

Abstract
A direct β-coupling of cyclic ketones with imines has been accomplished via the synergistic combination of photoredox catalysis and organocatalysis. Transient β-enaminyl radicals derived from ketones via enamine and oxidative photoredox catalysis readily combine with persistent α-amino radicals in a highly selective hetero radical–radical coupling. This novel pathway to γ-aminoketones is predicated upon the use of DABCO as both a base and an electron transfer agent. This protocol also formally allows for the direct synthesis of β-Mannich products via a chemoselective three-component coupling of aryl aldehydes, amines, and ketones.
The carbonyl group represents one of the most prevalent functional groups in organic synthesis.1 Most synthetic transformations of carbonyl groups capitalize on either (a) keto–enol tautomerism, which allows for the direct installation of various functional groups at the nucleophilic α-position, or (b) the polar nature of the C=O bond, which lends itself to numerous 1,2-functionalizations.1,2 The introduction of substituents at the β-carbonyl position has traditionally been limited to the conjugate addition of soft nucleophiles to α,β-unsaturated carbonyl compounds.1 In contrast, the direct β-functionalization of saturated carbonyl compounds represents an important challenge in organic synthesis that has attracted considerable interest from the chemical community over the past few years.3,4 In this context, our laboratory recently disclosed the 5π-electron (5πe−) activation mode for the direct β-arylation of saturated formyl and ketone systems (eq 1).4a More specifically, the synergistic merger of organocatalysis and photoredox catalysis enables the efficient coupling of two catalytically generated species, a 5πe− β-enaminyl radical, generated via single-electron oxidation of an enamine, and a persistent cyanoarene radical anion, formed by single-electron reduction of aryl nitriles. We have subsequently demonstrated the translation of this activation mode to two unprecedented β-carbonyl alkylations: the β-hydroxyalkylation of cyclic ketones via persistent ketyl radicals4b and the β-alkylation of aldehydes using Michael acceptors.4c
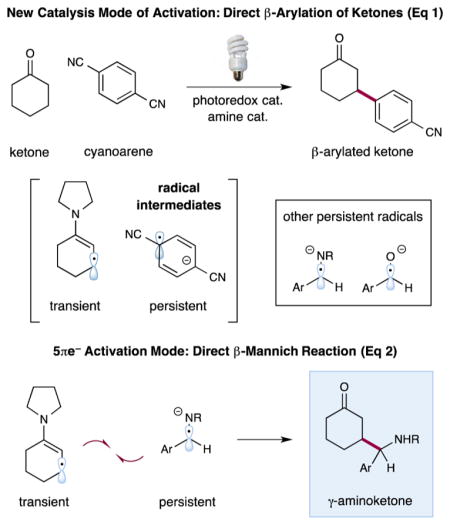
On the basis of these findings, we hypothesized that the persistent radical effect (PRE)5 might be employed to enable the selective heterocoupling of transient β-enaminyl radicals with persistent α-amino radicals derived from simple imines (eq 2) to directly forge β-aminoalkyl carbonyls (i.e., 1,4-aminoketones). The prevalence of 1,3- and 1,4-aminoketones and aminoalcohols in bioactive molecules renders this class of compounds a particularly attractive target for chemical synthesis.6,7 Whereas the Mannich reaction has traditionally been the method of choice for generating 1,3-aminoketones via α-functionalization of carbonyl compounds, an analogous method for the synthesis of 1,4-aminoketones is heretofore unknown.8 Herein, we report the successful completion of this goal through the development of a direct, photoredox organocatalytic β-aminoalkylation of saturated cyclic ketones, formally a β-Mannich protocol.
Design Plan
In accord with our previous reports on the β-functionalization of carbonyl compounds via transient β-enaminyl radicals, we envisioned that imines could be employed as viable precursors to 1,4-aminoketones through the mechanism outlined in Scheme 1. Irradiation of iridium photocatalyst 1 with visible light will generate the long-lived excited state 2, *IrIII(ppy)2(dtbbpy)+ (τ = 557 ns), which can undergo reductive quenching in the presence of an appropriate electron donor.9 Basedonan analysis of standard reduction potentials, we proposed that *IrIII 2 (E1/2 red = +0.7 V vs the saturated calomel electrode (SCE) in CH3CN) would readily oxidize 1,4-diazabicyclo[2.2.2]octane (DABCO; E1/2 red = +0.69 V vs SCE in CH3CN).9,10 This single-electron transfer (SET) step would result in simultaneous formation of the DABCO radical cation (DABCO•+) and the strongly reducing IrII(ppy)2(dtbbpy) (3) (E1/2 red= −1.5V vs SCE in CH3CN).9 SET from the electron-rich enamine 5 (E1/2 red = +0.4 V vs SCE in CH3CN)11—generated from a ketone and a suitable amine catalyst 4—to the radical cation DABCO•+ would deliver the enaminyl radical cation 6. While IrII 3 is a known reductant, electron transfer to a neutral imine was thought to be unlikely given the endergonic nature of this step (E1/2 red < −1.5 V vs SCE in CH3CN).12
Scheme 1.

Proposed β-Mannich Mechanism
However, the corresponding protonated imines should be susceptible to single-electron reduction when exposed to a thermodynamic driving force of ~1 V to readily forge the persistent α-amino radical 8. Concurrently, allylic deprotonation of 6 would provide the requisite β-enaminyl radical 7,13 which should rapidly undergo direct radical–radical coupling with the persistent α-amino radical 8 to yield γ-aminoketone enamine 9. Hydrolysis of enamine 9 would then regenerate the amine catalyst 4 and furnish the desired γ-aminoketone 10.
Results
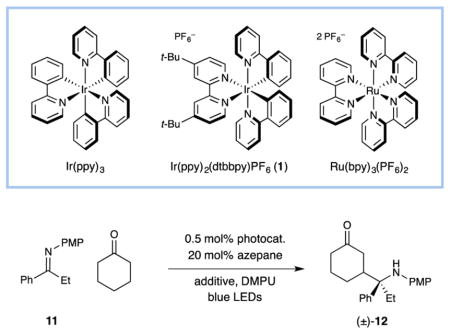
Our initial studies focused on the β-aminoalkylation of cyclohexanone with phenyl ethyl imine 11 under the action of a variety of photoredox catalysts (Table 1). Given the successful implementation of tris(2-phenylpyridinato-C2,N) iridium(III) [Ir(ppy)3] in our previous β-hydroxyalkylation studies,4b we were disappointed to find that this electron transfer catalyst was not successful in producing β-aminoalkyl ketone 12 in the presence of DABCO with blue LEDs as the light source (Table 1, entry 1). Moreover, Ru(bpy)3(PF6)2, a catalyst that exhibits a more oxidizing excited state than Ir(ppy)3 (+0.77 V and +0.31 V vs SCE in CH3CN, respectively),14,15 did not promote the desired coupling between imine 11 and cyclohexanone (entry 2). Fortunately, however, Ir(ppy)2(dtbbpy)PF6 was identified as a competent mediator of the desired β-aminoalkylation reaction, affording the functionalized ketone 12 in 75% yield (entry 3) as a 1:1 mixture of diastereomers.16 Remarkably, we did not observe any of the traditional α-substitution product (i.e., the Mannich adduct) under these electron-transfer-mediated conditions. It is important to note that control reactions, performed in the absence of Ir(ppy)2(dtbbpy)PF6 or light, resulted in no product formation (entries 4 and 5). Moreover, decreasing the DABCO loading from 1 to 0.5 equiv resulted in a significant decrease in efficiency (entry 6), with only low levels of conversion when this base was omitted (entry 7).17 Indeed, the critical role of DABCO in this coupling protocol was further illustrated via a series of Stern–Volmer fluorescence quenching experiments (Figure 1), which clearly demonstrate that both DABCO and preformed enamine 5 interact with the excited state of photocatalyst 1, while no significant change in photocatalyst emission was observed in the presence of imine 11. Taken together, these observations lend support to our proposed reductive quenching pathway (Scheme 1) wherein direct oxidation of enamine 5 by the DABCO radical cation is a viable electron transfer pathway. Finally, addition of a catalytic amount of trifluoroacetic acid led to an increase in the yield of the coupling adduct 12(entry 8)within a shorter reaction time(24 hvs 48 h), a result that is consistent with the proposed formation of a persistent radical 8 via reduction of a protonated imine.18
Table 1.
β-Aminoalkylation Optimization Studies
| |||
|---|---|---|---|
| entry | photocatalyst | additive | yielda |
| 1 | Ir(ppy)3 | DABCO (l equiv) | 0% |
| 2 | Ru(bpy)3(PF6)2 | DABCO (l equiv) | 0% |
| 3 | Ir(ppy)2(dtbbpy)PF6 | DABCO (l equiv) | 75% |
| 4 | none | DABCO (l equiv) | 0% |
| 5b | Ir(ppy)2(dtbbpy)PF6 | DABCO (1 equiv) | 0% |
| 6 | Ir(ppy)2(dtbbpy)PF6 | DABCO (0.5 equiv) | 66% |
| 7 | Ir(ppy)2(dtbbpy)PF6 | none | 35% |
| 8c | Ir(ppy)2(dtbbpy)PF6 | DABCO (1 equiv) + TFA (20 mol %) | 79% |
Yield determined after 48 h by 1H NMR using an internal standard (0.25 mmol of 11, 1.25 mmol of ketone).
Experiment was conducted in the dark.
Reaction was complete within 24 h. PMP = p-methoxyphenyl. See Supporting Information for experimental details.
Figure 1.

Stern–Volmer quenching studies with photocatalyst 1, imine 11, DABCO, and enamine 5.
Having identified optimal conditions for this photoredox catalyzed β-Mannich reaction, we first investigated the scope of the imine coupling partner. As summarized in Table 2, various ketone-derived imines furnish the corresponding γ-aminoketones with high levels of efficiency. Both diaryl and aryl alkyl ketimines (see 13–17) readily undergo coupling with cyclohexanone to forma series of products bearing tetrasubstituted carbon centers.19 Indeed, the capacity to form these sterically demanding 1,2-stereochemical dyads readily demonstrates the inherent value of radical–radical heterocoupling mechanisms as a route to traditionally difficult bond constructions. Perhaps less surprising, but equally useful, is the coupling of aldimines containing various substitution patterns in this β-aminoalkyl ketone-forming reaction (see 18–22).
Table 2.

|
Isolated yields (1.0 mmol imine, 5.0 mmol ketone). See Supporting Information for detailed experimental procedures.
Diastereomeric ratios (determined by 1H NMR analysis) were between 1:1 and 1.5:1.
With respect to the 5πe− β-enaminyl radical precursor, a range of substituted cyclohexanone derivatives was found to be readily tolerated, as illustrated in Table 3. For example, both 3-methyl and 4-methyl cyclohexanone provided the corresponding β-aminoalkyl adducts in good yield, albeit with effectively no diastereo-control (see 23 and 26, Table 3). Moreover, 4,4- and 3,3-dimethylcyclohexanones were amenable to β-aminoalkylation in useful yield (see 24 and 27). It should be noted that while 2-methylcyclohexanone underwent β-coupling using pyrrolidine as the organocatalyst (see 25), diminished levels of efficiency were observed in this case, presumably due to the accompanying low rate of enamine formation.20 Notably, cyclopentanones were also found to be suitable coupling partners for this new β-Mannich type coupling.21
Table 3.

|
Isolated yields (1.0 mmol of imine, 5.0–10 mmol of ketone). See Supporting Information for detailed experimental procedures.
Diastereomeric ratios (determined by 1H NMR analysis) were between 1:1 and 1.5:1.
LiBF4 (1 equiv) was added.
Pyrrolidine (20 mol %) was used in place of azepane.
Morpholine (40 mol %) was used in place of azepane.
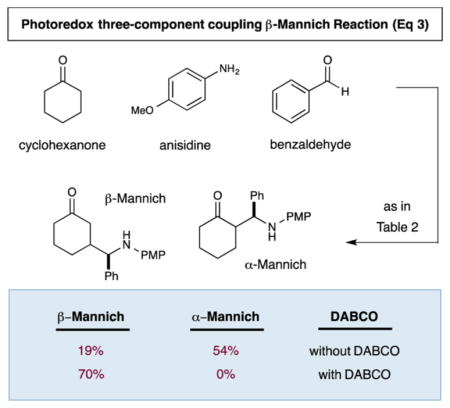
Finally, it has been long established that traditional α-Mannich reactions can often be conducted via a three-component coupling protocol that incorporates ketone, aldehyde, and amine substrates in a highly efficient condensation/C–C bond-forming pathway. To determine the capacity of our new photoredox mediated electron transfer mechanism to emulate the step and atom economy of the classical Mannich reaction, we sought to perform our β-aminoalkylation using cyclohexanone, benzaldehyde, and p-anisidine (eq 3). Indeed, employing a three-component coupling protocol, we were able to produce the formal β-Mannich adduct in a notable 70% yield. Perhaps more importantly, the traditional Mannich α-substitution products were not observed in this transformation unless DABCO was omitted, highlighting the capacity of photoredox catalysis to selectively partition transformations to nonclassical pathways. We fully expect that the viability of a photoredox mediated organocatalytic three-component coupling of imines with ketones should significantly expand the scope of coupling partners that can be employed in this β-functionalization reaction.
In summary, a practical and expedient β-ketone aminoalkylation reaction has been developed via the combination of photoredox and organocatalysis. This formal β-Mannich reaction can be conducted with imines derived from both aldehydes and ketones under mild conditions that are broadly functional group-tolerant. This strategy delivers products containing medicinally relevant, nitrogen-bearing tetrasubstituted carbon centers. Studies directed toward the development of an enantioselective variant of the β-aminoalkylation reaction are currently underway.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01GM078201-05 to D.W.C.M. and GM109536-01 to J.L.J.) and gifts from Merck and Amgen.
Footnotes
The authors declare no competing financial interest.
Experimental procedures, structural proofs, and spectral data for all new compounds are provided (PDF). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b05376.
References
- 1.Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part B: Reactions and Synthesis. Springer; New York: 2001. [Google Scholar]
- 2.(a) Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; (b) Allen AE, MacMillan DWC. Chem Sci. 2012;3:633. doi: 10.1039/C2SC00907B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Carreira EM, Kvaerno L. Classics in Stereoselective Synthesis. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 3.(a) Zaitsev VG, Shabashov D, Daugulis OJ. Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (b) Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Renaudat A, Jean-Gérard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. Angew Chem, Int Ed. 2010;49:7261. doi: 10.1002/anie.201003544. [DOI] [PubMed] [Google Scholar]; (d) Zhang S-L, Xie H-X, Zhu J, Li H, Zhang X-S, Li J, Wang W. Nat Commun. 2011;2:211. doi: 10.1038/ncomms1214. [DOI] [PubMed] [Google Scholar]; (e) Stowers KJ, Kubota A, Sanford MS. Chem Sci. 2012;3:3192. doi: 10.1039/C2SC20800H. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Leskinen MV, Yip KT, Valkonen A, Pihko PM. J Am Chem Soc. 2012;134:5750. doi: 10.1021/ja300684r. [DOI] [PubMed] [Google Scholar]; (g) Fu Z, Xu J, Zhu T, Leong WWY, Chi YR. Nat Chem. 2013;5:835. doi: 10.1038/nchem.1710. [DOI] [PubMed] [Google Scholar]; (h) Mo J, Shen L, Chi YR. Angew Chem, Int Ed. 2013;52:8588. doi: 10.1002/anie.201302152. [DOI] [PubMed] [Google Scholar]; (i) Huang Z, Dong G. J Am Chem Soc. 2013;135:17747. doi: 10.1021/ja410389a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593. doi: 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Petronijević FR, Nappi M, MacMillan DWC. J Am Chem Soc. 2013;135:18323. doi: 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Terrett JA, Clift MD, MacMillan DWC. J Am Chem Soc. 2014;136:6858. doi: 10.1021/ja502639e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Studer A. Chem—Eur J. 2001;7:1159. doi: 10.1002/1521-3765(20010316)7:6<1159::aid-chem1159>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 6.(a) Bäckvall JE, Schink HE, Renko ZD. J Org Chem. 1990;55:826. [Google Scholar]; (b) Fernández de la Pradilla R, Colomer I, Ureña M, Viso A. Org Lett. 2011;13:2468. doi: 10.1021/ol200718y. [DOI] [PubMed] [Google Scholar]; (c) Miyake Y, Nakajima K, Nishibayashi Y. Chem Commun. 2012;48:6966. doi: 10.1039/c2cc32745g. [DOI] [PubMed] [Google Scholar]; (d) Bertrand S, Hoffmann N, Pete JP. Eur J Org Chem. 2000:2227. doi: 10.1021/jo001166l. [DOI] [PubMed] [Google Scholar]
- 7.Augelli-Szafran CE, Wei HX, Lu D, Zhang J, Gu Y, Yang T, Osenkowski P, Ye W, Wolfe MS. Curr Alzheimer Res. 2010;7:207. doi: 10.2174/156720510791050920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reductive coupling of nitrones and N-sulfonyl imines with acrylates has been accomplished using SmI2. For example, see: ( Masson G, Cividino P, Py S, Vallée Y. Angew Chem, Int Ed. 2003;42:2265. doi: 10.1002/anie.200250480.Johannesen SA, Albu S, Hazell RG, Skrydstrup T. Chem Commun. 2004:1962. doi: 10.1039/b405141f.Desvergnes S, Py S, Vallée Y. J Org Chem. 2005;70:1459. doi: 10.1021/jo048237r.Peltier HM, McMahon JP, Patterson AW, Ellman JA. J Am Chem Soc. 2006;128:16018. doi: 10.1021/ja067177z.Cividino P, Py S, Delair P, Greene AEJ. Org Chem. 2007;72:485. doi: 10.1021/jo061976i.
- 9.Slinker JD, Gorodetsky AA, Lowry MS, Wang J, Parker S, Rohl R, Bernhard S, Malliaras GG. J Am Chem Soc. 2004;126:2763. doi: 10.1021/ja0345221. [DOI] [PubMed] [Google Scholar]
- 10.(a) Pischel U, Zhang X, Hellrun B, Haselbach E, Muller PA, Nau WN. J Am Chem Soc. 2000;122:2027. [Google Scholar]; (b) Mori Y, Sakaguchi W, Hayashi H. J Phys Chem A. 2000;104:4896. [Google Scholar]
- 11.Schoeller WW, Niemann J. J Chem Soc, Perkin Trans 2. 1988:369.(b) Azepane generates cyclohexenyl enamines that are more readily oxidized and more nucleophilic than the corresponding piperidine counterparts (see refs 4a, 4b, and 21b).
- 12.(a) Fry AJ. In: The Electrochemistry of the C=C, C=O and C=N Groups, Supplement A3: The Chemistry of Double-Bonded Functional Groups. Patai S, editor. J. Wiley & Sons; 1997. [Google Scholar]; (b) Fry AJ, Reed RG. J Am Chem Soc. 1969;91:6448. [Google Scholar]; (c) Fry AJ. Synthetic Organic Electrochemistry. 2. Chapter 6 Wiley; New York: 1989. [Google Scholar]; (d) Barnes JH, Triebe FM, Hawley MD. J Electroanal Chem. 1982;139:395. [Google Scholar]; (e) Root DK, Smith WH. J Electrochem Soc. 1982;129:1231. [Google Scholar]
- 13.Various radical cations have been shown to exhibit enhanced acidity relative to their ground state carbon acids; e.g., see: Bordwell FG, Cheng JP, Bausch MJ, Bares JE. J Phys Org Chem. 1988;1:209.Bordwell FG, Bausch MJ, Branca JC, Harrelson JA. J Phys Org Chem. 1988;1:225.Bordwell FG, Satish AV. J Am Chem Soc. 1992;114:10173.
- 14.(a) Kalyanasundaram K. Coord Chem Rev. 1982;46:159. [Google Scholar]; (b) Juris A, Balzani V, Belser P, von Zelewsky A. Helv Chim Acta. 1981;64:2175. [Google Scholar]
- 15.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143. [Google Scholar]
- 16.The lack of diastereoselection is consistent with a radical–radical coupling mechanism involving the unbiased approach of 8 to the planar π system of 7, and long transition state bonds.
- 17.Although DABCO functions as a catalyst (as evidenced by the result of entry 6, Table 1), use of a stoichiometric amount of DABCO allows for shorter reaction times and higher levels of efficiency. The requirement for a high concentration of DABCO may be a reflection of its dual role as an electron transfer agent and a base.
- 18.The presence of acid may facilitate both imine reduction and enamine formation. It should be noted that studies performed with hydrazones (for which direct addition of alkyl radicals is readily accomplished) yielded no addition products.
- 19.The scope of the imine coupling partner is apparently limited to imines for which reduction by 3 is favorable; alkyl imines (Ep < −3.0 V vs Ag/Ag+ in CH3CN [ref 12e]) were not suitable substrates.
- 20.The low rate of enamine formation with α-branched ketones is well documented: Sánchez D, Bastida D, Burés J, Isart C, Pineda O, Vilarrasa J. Org Lett. 2012;14:536. doi: 10.1021/ol203157s.
- 21.(a) Morpholine was the optimal organocatalyst for cyclopentanones, presumably due to the increased nucleophilicity of the corresponding enamine and 5πe− system. Relative reactivities of cyclopentanone-derived enamines towards electrophiles follow the trend morpholine > piperidine > pyrrolidine. See: Kempf B, Hampel N, Ofial AR, Mayr H. Chem—Eur J. 2003;9:2209. doi: 10.1002/chem.200204666.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.