

Abstract
Many aspects of the genomes of yeast species in the family Saccharomycetaceae have been well conserved during evolution. They have similar genome sizes, genome contents, and extensive collinearity of gene order along chromosomes. Gene functions can often be inferred reliably by using information from Saccharomyces cerevisiae. Beyond this conservative picture however, there are many instances where a species or a clade diverges substantially from the S. cerevisiae paradigm—for example, by the amplification of a gene family, or by the absence of a biochemical pathway or a protein complex. Here, we review clade-specific features, focusing on genomes sequenced in our laboratory from the post-WGD genera Naumovozyma, Kazachstania and Tetrapisispora, and from the non-WGD species Torulaspora delbrueckii. Examples include the loss of the pathway for histidine synthesis in the cockroach-associated species Tetrapisispora blattae; the presence of a large telomeric GAL gene cluster in To. delbrueckii; losses of the dynein and dynactin complexes in several independent yeast lineages; fragmentation of the MAT locus and loss of the HO gene in Kazachstania africana; and the patchy phylogenetic distribution of RNAi pathway components.
Keywords: evolution, comparative genomics, Kazachstania, Naumovozyma, Tetrapisispora, Torulaspora
The authors review species-specific evolutionary attributes of yeast genomes.
Graphical Abstract Figure.
The authors review species-specific evolutionary attributes of yeast genomes.
INTRODUCTION
Yeast species provide remarkable opportunities to study genomic evolution. In the two decades since the sequence of Saccharomyces cerevisiae was first reported (Goffeau et al. 1996), three major areas of research into yeast genome evolution have developed. First, studies such as mutation accumulation experiments have provided a view of mutational processes and rates at unprecedented resolution, both in Saccharomyces (Lynch et al. 2008; Nishant et al. 2010; Zhu et al. 2014) and in other yeasts (Polakova et al. 2009; Friedrich et al. 2015). Second, population genomics studies are providing extensive detail about genetic variation, both in S. cerevisiae and in its undomesticated relative S. paradoxus, which has led to inferences about the natural life cycle, the strength and direction of selection, and the nature of quantitative genetic variation of yeast phenotypes (Tsai et al. 2008; Liti et al. 2009; Schacherer et al. 2009; Fay 2013; Bergstrom et al. 2014; Liti 2015). Third, the comprehensive annotation and wealth of experimental information about the functions of S. cerevisiae genes has provided a solid starting point for the exploration of the genomes of related fungi (Sunnerhagen and Piskur 2006; Dujon 2010; Rozpedowska, Piskur and Wolfe 2011; Zarin and Moses 2014). This third area—yeast comparative genomics—is the focus of this review.
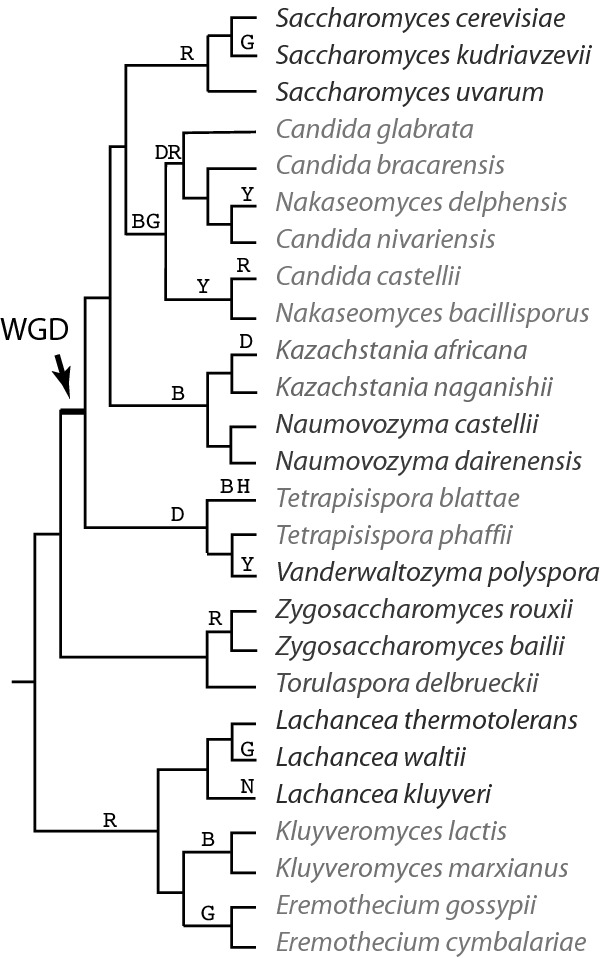
Saccharomyces cerevisiae is a member of the family Saccharomycetaceae (Fig. 1), which in turn is part of the subphylum Saccharomycotina (Kurtzman 2011). The most striking evolutionary event in the family was the occurrence of a whole-genome duplication (WGD) approximately 100–200 million years ago (Wolfe and Shields 1997). This event defines a clade called the post-WGD species, whose genomes contain evidence of this shared event, whereas outgroup species that diverged from the S. cerevisiae lineage before the WGD occurred are called non-WGD species. The ancestral organism that underwent WGD contained about 5000 genes. The WGD increased this number transiently to about 10 000 genes, but most of the extra copies of genes were not retained and instead they became ‘lost’—that is, one of the two genes in each pair became deleted, usually without any other rearrangements in the local area of chromosome (Byrnes, Morris and Li 2006; Scannell, Butler and Wolfe 2007a). Post-WGD species now typically contain about 5500 genes, which includes 500 pairs of genes (ohnologs) that were formed by the WGD; the other 4500 loci were not retained in duplicate and became single-copy again.
Figure 1.

Phylogenetic relationships of species in the family Saccharomycetaceae, and summary of some gene content differences. Letters above branches indicate inferred points of loss of the DAL (D), GAL (G), BNA (B), HIS (H), RNAi (R; loss of all Argonaute genes or all Dicer genes), NHEJ (N) and dynein (Y) pathways. ‘Chrs’, number of chromosomes. ‘DAL genes’ refers to DAL1,2,3,4,7 and DCG1. Asterisks indicate DAL clusters that also include DAL5. ‘GAL genes’ refers to GAL1/3, GAL4, GAL7, GAL10 and GAL80 (Hittinger, Rokas and Carroll 2004). The topology of the cladogram is from (Kurtzman 2003; Hedtke, Townsend and Hillis 2006; Gordon et al. 2011a; Gabaldon et al. 2013). Different colors indicate different genera. Note that Tetrapisispora does not appear to be monophyletic (Gordon et al. 2011a).
The order of genes along chromosomes (synteny) is strongly conserved within the family Saccharomycetaceae, although it is relatively poorly conserved in more distant comparisons between different families within Saccharomycotina (Dujon 2010). We developed a database and browser interface, the Yeast Gene Order Browser (YGOB—http://ygob.ucd.ie), as a resource for exploring synteny relationships among Saccharomycetaceae species (Byrne and Wolfe 2005). Synteny comparisons, both within genomes and between post-WGD and non-WGD genomes, provided the main evidence for WGD (Wolfe and Shields 1997; Dietrich et al. 2004; Dujon et al. 2004; Kellis, Birren and Lander 2004). Synteny conservation also enabled our laboratory, in 2009, to infer the approximate gene content and genome organization of the ancestral yeast organism that underwent WGD, and hence to study the chromosomal rearrangements, gene losses and gains that occurred in S. cerevisiae and other post-WGD species in their hundred-million-year descent from this ancestor (Gordon, Byrne and Wolfe 2009). We refer to this inferred genome as the ancestral genome. It is a description of the state of the genome immediately before the WGD occurred. Our ancestral genome reconstruction was inferred manually, whereas other groups used computer-assisted methods and obtained highly congruent results (Sankoff 2009).
The ancestral genome reconstruction provides a convenient reference point for studying the evolution of gene content and chromosome organization in Saccharomycetaceae. The ancestral genome consists of eight lists, each of which is the deduced order of genes along one of the eight ancestral chromosomes that later became duplicated by the WGD. Its genes are named according to their chromosome and position, for example, Anc_2.345 indicates the 345th gene along ancestral chromosome 2. In any non-WGD species in the Saccharomycetaceae, the gene order along any section of chromosome is usually similar to the ancestral order. Discontinuities correspond to genomic rearrangements, either in the non-WGD species or on the lineage of the ancestor (i.e. the post-WGD lineage prior to the occurrence of the WGD). In any post-WGD species, there are two genomic regions corresponding to each chromosomal region in the ancestor. Each of these regions contains a subset of the ancestral genes, usually without rearrangement of gene order, and some ancestral genes remain in duplicate (ohnologs) and thus appear on both chromosomal regions of the post-WGD species.
We now have a genome sequence from at least one species in almost every known genus of the family Saccharomycetaceae as defined by Kurtzman (2011) (Fig. 1). The only genera classified in this family that have not yet been sequenced are Zygotorulaspora (a sister clade to Torulaspora and Zygosaccharomyces; Kurtzman 2003) and Cyniclomyces whose phylogenetic position is uncertain and which may be basal to the family (Boundy-Mills and Miller 2011). Outside this family (Kurtzman 2003; Kurtzman and Robnett 2013), we have relatively limited genomic data from the closest outgroup genera such as Hanseniaspora/Kloeckera (Giorello et al. 2014), Saccharomycodes, Wickerhamomyces (Schneider et al. 2012a,b) and Cyberlindnera (Tomita et al. 2012; Freel et al. 2014).
Although genome evolution in the sequenced Saccharomycetaceae species has largely been conservative, with similar genes being arranged in similar ways along the chromosomes of each species, the exceptions to this rule—the differences between the species—can often be of interest and can point to differences in the biology of the species that own the genomes. In this review, we focus on aspects that are unique to the genome of a particular species or genus. We focus in particular on seven genomes that we sequenced in 2011, from three post-WGD genera (Naumovozyma, Kazachstania and Tetrapisispora) and one non-WGD genus (Torulaspora). We have previously described the evolution of the mating-type (MAT) loci of these species (Gordon et al. 2011a), but here we comment on some of their other features. Summary statistics of these genomes is given in Table 1.
Table 1.
Properties of the sequenced genomes.
| Naumovozyma | Naumovozyma | Kazachstania | Kazachstania | Tetrapisispora | Tetrapisispora | Torulaspora | |
|---|---|---|---|---|---|---|---|
| castellii | dairenensis | africana | naganishii | phaffii | blattae | delbrueckii | |
| CBS 4309 | CBS 421 | CBS 2517 | CBS 8797 | CBS 4417 | CBS 6284 | CBS 1146 | |
| Protein-coding genes | 5648 | 5548 | 5378 | 5321 | 5253 | 5389 | 4972 |
| Ancestrala | 5199 | 5100 | 5018 | 4987 | 4991 | 4895 | 4611 |
| Singletonsb | 365 | 368 | 314 | 297 | 237 | 479 | 256 |
| Median length (a.a.) | 412 | 428 | 400 | 410 | 419 | 439 | 405.5 |
| tRNA genes | 270 | 264 | 267 | 159 | 205 | 327 | 193 |
| Chromosomes | 10 | 11 | 12 | 13 | 16c | 10 | 8 |
| Genome size (Mb) | 11.2 | 13.5 | 11.1 | 10.8 | 12.1 | 14.0 | 9.2 |
aNumber of protein-coding genes with an ortholog in the ancestral genome reconstruction.
bNumber of protein-coding genes without a syntenic ortholog in any other species.
cExcluding a short unmapped 17th scaffold.
Saccharomycetaceae genomes
We sampled three post-WGD genera that had not previously been extensively studied: Naumovozyma, Kazachstania and Tetrapisispora (Kurtzman 2003). Together with the recent sequencing of genomes in the Nakaseomyces clade (including Candida glabrata and its asexual relatives) by Gabaldon et al. (2013), these data mean that we now have a genome sequence for every known post-WGD genus, and multiple genomes for all post-WGD genera except Vanderwaltozyma (Fig. 1). The genome of Naumovozyma castellii had been sequenced to draft level by Cliften et al. (2003, 2006) and it had been shown to be a post-WGD species with differential gene loss as compared to S. cerevisiae (Langkjaer et al. 2003; Scannell, Butler and Wolfe 2007a). We completed the genome sequence of the type strain of N. castellii (CBS 4309; this species was previously also called Saccharomyces castellii and Naumovia castellii), and also sequenced its congener N. dairenensis (CBS 421). Until recently, these were the only two species known in the genus Naumovozyma (Liu et al. 2012a).
The genus Kazachstania is much more species rich and we sequenced two representatives, Kazachstania naganishii (CBS 8797) and K. africana (CBS 2517), that span the phylogenetic breadth of this genus (Vaughan-Martini, Lachance and Kurtzman 2011). Kazachstania naganishii was previously known as S. exiguus strain Yp74L-3, before it was realized to be a separate species distinct from S. exiguus (which itself is now called K. exigua). Extensive methods for genetic manipulation of K. naganishii have been developed by Hisatomi, Kubota and Tsuboi (1999a,b) and Sugihara et al. (2011).
We sequenced two Tetrapisispora species because this genus was known to be sister to Vanderwaltozyma, representing the post-WGD lineage most distantly related to S. cerevisiae. Analysis of the genome of Vanderwaltozyma polyspora (obsolete name: Kluyveromyces polysporus) showed that it had undergone extensive loss of duplicate genes independently of the post-WGD gene losses in S. cerevisiae (Scannell et al. 2007b). We again chose two species spanning the phylogenetic breadth of the genus: Tetrapisispora phaffii (CBS 4417) and T. blattae (CBS 6284). A transformation system for T. phaffii has been developed (Oro et al. 2014). Tetrapisispora blattae is an outgroup to all other known species of Tetrapisispora (Lachance 2011). Surprisingly, we found by phylogenomic analysis that the genus Tetrapisispora is not monophyletic: T. phaffii and V. polyspora are more closely related to each other than either of them is to T. blattae (Gordon et al. 2011a). This relationship is shown in Fig. 1.
At 14.0 Mb excluding the rDNA locus, the T. blattae genome is relatively large for a post-WGD species. The genome contains some unusually large regions of non-coding DNA (for example, flanking the MAT genes; Gordon et al. 2011a). It is also characterized by the presence of regions of amino acid repeats internal to many proteins. An extreme example is its ortholog of S. cerevisiae Yel1 (a GEF required for localization of Arf3 to the bud neck), which is more than twice the length of the orthologs in all other Saccharomycetaceae (2001 residues compared to 687 in S. cerevisiae). Across the whole proteome, T. blattae proteins have a longer median length than any of the other species discussed here (Table 1). But at the same time, the T. blattae genome lacks orthologs of some well-known large genes: it has IRA1 but not IRA2, TOR1 but not TOR2, SPH1 but not SPA2. In each of these cases, the lost gene is one member of an ohnolog pair in S. cerevisiae.
We also sequenced Torulaspora delbrueckii (CBS 1146), a stress-tolerant yeast that is associated with winemaking (Albertin et al. 2014). Torulaspora, Zygosaccharomyces and a third genus Zygotorulaspora comprise the clade of non-WGD species that is the closest outgroup to the post-WGD species (Fig. 1) (Kurtzman 2003). The genomes of two Zygosaccharomyces species, Z. rouxii and Z. bailii, were sequenced by Souciet et al. (2009) and Galeote et al. (2013), respectively. So far, no Zygotorulaspora genome has been sequenced. Outside this small clade, there is a larger clade of better known non-WGD genera: Kluyveromyces, Lachancea and Eremothecium (Ashbya).
Clade-specific gene families, amplifications and transposable elements
Clade-specific gene amplifications are of interest because they can indicate recent directional evolutionary pressure on genomes. Saccharomyces cerevisiae, for example, contains more hexose transporters than other Saccharomycetaceae, and this may indicate adaptation to the ‘fermentative lifestyle’ (Piskur et al. 2006; Merico et al. 2007; Lin and Li 2011). In many cases, however, the amplifications are of ‘orphan’ genes that lack homologs in other species and whose functions are unknown. There is at least one such orphan family in S. cerevisiae itself: a family of five highly divergent but related genes including ABM1. These genes are of unknown function and appear as recent insertions into the Saccharomyces genome when compared to the ancestral genome (Gordon, Byrne and Wolfe 2009).
Yeasts other than S. cerevisiae also contain species-specific or clade-specific gene amplifications. In many of these cases, however, it is difficult to say anything about the functions of the amplified genes because they are completely unknown and the genes lack homologs or even recognizable protein domains. It is often not even clear whether the amplified genes have ‘normal’ cellular roles or if they are mobile genetic elements. For example, the N. castellii genome has two large orphan gene families, as first described by Cliften et al. (2006). Neither of these families has homologs in the congeneric species N. dairenensis. One of these families, exemplified by NCAS0A14090, has 19 members. It was recently identified as a transposase of the hAT family, part of a mobile element that was named Roamer (Sarilar, Bleykasten-Grosshans and Neuveglise 2015). The other orphan family, exemplified by NCAS0G03840, has 24 members and lacks any identifiable protein domains. Despite being species-specific, this family is highly divergent in sequence with only 27% amino acid sequence identity between its most diverse members. Similarly T. phaffii has a diverse nine-member family, again completely species-specific, exemplified by TPHA0N00100. Amino acid sequence identity between the two most divergent members is only 24%. This T. phaffii family is telomeric whereas the N. castellii one is not. The existence of such diverse gene families within a single species raises some interesting (but currently unanswerable) evolutionary questions about how they originated and how the members of the family became so different in sequence. In other cases orphan gene families are genus-specific, for example a family with 5 members in N. castellii (e.g. NCAS0G01740) and 15 members in N. dairenensis (e.g. NDAI0G00110). In this last example, many of the N. dairenensis members of the family are telomeric, whereas the five N. castellii genes are not telomeric but are all clustered within a 40-kb region on chromosome 7.
We identified a singleton gene in T. blattae (TBLA0D02000, 1032 amino acids) with similarity to DNA transposases of the MULE (Mutator-like element) family. MULEs are members of the Mutator superfamily of DNA transposons (class II transposable elements) (Neuveglise et al. 2005; Wicker et al. 2007). The T. blattae gene has highest similarity to the α3 gene of K. lactis, which functions in mating-type switching in that species (Barsoum, Martinez and Astrom 2010; Rajaei et al. 2014) and is the only other MULE-like transposase in the family Saccharomycetaceae. Complete MULE elements have terminal inverted repeats (TIRs) as well as a transposase gene, but the only described MULE in subphylum Saccharomycotina that is complete and active in transposition is the Mutyl element of Yarrowia lipolytica (Neuveglise et al. 2005). Neither TBLA0D02000 nor K. lactis α3 is flanked by TIRs. We also found truncated non-syntenic homologs of TBLA0D02000 in other Saccharomycetaceae: TPHA0G02110 in T. phaffii (270 residues lacking the conserved transposase domain), and several possible pseudogene fragments in N. castellii and N. dairenensis.
Mating genes, pheromones and receptors
The MATα1 gene of N. dairenensis contains two identical tandem copies of a 20-bp repeat sequence, which cause a frameshift by comparison to the α1 genes of other species. It is therefore possible that α1 is a pseudogene in this species. Losses of genes from the MAT locus have previously been identified in the CTG clade of Candida species (Logue et al. 2005; Butler 2010), but are uncommon in the Saccharomycetaceae where all other species have intact α1, α2 and a1 genes. The a2 gene, coding for an HMG-domain activator of transcription of a-specific genes (Tsong et al. 2003, 2006), is present in all non-WGD Saccharomycetaceae but was lost in the common ancestor of all post-WGD species, more or less concurrently with the WGD (Butler et al. 2004).
In K. naganishii, the HMR locus, containing a silenced copy of the mating-type a information, is not located near a telomere as in all other species. Instead, it is located between orthologs of the S. cerevisiae genes YKR011C and TOF2, more than 300 kb from the nearest telomere. Comparison of the MAT, HML and HMR structures in this species (Fig. 2) shows that the copy of the a1 gene at HMR is truncated at the 5′ end, which is unusual. The way these loci are organized in K. naganishii makes silencing of transcription at HMR unnecessary, because the copy of the a1 gene at HMR has no promoter or start codon. During mating-type switching, a copy of the 3′ part of the a1 gene from HMR is inserted beside the complete 5′ part of the gene at the MAT locus to assemble a full-length and functional a1 gene. The lack of requirement for silencing has apparently removed the need for HMR to be located beside a telomere, allowing HMR to move to a new chromosomal site in this species. However, in S. cerevisiae chromatin modification at HMR has a dual role: as well as silencing transcription, it also prevents the Ho endonuclease cleaving HMR (Haber 2012). Therefore, it is unclear what prevents K. naganishii Ho endonuclease from cleaving its HMR, and laboratory experiments will be necessary to discover whether there are chromatin modifications at K. naganishii HMR.
Figure 2.

Non-telomeric HMR locus in K. naganishii. The organization of the MAT, HML and HMR loci and the triplicated X and Z repeat regions is shown schematically. HMR is 322 kb from one telomere and 983 kb from the other. The copy of the a1 gene at HMR lacks exon 1 and therefore does not need to be transcriptionally repressed by chromatin modification. HML is close to a telomere and transcription of HMLα1 and HMLα2 is probably repressed by Sir proteins. Gene copies with names in parentheses are incomplete.
Similarly, in T. blattae the HML locus is not beside a telomere. This change is the result of a rearrangement that occurred between the HML locus and the ancestral telomere. The T. blattae HML genes (HMLα1 = Anc_1.1 = TBLA0A07590 and HMLα2 = Anc_1.2 = TBLA0A07600) retain linkage to the end of ancestral chromosome 1 on one side (they are beside Anc_1.4 and Anc_1.5), but on the other side, where a telomere is found in most other species, they are instead adjacent to genes Anc_7.365 (PEX13) and Anc_7.366 (MMR1) from the middle of ancestral chromosome 7. Consequently, T. blattae HML is more than 800 kb from the nearest telomere. It is still on the same chromosome as the MAT locus as seen in all Saccharomycetaceae (Gordon et al. 2011a). Tetrapisispora blattae has a low number of chromosomes for a post-WGD species (10 chromosomes; Fig. 1), and this example of a telomere fusion is one of several that have occurred in the T. blattae genome. Unlike the situation in K. naganishii, Sir proteins are probably still required for silencing of the non-telomeric HML in T. blattae because the copy of the α1 gene at this locus is full length and intact (HMLα2 is truncated at the 3′ end).
In K. africana, the ability to switch mating types appears to have been lost. This species has no HO endonuclease gene. Instead of the usual arrangement of three MAT-like loci (MAT, HML and HMR), this species has only two MAT-like loci which we refer to as MATa and MATα (Fig. 3). Neither of these loci appear to be an HML or HMR-type silent cassette because they have no X or Z repeat sequences that could allow DNA exchange to occur between the two loci during mating-type switching. The situation in K. africana appears to have arisen by chromosome breakage and rearrangement; its MATα and MATa loci share synteny with the left and right sides, respectively, of the MAT locus in the related species K. naganishii which has an organization resembling S. cerevisiae. We are confident that our assembly of the K. africana genome is structurally correct across the MATa and MATα loci because our sequencing strategy generated pairs of sequence reads from the ends of clones in four genomic libraries with large insert sizes (averaging 3, 7, 19 and 20 kb). The chromosome 4 and 7 rearrangements are supported by 134 and 160 unique pairs of reads, respectively, with no pairs supporting an unrearranged configuration. The type strain of K. africana (CBS 2517), which is the strain we sequenced, has been described as diploid and capable of sporulation (Vaughan-Martini, Lachance and Kurtzman 2011). However, we have not examined any other strains of K. africana.
Figure 3.

Evolutionary rearrangement at the K. africana MAT locus. Kazachstania africana (orange) has a MATa-like locus on chromosome 7 and a MATα-like locus on chromosome 4. It has no apparent HML or HMR loci, no Z or X repeats, and no HO endonuclease gene. For comparison, both idiomorphs (MATα and MATa) of the MAT locus in the related species K. naganishii are shown (blue). HXT indicates a cluster of hexose transporter genes.
One possible scenario to explain the rearrangement in K. africana is that a MAT chromosome broke into two pieces during an attempt to switch mating types. One piece, consisting of the right-hand part of the MAT chromosome (as drawn in Fig. 3) and the MATa1 gene, gained a new telomere to form K. africana chromosome 7. The other piece, consisting of the left-hand part of the MAT chromosome and the MATα genes, became fused end to end with another chromosome corresponding to K. naganishii chromosome 12 (Fig. 3). After this rearrangement occurred, the species was unable to switch mating types so the HML, HMR and HO loci all became unnecessary and were lost. Other scenarios, such as loss of HO before loss of HML and HMR and rearrangement of the MAT locus, are also plausible. Importantly, in the absence of any experimental data from K. africana it remains unclear how cell types are specified in this species, particularly whether MATa and MATα genes are both transcribed in haploid cells.
All species of Saccharomycetaceae contain homologs of the S. cerevisiae genes for mating pheromones (α-factor MFα and a-factor MFa) and their receptors (Ste2 and Ste3). The a-factor genes seem to be remarkably mobile: among 23 species we examined, MFa genes were found at 19 different genomic locations (Fig. 4A; see also OhEigeartaigh et al. 2011). Some of this diversity is due to high levels of gene duplication (for example N. castellii and T. blattae each have five MFa genes). The diversity of locations seen even in species with only one MFa gene may be due to cycles of gene duplication and loss from the original location. The S. cerevisiae MFA2 gene is at a position (Anc_2.114) that appears to be the ancestral MFa site for all post-WGD species and their close non-WGD relatives Z. rouxii and To. delbrueckii. Other non-WGD species show MFa genes at four different sites and it is unclear which of these sites is ancestral to the non-WGD clade (Fig. 4A). Notably, in many cases where an MFa gene has a location unique to a single species or genus, the location is also a site of rearrangement in that clade relative to the ancestral gene order (indicated by two ‘nearest ancestral’ genes in Fig. 4A). This suggests that the duplication of MFa genes may have occurred at the same time as the chromosomal rearrangement. The high mobility of MFa genes may be related to their small size, with a coding region of only ∼100 bp (OhEigeartaigh et al. 2011).
Figure 4.

Pheromone genes in Saccharomycetaceae species. Species named in red are non-WGD species. (A) MFa genes. Complete a-factor proteins sequences are aligned and arranged into groups of orthologs according to their genomic location. For each gene, the nearest ‘Anc’ gene in the ancestral numbering system (Gordon et al. 2009) is shown. Genes with two Anc numbers are located at points of rearrangement relative to the ancestral genome. Species marked with asterisks have only one MFa gene. (B) MFα genes. For each gene, the number of mature pheromone repeat units and the amino acid sequence of the most common repeat unit is shown.
In contrast to the MFa genes, genes for α-pheromone and the Ste2/Ste3 pheromone receptors show more sedate modes of evolution. The α-pheromone gene(s) code for precursor proteins containing one to eight repeats of an active peptide that is 13 amino acid residues long in most species (Fig. 4B). Exceptions to this pattern are Ashbya gossypii whose MFα gene (AAR163C) codes for a single copy of a 12-mer peptide that has been shown to be functional (Wendland, Dunkler and Walther 2011), and To. delbrueckii whose MFα gene (TDEL0G01600) appears to code for three copies of a 12-mer peptide (Fig. 4B). Only two duplications of MFα genes can be inferred, apart from the retention of two ohnologs of the gene at its ancestral location (Anc_6.185) in many species after WGD. One duplication produced a second copy in an ancestor of Naumovozyma species, and the other produced a second copy in an ancestor of the Eremothecium/Lachancea clade (Wendland, Dunkler and Walther 2011). Interestingly, neofunctionalization of extra copies of pheromone genes appears to have occurred, separately in A. gossypii and N. dairenensis, after these two duplications. In each case, a gene was formed (AFL062W and NDAI0F02280) that codes for an N-terminal secretion signal similar to a pheromone precursor protein, but the gene does not code for any pheromone-like repeats. The current functions of these genes are unknown. The STE2 and STE3 pheromone receptor genes are single copy in all species, except V. polyspora which retained two STE2 ohnologs after WGD, and they have not moved from their ancestral locations in any species.
Gene clusters
The DAL cluster of six genes, coding for the allantoin catabolism pathway, is the largest metabolic gene cluster in S. cerevisiae (Wong and Wolfe 2005; Naseeb and Delneri 2012). We previously showed that there is also a DAL cluster in N. castellii, but not in any non-WGD species, and that this cluster was assembled by relocation of genes after the WGD (Wong and Wolfe 2005). Naumovozyma dairenensis contains a DAL cluster with identical gene content and order to N. castellii. In K. naganishii, the cluster has expanded even more by the incorporation of a seventh gene, the allantoin transporter DAL5/KNAG0D03140, into the cluster. Seven-gene DAL clusters including DAL5 have also been found in Nakaseomyces bacillisporus and C. castellii (Gabaldon et al. 2013). The phylogenetic distribution of the seven-gene cluster, and the fact that DAL5 is at a different place in the clusters, suggests that DAL5 was recruited into the cluster by two separate events in the Kazachstania and Nakaseomyces genera.
The DAL cluster in K. naganishii is not located at a site orthologous to the DAL clusters in S. cerevisiae and N. castellii, but instead is at a site that corresponds to a point of rearrangement between an ancestral telomere (Anc_3.581) and an internal chromosomal site (Anc_4.55). In stark contrast to the K. naganishii cluster, the DAL genes are completely absent from the genomes of its congener K. africana and both of the Tetrapisispora species, as well as the previously reported absences from V. polyspora (Scannell et al. 2007b) and four species in the C. glabrata/Nakaseomyces clade (Gabaldon et al. 2013) (Fig. 1). Thus, the DAL genes are either tightly clustered in the genome or completely absent from the genome, in all post-WGD species, whereas they are scattered around the genome in all non-WGD species. It appears that the duplication of the gene pairs DAL7/MLS1 and DAL4/FUR4, which occurred as part of the WGD, facilitated a major reorganization of the DAL pathway, enabling both the formation of a cluster and multiple subsequent losses of that cluster.
Similar to the DAL genes, the GAL genes for galactose catabolism form a cluster in many yeast species but are completely absent in others (Hittinger, Rokas and Carroll 2004; Slot and Rokas 2010) (Fig. 1). When present in Saccharomycetaceae, this cluster is usually found in a conserved syntenic location corresponding to the position of S. cerevisiae GAL1/3, GAL7 and GAL10 (Anc_3.217–3.219). In contrast, we found that To. delbrueckii has no GAL genes at this ancestral location but instead has a large cluster of GAL genes near the telomere of chromosome 5 (Fig. 5A). The telomeric cluster spans 22 kb and contains two GAL1 genes (74% amino acid sequence identity), two GAL10 genes (79% identity) and one GAL7 gene. In addition, it contains one copy each of the genes GAL4 (transcription activator) and GAL2 (galactose permease) which do not form part of the cluster in other Saccharomycetaceae. The To. delbrueckii cluster also contains genes for additional enzymatic steps in the Leloir pathway: it has homologs of S. cerevisiae MEL1 (secreted alpha-galactosidase, which converts extracellular melibiose into galactose and glucose), and PGM1 (phosphoglucomutase, the glycolytic step immediately downstream of the Gal7 step). The cluster also has a homolog of K. lactis HGT1 (high-affinity glucose transporter; Billard et al. 1996). The 10-gene cluster therefore contains all the genes necessary for conversion of extracellular melibiose into glucose-6-phosphate. Although its telomeric location might suggest that the cluster was gained by horizontal gene transfer into To. delbrueckii, phylogenetic analysis does not provide clear support for this hypothesis (Fig. 5B); the To. delbrueckii genes are of Saccharomycetaceae origin and its GAL7 and GAL4 genes group with Zygosaccharomyces (which has a GAL1-GAL10-GAL7 cluster at the ancestral location) as expected in the absence of horizontal transfer. Torulaspora delbrueckii also contains a third GAL10 gene (TDEL0G04910) near another telomere; all three Gal10 proteins contain the fused epimerase and mutarotase domains typical of Saccharomycetaceae (Slot and Rokas 2010). The multiple GAL10 and GAL1 genes appear to have originated by duplications that occurred within the genus Torulaspora (Fig. 5B). It is also notable that To. delbrueckii has no homolog of GAL80 (an inhibitor of GAL4).
Figure 5.

(A) Large GAL gene cluster near the telomere of To. delbrueckii chromosome 5. (B) Phylogenetic trees of GAL1/GAL3, GAL7, GAL10 and GAL4 genes. Sequences from To. delbrueckii (TDEL) are highlighted in red. Sequences from Zygosaccharomyces (ZYRO—Z. rouxii and ZYBA—Z. bailii; Souciet et al. 2009; Galeote et al. 2013), the sister taxon to Torulaspora, are highlighted in blue. Amino acid sequences were aligned using Clustal Omega, filtered with Gblocks and trees were constructed using PhyML, all as implemented in Seaview (Gouy, Guindon and Gascuel 2010). Thin lines indicate branches with aLRT (approximate likelihood ratio test) support values below 80%. Trees were rooted using Pachysolen tannophilus (Ptan) GAL genes (Liu et al. 2012b).
RNAi pathway genes
There is no RNA interference pathway in S. cerevisiae, but there is one in N. castellii and V. polyspora (Drinnenberg et al. 2009). The primary actors in this pathway are Dicer and Argonaute proteins. The genes coding for these proteins have a patchy phylogenetic distribution (Fig. 1), and it has been proposed that the presence of an RNAi system is inversely correlated with the presence of double-strand RNA killer viruses (Drinnenberg, Fink and Bartel 2011). Argonaute and Dicer genes are present in only 1 species (To. delbrueckii) out of the 11 non-WGD Saccharomycetaceae species whose genomes have been sequenced, although they are present in C. albicans and many other outgroup fungi (Alexandersson and Sunnerhagen 2005). Among the post-WGD species, Argonaute is present in all studied species of two very divergent clades—the Vanderwaltozyma/Tetrapisispora clade and the Naumovozyma/Kazachstania clade. Argonaute has been lost in the Saccharomyces genus and in all studied species of the Nakaseomyces/C. glabrata clade (Fig. 1) with the sole exception of Na. bacillisporus. It is also of interest to note that two species retained both of the ohnolog copies of the Argonaute gene after the WGD, which suggests that some functional divergence or specialization within the RNAi pathway may have occurred in these species (N. dairenensis and T. blattae). The phylogenetic distribution of Dicer genes closely follows that of Argonaute, except that there are two species that retain Dicer but not Argonaute (C. castellii and S. uvarum, the sole member of the genus Saccharomyces that has any RNAi component).
Missing genes and pathways
Comparison of the genome sequences to the ancestral genome reveals some examples of losses of complete biochemical pathways or protein complexes. The most dramatic of these is the absence of the histidine biosynthesis pathway in T. blattae, which lacks six genes (HIS1, HIS2, HIS3, HIS4, HIS5 and HIS7) from this seven-gene pathway. Tetrapisispora blattae is known to be unable to grow on minimal media without histidine (Lachance 2011). The only gene it retains in the pathway is HIS6. It is possible that this auxotrophy reflects the natural environment of T. blattae because the only two known strains of this species were isolated from cockroaches, but it is unclear whether T. blattae is an intestinal symbiont of cockroaches or merely cockroach associated (Lachance 2011).
Almost all genes of the dynein/dynactin pathway (Winey and Bloom 2012), which in S. cerevisiae controls movement of the nucleus into daughter cells during budding, are missing in four species: V. polyspora (first reported by Scannell et al. 2007b), Na. delphensis, Na. bacillisporus and C. castellii. The 11 missing genes are DYN1 (=DHC1), DYN3, ARP1, ARP10, JNM1, NIP100, PAC1, PAC11, NDL1, LDB18 and KIP1. The latter two species are also missing a 12th gene, KIP2. The topology of the species tree (Fig. 1) shows that loss of 11–12 genes from this pathway has occurred independently three times. Some of the pathway components, particularly NDL1 and KIP1, have also been lost repeatedly in many other Saccharomycetaceae species without complete collapse of the pathway (Fig. 1).
Losses of BNA genes in the seven-gene pathway for de novo NAD synthesis have previously been reported in C. glabrata and other members of the Nakaseomyces genus (Domergue et al. 2005; Gabaldon et al. 2013). The BNA pathway is also missing in the genera Kluyveromyces, Kazachstania and Naumovozyma, and in T. blattae (Fig. 1). Most species that have lost the pathway retain BNA3. However T. blattae has lost BNA3 as well as the other six genes. Two species (N. dairenensis and C. castellii) retain BNA6 as well as BNA3, but have lost the other five genes. Candida castellii BNA6 is present at the gene's ancestral location (Anc_3.563), whereas N. dairenensis BNA6 has transposed to a site of species-specific genomic rearrangement. Phylogenetic analysis indicated accelerated evolution in both these BNA6 genes, but no evidence of horizontal gene transfer (data not shown). These gene distributions indicate that BNA1,2,4,5,7 have been lost on a minimum of four independent occasions (marked by ‘B’ in Fig. 1), and BNA6 has been lost at least six times, during Saccharomycetaceae evolution.
As previously noted (Gordon, Byrne and Wolfe 2011b), L. kluyveri lacks four genes essential for non-homologous end joining (NHEJ), a DNA repair pathway that is normally used to repair double-strand DNA breaks in situations where homologous recombination is not possible, such as in haploid cells. These genes are DNL4 (DNA ligase IV), POL4 (DNA polymerase IV), LIF1 (ligase interacting factor) and NEJ1 (a regulator of NHEJ) (Deshpande and Wilson 2007). The absence of these genes is possibly a factor in the low level of rearrangement seen in the L. kluyveri genome as compared to other Saccharomycetaceae (Gordon, Byrne and Wolfe 2011b). In contrast, the NHEJ pathway is intact in all 25 other species shown in Fig. 1, including two other Lachancea species.
In summary, the tree in Fig. 1 indicates that there have been multiple independent losses, in different clades, of many groups of functionally related genes: three independent losses of the DAL genes, four losses of GAL genes, four losses of BNA genes, one loss of HIS genes, five losses of the RNAi pathway, one loss of NHEJ and three losses of the dynein/dynactin complex. Almost every species in the tree has been affected by one or other of these losses, which are dramatic but evidently not catastrophic.
CONCLUSION
These examples of interspecies functional variation highlight the shortcomings of S. cerevisiae, or indeed any single species, as a model for the biology of a wider taxonomic clade such as the Saccharomycetaceae. The RNAi pathway serves as a case in point: the functions of the Argonaute and Dicer genes in N. castellii would never have been discovered if S. cerevisiae was the only model organism, no matter how intensively S. cerevisiae was studied. These genes were saved from the anonymity of being labeled as ‘conserved hypothetical’ genes because they had homologs in organisms such as Caenorhabditis and Schizosaccharomyces in which RNAi had already been discovered. By extension, it is very probable that some other important biochemical pathways, protein complexes and even biological processes that are widely conserved across many yeast species remain undiscovered because they are both (i) absent in the model organism S. cerevisiae, and (ii) yeast specific so their functions cannot be inferred from other eukaryotes.
Our understanding of Saccharomycetaceae genome evolution is also incomplete because for most clades except Saccharomyces we only have one genome sequence per species. Population genomics has revealed that there is extensive intraspecies polymorphism of gene content, particularly at telomeric regions, leading to the concept of the pan-genome as the complete set of genes that exists within a species even if no individual member of the species contains them all (Song et al. 2015). Saccharomyces kudriavzevii provides a spectacular example of intraspecies polymorphism, with the GAL genes being intact in Portuguese isolates but pseudogenized in Japanese isolates, the result of a balanced polymorphism at multiple separate genomic loci (Hittinger et al. 2010). It is possible that a similar situation of intraspecies presence/absence polymorphism could pertain to other sets of genes that appear to have multiple independent losses in Fig. 1, such as the DAL and BNA genes, and that we simply have not yet sampled enough individuals from the lineages showing apparent losses.
Just as the lab strain S288c has turned out to be an imperfect representative of the species S. cerevisiae, with unrepresentative alleles at loci such as FLO8 and MKT1 (Liu, Styles and Fink 1996; Lewis et al. 2014), the species S. cerevisiae is also an imperfect representative of the family Saccharomycetaceae. That does not however mean that any other species could be a better model organism. Instead, we may benefit by extending the concept of the pan-genome to higher taxonomic levels to describe the complete set of biological molecules, complexes and processes that exists within Saccharomycetaceae, even if no single species contains the whole set. Ideally, we would then like to ask the evolutionary reasons why particular parts of the pan-genome are missing in particular lineages. However, our ability to answer such questions is severely limited by the elephant in the room: our lack of knowledge about the natural environments in which the different yeast species have evolved and the niches (if any) to which they are adapted (Goddard and Greig 2015).
Acknowledgments
This paper is dedicated to the memory of our friend Jure Piškur, a pioneer of yeast comparative genomics.
FUNDING
Research in our group is supported by the European Research Council (Advanced Grant 268893) and Science Foundation Ireland (13IA1910).
Conflict of interest. None declared.
REFERENCES
- Albertin W, Chasseriaud L, Comte G, et al. Winemaking and bioprocesses strongly shaped the genetic diversity of the ubiquitous yeast Torulaspora delbrueckii. PLoS One. 2014;9:e94246. doi: 10.1371/journal.pone.0094246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandersson M, Sunnerhagen P. Comparative genomics and gene finding in fungi. In: Sunnerhagen P, Piskur J, editors. Comparative Genomics Using Fungi as Models. Berlin: Springer; 2005. [Google Scholar]
- Barsoum E, Martinez P, Astrom SU. Alpha3, a transposable element that promotes host sexual reproduction. GeneDev. 2010;24:33–44. doi: 10.1101/gad.557310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom A, Simpson JT, Salinas F, et al. A high-definition view of functional genetic variation from natural yeast genomes. Mol Biol Evol. 2014;31:872–88. doi: 10.1093/molbev/msu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billard P, Menart S, Blaisonneau J, et al. Glucose uptake in Kluyveromyces lactis: role of the HGT1 gene in glucose transport. J Bacteriol. 1996;178:5860–6. doi: 10.1128/jb.178.20.5860-5866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boundy-Mills K, Miller MW. Cyniclomyces van der Walt & D.B. Scott (1971) In: Kurtzman CP, Fell JW, Boekhout T, editors. The Yeasts, A Taxonomic Study. Vol. 2. Amsterdam: Elsevier; 2011. pp. 357–60. [Google Scholar]
- Butler G. Fungal sex and pathogenesis. Clin Microbiol Rev. 2010;23:140–59. doi: 10.1128/CMR.00053-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Kenny C, Fagan A, et al. Evolution of the MAT locus and its Ho endonuclease in yeast species. P Natl Acad Sci USA. 2004;101:1632–7. doi: 10.1073/pnas.0304170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes JK, Morris GP, Li WH. Reorganization of adjacent gene relationships in yeast genomes by whole-genome duplication and gene deletion. Mol Biol Evol. 2006;23:1136–43. doi: 10.1093/molbev/msj121. [DOI] [PubMed] [Google Scholar]
- Byrne KP, Wolfe KH. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 2005;15:1456–61. doi: 10.1101/gr.3672305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–6. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Cliften PF, Fulton RS, Wilson RK, et al. After the duplication: gene loss and adaptation in Saccharomyces genomes. Genetics. 2006;172:863–72. doi: 10.1534/genetics.105.048900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Wilson TE. Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair. 2007;6:1507–16. doi: 10.1016/j.dnarep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich FS, Voegeli S, Brachat S, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 2004;304:304–7. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- Domergue R, Castano I, De Las Penas A, et al. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science. 2005;308:866–70. doi: 10.1126/science.1108640. [DOI] [PubMed] [Google Scholar]
- Drinnenberg IA, Fink GR, Bartel DP. Compatibility with killer explains the rise of RNAi-deficient fungi. Science. 2011;333:1592. doi: 10.1126/science.1209575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Weinberg DE, Xie KT, et al. RNAi in budding yeast. Science. 2009;326:544–50. doi: 10.1126/science.1176945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–24. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Fay JC. The molecular basis of phenotypic variation in yeast. Curr Opin Genet Dev. 2013;23:672–7. doi: 10.1016/j.gde.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freel KC, Sarilar V, Neuveglise C, et al. Genome sequence of the yeast Cyberlindnera fabianii (Hansenula fabianii) Genome Announc. 2014;2:e00638-14. doi: 10.1128/genomeA.00638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich A, Jung P, Reisser C, et al. Population genomics reveals chromosome-scale heterogeneous evolution in a protoploid yeast. Mol Biol Evol. 2015;32:184–92. doi: 10.1093/molbev/msu295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Martin T, Marcet-Houben M, et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics. 2013;14:623. doi: 10.1186/1471-2164-14-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeote V, Bigey F, Devillers H, et al. Genome sequence of the food spoilage yeast Zygosaccharomyces bailii CLIB 213T. Genome Announc. 2013;1:e00606-13. doi: 10.1128/genomeA.00606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorello FM, Berna L, Greif G, et al. Genome sequence of the native apiculate wine yeast Hanseniaspora vineae T02/19AF. Genome Announc. 2014;2:e00530-14. doi: 10.1128/genomeA.00530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard MR, Greig D. Saccharomyces cerevisiae: a nomadic yeast with no niche? FEMS Yeast Res. 2015;15:fov009. doi: 10.1093/femsyr/fov009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, et al. Life with 6000 genes. Science. 1996;274:546, 563–7. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Gordon JL, Armisen D, Proux-Wera E, et al. Evolutionary erosion of yeast sex chromosomes by mating-type switching accidents. P Natl Acad Sci USA. 2011a;108:20024–9. doi: 10.1073/pnas.1112808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. Additions, losses and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009;5:e1000485. doi: 10.1371/journal.pgen.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011b;7:e1002190. doi: 10.1371/journal.pgen.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Haber JE. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics. 2012;191:33–64. doi: 10.1534/genetics.111.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedtke SM, Townsend TM, Hillis DM. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst Biol. 2006;55:522–9. doi: 10.1080/10635150600697358. [DOI] [PubMed] [Google Scholar]
- Hisatomi T, Kubota K, Tsuboi M. The autonomously replicating sequence (ARS) of the yeast Saccharomyces exiguus Yp74L-3. Curr Microbiol. 1999a;38:122–5. doi: 10.1007/s002849900414. [DOI] [PubMed] [Google Scholar]
- Hisatomi T, Takahashi K, Oomoto T, et al. Construction of an effective host-vector system for the yeast Saccharomyces exiguus Yp74L-3. Biosci Biotech Bioch. 1999b;63:847–50. doi: 10.1271/bbb.63.847. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Goncalves P, Sampaio JP, et al. Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature. 2010;464:54–8. doi: 10.1038/nature08791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Carroll SB. Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. P Natl Acad Sci USA. 2004;101:14144–9. doi: 10.1073/pnas.0404319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–24. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP. Phylogenetic circumscription of Saccharomyces, Kluyveromyces and other members of the Saccharomycetaceae, and the proposal of the new genera Lachancea, Nakaseomyces, Naumovia, Vanderwaltozyma and Zygotorulaspora. FEMS Yeast Res. 2003;4:233–45. doi: 10.1016/S1567-1356(03)00175-2. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP. Discussion of teleomorphic and anamorphic ascomycetous yeasts and yeast-like taxa. In: Kurtzman CP, Fell JW, Boekhout T, editors. The Yeasts, A Taxonomic Study. 5th edn. Vol. 2. Amsterdam: Elsevier; 2011. pp. 293–307. [Google Scholar]
- Kurtzman CP, Robnett CJ. Relationships among genera of the Saccharomycotina (Ascomycota) from multigene phylogenetic analysis of type species. FEMS Yeast Res. 2013;13:23–33. doi: 10.1111/1567-1364.12006. [DOI] [PubMed] [Google Scholar]
- Lachance MA. Tetrapisispora Ueda-Nishimura & Mikata emend. Kurtzman (2003) In: Kurtzman CP, Fell JW, Boekhout T, editors. The Yeasts, A Taxonomic Study. Amsterdam: Elsevier; 2011. pp. 859–66. [Google Scholar]
- Langkjaer RB, Cliften PF, Johnston M, et al. Yeast genome duplication was followed by asynchronous differentiation of duplicated genes. Nature. 2003;421:848–52. doi: 10.1038/nature01419. [DOI] [PubMed] [Google Scholar]
- Lewis JA, Broman AT, Will J, et al. Genetic architecture of ethanol-responsive transcriptome variation in Saccharomyces cerevisiae strains. Genetics. 2014;198:369–82. doi: 10.1534/genetics.114.167429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Li WH. Expansion of hexose transporter genes was associated with the evolution of aerobic fermentation in yeasts. Mol Biol Evol. 2011;28:131–42. doi: 10.1093/molbev/msq184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G. The fascinating and secret wild life of the budding yeast S. cerevisiae. Elife. 2015;4:e05835. doi: 10.7554/eLife.05835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Styles CA, Fink GR. Saccharomyces cerevisiae S288C has a mutation in FLO8, a gene required for filamentous growth. Genetics. 1996;144:967–78. doi: 10.1093/genetics/144.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WQ, Han PJ, Qiu JZ, et al. Naumovozyma baii sp. nov., an ascomycetous yeast species isolated from rotten wood in a tropical forest. Int J Syst Evol Micr. 2012a;62(Pt 12):3095–8. doi: 10.1099/ijs.0.044909-0. [DOI] [PubMed] [Google Scholar]
- Liu X, Kaas RS, Jensen PR, et al. Draft genome sequence of the yeast Pachysolen tannophilus CBS 4044/NRRL Y-2460. Eukaryot Cell. 2012b;11:827. doi: 10.1128/EC.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue ME, Wong S, Wolfe KH, et al. A genome sequence survey shows that the pathogenic yeast Candida parapsilosis has a defective MTLa1 at its mating type locus. Eukaryot Cell. 2005;4:1009–17. doi: 10.1128/EC.4.6.1009-1017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. P Natl Acad Sci USA. 2008;105:9272–7. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico A, Sulo P, Piskur J, et al. Fermentative lifestyle in yeasts belonging to the Saccharomyces complex. FEBS J. 2007;274:976–89. doi: 10.1111/j.1742-4658.2007.05645.x. [DOI] [PubMed] [Google Scholar]
- Naseeb S, Delneri D. Impact of chromosomal inversions on the yeast DAL cluster. PLoS One. 2012;7:e42022. doi: 10.1371/journal.pone.0042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuveglise C, Chalvet F, Wincker P, et al. Mutator-like element in the yeast Yarrowia lipolytica displays multiple alternative splicings. Eukaryot Cell. 2005;4:615–24. doi: 10.1128/EC.4.3.615-624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant KT, Wei W, Mancera E, et al. The baker's yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 2010;6:e1001109. doi: 10.1371/journal.pgen.1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhEigeartaigh SS, Armisen D, Byrne KP, et al. Systematic discovery of unannotated genes in 11 yeast species using a database of orthologous genomic segments. BMC Genomics. 2011;12:377. doi: 10.1186/1471-2164-12-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oro L, Zara S, Fancellu F, et al. TpBGL2 codes for a Tetrapisispora phaffii killer toxin active against wine spoilage yeasts. FEMS Yeast Res. 2014;14:464–71. doi: 10.1111/1567-1364.12126. [DOI] [PubMed] [Google Scholar]
- Piskur J, Rozpedowska E, Polakova S, et al. How did Saccharomyces evolve to become a good brewer? Trends Genet. 2006;22:183–6. doi: 10.1016/j.tig.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Polakova S, Blume C, Zarate JA, et al. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. P Natl Acad Sci USA. 2009;106:2688–93. doi: 10.1073/pnas.0809793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaei N, Chiruvella KK, Lin F, et al. Domesticated transposase Kat1 and its fossil imprints induce sexual differentiation in yeast. P Natl Acad Sci USA. 2014;111:15491–6. doi: 10.1073/pnas.1406027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozpedowska E, Piskur J, Wolfe K. Genome sequences of Saccharomycotina: Resources and applications in phylogenomics. In: Kurtzman CP, Boekhout T, Fell JW, editors. The Yeasts, A Taxonomic Study. Amsterdam: Elsevier; 2011. pp. 145–57. [Google Scholar]
- Sankoff D. Reconstructing the history of yeast genomes. PLoS Genet. 2009;5:e1000483. doi: 10.1371/journal.pgen.1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarilar V, Bleykasten-Grosshans C, Neuveglise C. Evolutionary dynamics of hAT DNA transposon families in Saccharomycetaceae. Genome Biol Evol. 2015;7:172–90. doi: 10.1093/gbe/evu273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Butler G, Wolfe KH. Yeast genome evolution—the origin of the species. Yeast. 2007a;24:929–42. doi: 10.1002/yea.1515. [DOI] [PubMed] [Google Scholar]
- Scannell DR, Frank AC, Conant GC, et al. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. P Natl Acad Sci USA. 2007b;104:8397–402. doi: 10.1073/pnas.0608218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DM, et al. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;458:342–5. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Andrea H, Blom J, et al. Draft genome sequence of Wickerhamomyces ciferrii NRRL Y-1031 F-60-10. Eukaryot Cell. 2012a;11:1582–3. doi: 10.1128/EC.00258-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Rupp O, Trost E, et al. Genome sequence of Wickerhamomyces anomalus DSM 6766 reveals genetic basis of biotechnologically important antimicrobial activities. FEMS Yeast Res. 2012b;12:382–6. doi: 10.1111/j.1567-1364.2012.00791.x. [DOI] [PubMed] [Google Scholar]
- Slot JC, Rokas A. Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. P Natl Acad Sci USA. 2010;107:10136–41. doi: 10.1073/pnas.0914418107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Dickins BJ, Demeter J, et al. AGAPE (Automated Genome Analysis PipelinE) for pan-genome analysis of Saccharomyces cerevisiae. PLoS One. 2015;10:e0120671. doi: 10.1371/journal.pone.0120671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souciet JL, Dujon B, Gaillardin C, et al. Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 2009;19:1696–709. doi: 10.1101/gr.091546.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara C, Hisatomi T, Kodama T, et al. The GAL10 gene is located 40 kbp away from the GAL7-GAL1 region in the yeast Kazachstania naganishii. Curr Microbiol. 2011;63:366–71. doi: 10.1007/s00284-011-9988-z. [DOI] [PubMed] [Google Scholar]
- Sunnerhagen P, Piškur J (eds.) Topics in Current Genetics. Vol. 15. Berlin: Springer; 2006. Comparative genomics using fungi as models. [Google Scholar]
- Tomita Y, Ikeo K, Tamakawa H, et al. Genome and transcriptome analysis of the food-yeast Candida utilis. PLoS One. 2012;7:e37226. doi: 10.1371/journal.pone.0037226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai IJ, Bensasson D, Burt A, et al. Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. P Natl Acad Sci USA. 2008;105:4957–62. doi: 10.1073/pnas.0707314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsong AE, Miller MG, Raisner RM, et al. Evolution of a combinatorial transcriptional circuit: a case study in yeasts. Cell. 2003;115:389–99. doi: 10.1016/s0092-8674(03)00885-7. [DOI] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, et al. Evolution of alternative transcriptional circuits with identical logic. Nature. 2006;443:415–20. doi: 10.1038/nature05099. [DOI] [PubMed] [Google Scholar]
- Vaughan-Martini A, Lachance MA, Kurtzman CP. Kazachstania Zubkova (1971) In: Kurtzman CP, Fell JW, Boekhout T, editors. The Yeasts, a Taxonomic Study. Vol. 2. Amsterdam: Elsevier; 2011. pp. 439–70. [Google Scholar]
- Wendland J, Dunkler A, Walther A. Characterization of alpha-factor pheromone and pheromone receptor genes of Ashbya gossypii. FEMS Yeast Res. 2011;11:418–29. doi: 10.1111/j.1567-1364.2011.00732.x. [DOI] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Winey M, Bloom K. Mitotic spindle form and function. Genetics. 2012;190:1197–224. doi: 10.1534/genetics.111.128710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–13. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Wong S, Wolfe KH. Birth of a metabolic gene cluster in yeast by adaptive gene relocation. Nat Genet. 2005;37:777–82. doi: 10.1038/ng1584. [DOI] [PubMed] [Google Scholar]
- Zarin T, Moses AM. Insights into molecular evolution from yeast genomics. Yeast. 2014;31:233–41. doi: 10.1002/yea.3018. [DOI] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, et al. Precise estimates of mutation rate and spectrum in yeast. P Natl Acad Sci USA. 2014;111:E2310–8. doi: 10.1073/pnas.1323011111. [DOI] [PMC free article] [PubMed] [Google Scholar]