; the order in which the groups were used as input was arbitrary and one group’s input did not impact any subsequent group’s findings. The cluster miRNA groups contained 12 miRNAs; however, in the alternative spelling group, there were 14 names, as two miRNAs had two alternative names each. In this case, rather than creating two separate alternative spelling groups, all forms of the name were included in the input group.
Table 3.
MiRNA nomenclature and linking in tools. This table displays the groupings of the miRNAs used as input to the evaluated tools (which were clustered based on expression in CRC tumors). There are three groups: 1) mature miRNA exact spelling (using ‘miR’ in the name and the most current and specific version, ie, ‘-3p’); 2) gene miRNA exact spelling (using ‘mir’ in the name and the most specific version, ie, ‘-3p’); and 3) alternative spelling (this is the older version of the miRNA’s nomenclature). Each miRNA was tried individually (if available) and as a group (if available); this means that each miRNA was input twice in total and the ‘input group #’ column reflects this. For each tool, it is recorded under the tool column if the tool recognized the miRNA as a valid miRNA (‘Recognized’) and had curated pathway or process data for that miRNA (‘Curated’). A cell value of ‘Y, Y’ for example means that that miRNA (row) was both ‘Recognized’ and ‘Curated’ by that tool (column). If the tool could not be executed an ‘N/A’ is recorded in that column.
| CATEGORY | miRNA | INPUT GROUP # | RESULTS (RECOGNIZED, CURATED) | |||||
|---|---|---|---|---|---|---|---|---|
| EMPIRICAL GO | miRó | miRMaid1 | PhenomiR | miRPath | IPA | |||
| Mature miRNA exact spelling | hsa-miR-106b-5p | 1, 4 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 |
| hsa-miR-25-3p | 1, 5 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-93-5p | 1, 6 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-221-3p | 1, 7 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-miR-17-5p | 1, 8 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-miR-20a-5p | 1, 9 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-20b-5p | 1, 10 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-92a-3p | 1, 11 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-miR-203a | 1, 12 | N/A | Y, N | N, N | N, N | N, N* | Y, Y* | |
| hsa-miR-19a-3p | 1, 13 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-7-5p | 1, 14 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-miR-224-5p | 1, 15 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| Gene miRNA exact spelling | hsa-mir-106b-5p | 2, 16 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 |
| hsa-mir-25-3p | 2, 17 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-93-5p | 2, 18 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-221-3p | 2, 19 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-mir-17-5p | 2, 20 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-mir-20a-5p | 2, 21 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-20b-5p | 2, 22 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-92a-3p | 2, 23 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| hsa-mir-203a | 2, 24 | N/A | Y, N | N, N | N, N | N, N* | Y, Y* | |
| hsa-mir-19a-3p | 2, 25 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-7-5p | 2, 26 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y4 | |
| hsa-mir-224-5p | 2, 27 | N/A | Y, N | Y, N | N, N | Y, Y | Y, Y | |
| Alternative ID spellings | hsa-miR-106b | 3, 28 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 |
| hsa-miR-25 | 3, 29 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-93 | 3, 30 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-221 | 3, 31 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y* | |
| hsa-miR-17 | 3, 32 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y* | |
| hsa-miR-20a | 3, 33 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-20 | 3, 34 | N/A | Y, Y* | N, N | Y, Y3 | Y, Y* | Y, Y4 | |
| hsa-miR-20b | 3, 35 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-92a | 3, 36 | N/A | Y, Y | N, N | Y, Y | Y, Y* | Y, Y* | |
| hsa-miR-92 | 3, 37 | N/A | Y, Y* | N, N | Y, Y3 | Y, Y* | Y, Y* | |
| hsa-miR-203a-3p | 3, 38 | N/A | Y, N | N, N | N, N | N, N* | Y, Y | |
| hsa-miR-19a | 3, 39 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-7 | 3, 40 | N/A | Y, Y | N, N | Y, Y | Y, Y* | Y, Y4 | |
| hsa-miR-224 | 3, 41 | N/A | Y, Y | N, N2 | Y, Y | Y, Y* | Y, Y4 | |
| miRNA group type | Linked to tool | |||||||
| Cluster 1 (Y/N, %) | N/A | N | N | N | Y, 92% | Y,5 58% | ||
| Cluster 2 (Y/N, %) | N/A | N | N | N | Y, 92% | Y,5 58% | ||
| Cluster 3 (Y/N, %) | N/A | N | N | N | N, 0% | Y,5 58% | ||
| Individual (#, %) ALL clusters | N/A | 13, 34% | 0, 0% | 13, 34% | 35, 92% | 7, 58% | ||
| Individual (#, %) ONE cluster | N/A | 13, 93% | 0, 0% | 13, 93% | 13, 93% | 7, 58% | ||
Notes:
Provided alternative options for spelling.
Questions involving the miRNA-linking of miRMaid were answered using the web-based server’s search tool.
These miRNAs were recognized as gene miRNAs (or precursors) and not mature miRNAs.
These results included any entry that has this miRNA in the title (ie, searching for ‘hsa-miR-20’ returns all entries including miRNAs such as hsa-miR-20b or -205).
Provided annotation for miRNAs with the same seed sequence, which is typically from the same family but also includes other species.
As IPA linked the miRNAs to miRNA families, only the families were counted.
If a barrier to tool execution was encountered, which was not described by an evaluation criterion and was not readily solved by either the tool manual or the tool site, it was documented and evaluation was discontinued; “U”s were then added for all unanswered criteria. Criteria that could be answered by visiting the tool’s site or reading the tool’s paper were filled in “Y” or “N” as appropriate.
Results
MiRNA linking
MiRNA linking is defined here as a tool both recognizing a miRNA and having biological process or pathway information. Individual miRNA linking by spelling variations can be seen in Table 3; linking is shown as a percent calculated from the number of ”Y, Y” (being both recognized and curated by the tool) answers divided by all other answers (“N, N”; “Y, N”; or “N/A). In terms of miRNA linking, miRPath performed the best of all the tools, with 92% of all miRNAs linking overall. Ingenuity performed the next best, with 58% of all miRNA attempts linking, although this number is ambiguous as IPA links all the input miRNAs to a single miRNA of the same seed sequence and annotates the list based only on the sublist of miRNAs. This group of miRNAs is typically a miRNA family, although it includes miRNAs from all species. In the list of input miRNAs, seven miRNAs happened to be those that are the seed sequence, and these were picked up by IPA. The other miRNAs in the list were grouped into the seed sequence family of the seven miRNAs. As such, only the miRNAs that directly linked were counted. Even though IPA recognized all the input miRNAs and accounted for them with gene and pathway information, not all the original miRNAs were in the final analysis and some miRNAs from other species were in the analysis. Because of this, all the miRNAs that linked to a different miRNA were given a Y, Y score in Table 4, but the overall summary for miRNAs that truly linked was 58%. Both IPA and miRPath were able to link most of the input forms to the correct, most current, miRNA information, took in individual and cluster data, provided validated and predicted target information, and provided pathways. MiRó and PhenomiR performed similarly, and worse than IPA, with 34% of overall miRNAs linking. Neither of these tools accepted cluster data. Both tools performed best with the “Alternative ID Spellings” group, finding and containing information on all but one miRNA (hsa-miR-203a-3p). MiRó was able to recognize all the miRNAs that were used as input from all three spelling groups, but they only had information linked to the Alternative ID Spelling. PhenomiR was unable to recognize any spelling aside from the Alternative ID Spelling. MiRMaid and Empirical GO were not completely analyzed. MiRMaid performs similar to miRBase when it is not run through Ruby and is only executed from the graphical user interface; as such, only individual miRNAs could be queried and limited information was gleaned from the tool execution. Empirical GO required multiple text files and software to be downloaded and additional files to be generated subsequently; all these steps created barriers to the evaluation of the tool in this study.
Tool performance based on criteria
Evaluation of the tools based on the 13 criteria showed that miRPath and IPA scored the highest (Table 4). Major limitations of other tools included lack of tutorials on the web, needing additional tools to complete the analysis, general lack of flexibility and interpretability, and difficulty using the tool.
Results with different tissues and clusters
The different miRNA input sets were run using miRPath to compare for the specificity in determining pathway output for different tissue types (Table 5). Comparison of CRC tumor cluster pathways with those identified from other sets of miRNAs showed that 13 pathways were in both the CRC tumor cluster and CRC random nonclustered miRNAs. Five of these pathways also were identified in normal nonclustered expressed miRNAs, and six were identified in breast tumor randomly upregulated miRNAs. The fewest pathways were identified for the normal nonclustered expressed tissue, with five pathways identified, and the next fewest pathways were identified for the breast tumor randomly upregulated miRNAs, with seven common pathways identified.
Table 5.
MiRPath pathway results overlap by tissue type. This table displays the number of pathway results shared between any two miRNA clusters used as input. For example, the group of miRNAs that were clustered in CRC tumors and the miRNAs that were randomly selected from CRC tumors share 13 pathways in common. There were 33 pathways total for CRC clustered miRNAs and 17 total pathways for random CRC miRNAs.
| CRC TUMOR CLUSTER | CRC TUMOR RANDOM | CRC NORMAL RANDOM | BREAST TUMOR RANDOM | |||||
|---|---|---|---|---|---|---|---|---|
| N | N/TOTAL | N | N/TOTAL | N | N/TOTAL | N | N/TOTAL | |
| CRC tumor cluster | 13 | 0.76 | 5 | 1.00 | 6 | 0.86 | ||
| CRC tumor random | 13 | 0.39 | 3 | 0.60 | 5 | 0.71 | ||
| CRC normal random | 5 | 0.15 | 3 | 0.18 | 1 | 0.14 | ||
| Breast tumor random | 6 | 0.18 | 5 | 0.29 | 1 | 0.20 | ||
| Total | 33 | 17 | 5 | 7 | ||||
Discussion
Our evaluation suggests that miRPath performs the best in terms of usability and effectiveness in the evaluation of multiple miRNAs to determine biological pathways. This tool is free, allows individual and cluster miRNA input, recognizes multiple nomenclatures for the majority of miRNAs tested, allows for both predicted and validated targets and allows them to be viewed separately or simultaneously, links to external sites for gene and pathway information, and gives genes and biological pathway results. One potential source of error in using this tool is that it is based on miRBase 18, while the most current version is 21 (as of June 2014). MiRBase 21 is mapped to the newer human genome assembly GRCh38, which has resulted in the removal of some duplicate entries that are now mapped to a single locus. Additionally, miRBase 21 has an increased number of mature miRNAs, including 2,588 miRNAs, whereas miRBase blog entries put miRBase 18 at 1,921 distinct miRNAs. Another possible disadvantage to miRPath is that it performs an enrichment analysis based on the hypergeometric distribution, which has been contested as the correct approach by Bleazard et al.15
Several tools displayed similar limitations, including lack of flexibility within the program. This included not being able to find validated targets, not being able to use various nomenclatures, or providing feedback to the researcher in terms of linkage and interpretability of findings (Tables 4 and 5). Other problems encountered were poor documentation, lack of pathway output, or needing to use multiple tools to get the pathway information being sought. Lack of ongoing updating of databases was a fairly consistent problem across databases. This problem leads to other problems including inconsistencies in nomenclature for linking and lack of incorporation of current knowledge into regarding pathways.
Our evaluation of the pathways was based on a predetermined set of criteria that were based on the literature and clearly defined prior to the start of the evaluation. Utilization of a set of criteria allowed us to consistently and objectively evaluate these tools. As no gold standard currently exists for this type of tool or evaluation, the list of measured variables is not comprehensive and some criteria reflect the specific needs of the investigators (as opposed to usability and effectiveness in general). Some criteria of potential interest were not included. One example would be evaluation of tool speed because this criterion is dependent on variables such as Internet connection, software version, and computer processing speed. We felt that we would not be able to obtain accurate results, and any results we found would not be informative across readers with different circumstances. We also elected to have criteria scored by either a yes or no response. While in some instances, more knowledge could be beneficial, such as type of summary statistics included, we did not believe that further evaluation of types of statistics included could be objectively evaluated. While only one individual evaluated the tools, which could lead to bias, we felt that having one reviewer assured that the criteria were being applied in the same manner. As the criteria were written to be as objective and precise as possible, we do not believe this should generate answers that would vary from user to user, thus having one evaluator should not influence the results. Empirical GO and miRMaid require more sophisticated programming and/or third-party software to execute and may have performed better among individuals with a strong computer programming background.
As only 12 distinct miRNAs were employed in the evaluation of the selected tools, the general effectiveness of the tools may not be determined as well as if a larger group of miRNAs was used as some of the miRNAs may not link simply due to chance; however, there is no guarantee that a larger cluster would contain miRNAs with a better chance of linking across all tools. The cluster size of 12 was used because this group of miRNAs was shown to be statistically significantly clustered together, and as such, it is of biological relevance and represents a typical input group. Because the miRNAs were input to the tools in both cluster and individual formats, as well as with different spellings, this cluster size was manageable, and we do not think this is a significant limitation to the results of the study.
Our results further suggest that even the best performing tools may not be specific to identifying functions and pathways associated with miRNA clusters specific to any one tumor. Since miRNAs have many functions and are associated with many genes, it is not unexpected that pathways identified as being associated with miRNAs in one cancer would also be associated with other cancers. This most likely reflects the need for additional tools to help identify specific characteristics of clusters of miRNAs associated with specific tumors that can be utilized to inform potential screening and treatment modalities.
In summary, while most bioinformatics tools that were evaluated performed well in certain areas, they all had other areas that did not perform as well. MiRPath and IPA performed the best and generated an array of biological pathways associated with the miRNA cluster of interest. The need to update existing tools and refine them in terms of specificity could enhance our understanding of how miRNAs function in the carcinogenic process.
Acknowledgments
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute. The authors would like to acknowledge Dr. Karen Eilbeck’s insight and suggestions.
Footnotes
ACADEMIC EDITOR: J.T. Efird, Editor in Chief
PEER REVIEW: Seven peer reviewers contributed to the peer review report. Reviewers’ reports totaled 2,392 words, excluding any confidential comments to the academic editor.
FUNDING: This study was supported by NCI grants CA163683 and CA48998. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: LM, MS. Analyzed the data: LM. Wrote the first draft of the manuscript: LM. Contributed to the writing of the manuscript: LM, MS, RW. Agree with manuscript results and conclusions: LM, MS, RW. Jointly developed the structure and arguments for the paper: LM, MS, RW. Made critical revisions and approved final version: LM, MS, RW. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Murray BS, Choe SE, Woods M, Ryan TE, Liu W. An in silico analysis of microRNAs: mining the miRNAome. Mol Biosyst. 2010;6(10):1853–62. doi: 10.1039/c003961f. [DOI] [PubMed] [Google Scholar]
- 3.Arora S, Rana R, Chhabra A, Jaiswal A, Rani V. miRNA-transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics. 2013;288(3–4):77–87. doi: 10.1007/s00438-013-0734-z. [DOI] [PubMed] [Google Scholar]
- 4.Gartel AL, Kandel ES. miRNAs: little known mediators of oncogenesis. Semin Cancer Biol. 2008;18(2):103–10. doi: 10.1016/j.semcancer.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Nam S, Li M, Choi K, Balch C, Kim S, Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009;37(Web Server issue):W356–62. doi: 10.1093/nar/gkp294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg RA. The Biology of Cancer. 2nd ed. Garland Science; New York, NY: 2013. [Google Scholar]
- 7.Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11(7):537–61. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coex-pression with neighboring miRNAs and host genes. RNA. 2005;11(3):241–47. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laganà A, Forte S, Giudice A, et al. miRo: a miRNA knowledge base. Database. 2009;2009:ba008. doi: 10.1093/database/bap008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komili S, Silver PA. Coupling and coordination in gene expression processes: a systems biology view. Nat Rev Genet. 2008;9(1):38–48. doi: 10.1038/nrg2223. [DOI] [PubMed] [Google Scholar]
- 11.Claverie JM. Computational methods for the identification of differential and coordinated gene expression. Hum Mol Genet. 1999;8(10):1821–32. doi: 10.1093/hmg/8.10.1821. [DOI] [PubMed] [Google Scholar]
- 12.Nazarov PV, Reinsbach SE, Muller A, et al. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013;41(5):2817–31. doi: 10.1093/nar/gks1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slattery ML, Fitzpatrick FA. Convergence of hormones, inflammation, and energy-related factors: a novel pathway of cancer etiology. Cancer Prev Res. 2009;2(11):922–30. doi: 10.1158/1940-6207.CAPR-08-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15(6):321–33. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bleazard T, Lamb JA, Griffiths-Jones S. Bias in microRNA functional enrichment analysis. Bioinformatics. 2015;31(10):1592–8. doi: 10.1093/bioinformatics/btv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobsen A, Krogh A, Kauppinen S, Lindow M. miRMaid: a unified programming interface for microRNA data resources. BMC Bioinformatics. 2010;11:29. doi: 10.1186/1471-2105-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J, Dang J, Borchert GM, et al. OMIT: dynamic, semi-automated ontology development for the microRNA domain. PLOS One. 2014;9(7):1–16. doi: 10.1371/journal.pone.0100855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slattery ML, Herrick JS, Mullany LE, et al. An evaluation and replication of miRNAs with disease stage and colorectal cancer-specific mortality. Int J Cancer. 2015;137(2):428–38. doi: 10.1002/ijc.29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruepp A, Kowarsch A, Schmidl D, et al. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010;11(1):R6. doi: 10.1186/gb-2010-11-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruepp A, Kowarsch A, Theis F. PhenomiR: microRNAs in human diseases and biological processes. Methods Mol Biol. 2012;822:249–60. doi: 10.1007/978-1-61779-427-8_17. [DOI] [PubMed] [Google Scholar]
- 21.Hsu SD, Tseng YT, Shrestha S, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42(Database issue):D78–85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43(Database issue):D153–9. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong N, Wang X. miRDB: an online resource for microRNA target predicted and functional annotations. Nucleic Acid Res. 2014;43(D1):D146–52. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vlachos IS, Kostoulas N, Vergoulis T, et al. DIANA miRPath 2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40(Web Server issue):W498–504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(Database issue):D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Consortium TGO. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartlett JC, Ishimura Y, Kloda LA. Why choose this one? Factors in scientists’ selection of bioinformatics tools. Inform Res. 2011;16(1):1–16. [Google Scholar]
- 31.Bottomley S. Bioinformatics: guide for evaluating bioinformatic software. Drug Discov Today. 1999;4(5):240–3. doi: 10.1016/s1359-6446(99)01352-5. [DOI] [PubMed] [Google Scholar]
- 32.Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–9. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham F, Amode MR, Barrell D, et al. Ensembl 2015. Nucleic Acids Res. 2015;43(Database issue):D662–9. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baumgartner WA, Jr, Cohen KB, Fox LM, Acquaah-Mensah G, Hunter L. Manual curation is not sufficient for annotation of genomic databases. Bioinformatics. 2007;23(13):i41–8. doi: 10.1093/bioinformatics/btm229. [DOI] [PMC free article] [PubMed] [Google Scholar]