

Abstract
Electrochemistry provides a powerful tool for the late-stage functionalization of complex lactams. A two-stage protocol for converting lactams, many of which are preparable through the intramolecular Schmidt reaction of keto azides, is presented. In the first step, anodic oxidation in MeOH using a repurposed power source provides a convenient route to lactams bearing a methoxy group adjacent to nitrogen. Treatment of these intermediates with a Lewis acid in DCM permits the regeneration of a reactive acyliminium ion that is then reacted with a range of nucleophilic species.
Keywords: Anodic oxidation, N-acyliminium ion, Lactam, Diversity-oriented synthesis
Late-stage functionalization of complex molecules is an important strategy in both natural product[1] and diversity-oriented synthesis (DOS) programs.[2] A body of creative work toward this end has been steadily building, including metal-mediated C–H activation chemistry,[3] photochemical methods,[4] and electrochemical oxidation reactions.[5] In particular, the application of electrochemistry in organic synthesis has benefited from advances such as cation pool[6] and flow chemistry techniques,[7] Here, we introduce a simple and inexpensive way of carrying out electrochemical oxidations and demonstrate its utility in diversifying polycyclic lactams.
We have developed useful routes to complex lactams using the intramolecular azido-Schmidt reaction, such as the Diels–Alder/Schmidt sequence affording 1a in Scheme 1.[8] Having adapted this chemistry to diversity-oriented synthesis by using the ketone as a fulcrum for analog synthesis,[9] we felt that a useful alternative would be to generate analogs by modifying the normally unreactive amide linkage. Specifically, we imagined that converting the lactam products into acyliminium ions like 2 would lead to broadly useful intermediates for downstream manipulation. Since many of the chemical oxidants traditionally used for such oxidations,[10] such as ceric ammonium nitrate and dichlorodicyanoquinone, are highly toxic or poorly compatible with other functional groups, we considered electrochemical oxidation as an attractive alternative.[11] Operationally, these reactions are carried out in methanol, where anodic oxidation leads to an iminium ion that is trapped by solvent, e.g., 2 → 1b. Having stored the higher oxidation state as 1b, regeneration of 2 can be effected by treatment with a Lewis or protic acid in a non-participating solvent, where it can be trapped by another nucleophile.
Scheme 1.

Strategy for the modification of polycyclic lactams.
Similar electrochemical oxidations have involved proline or pyrrolidinone derivatives, often in the context of peptidomimetic synthesis.[5, 12] In contrast, we are aware of only two examples where bicyclic lactams were used as substrates.[13] Accordingly, a primary goal of the present project was to show the utility of this approach in more complex settings. An important secondary goal was to develop an accessible anodic oxidation method for mainstream laboratory use. In addition to some of the aforementioned efforts toward real-world electrochemistry, Moeller has used 6-volt lantern batteries connected in series or photovoltaic cells for organic electrochemical transformations,[14] and Boydston and coworkers demonstrated the organocatalyzed anodic oxidation of aldehydes to esters powered by D-cell batteries.[15] Here, we report the design and construction of an improvised device for undivided cell electrochemistry using a mobile phone recharger.
We first assembled a simple electrochemical setup repurposing a cell phone charger as the DC power source (Figure 1). Such power sources with different voltage and current outputs are ubiquitous in our technology-driven society. These power sources are sold as accessories with most portable electronic devices and typically outlive the useful life of the device itself. In many cases, simply connecting the output wires from the power source to the electrodes in the reaction cell is all that is necessary to fabricate a useful electrochemical setup. If the current output from the power source is higher than desired, it can be reduced by connecting resistors to the circuit in series, as shown in Figure 1a. Other convenient modifications are the attachment of the lead wires to alligator clips, allowing easier connection to the electrodes (Figure 1b) and the use of #7 mechanical pencil lead refills as electrodes (Figure 1c). This last example has the advantages of further removing the need for any specialized supplies and more importantly, the small diameter electrodes allow for microscale electrochemical oxidations (reaction volumes < 1 mL). CAUTION: we recommend common-sense precautions when preparing similar devices to avoid electric shock (see Supporting Information for best practice precautions in carrying out these reactions).
Figure 1.

Improvised electrochemical devices: (a) initial 30 mA prototype device, (b) alternate 800 mA device, and (c) microscale set-up using # 7 pencil leads as electrodes. See Supporting Information for larger photographs and details for the fabrication and use of these devices.
We first confirmed the ability of the DC power source to perform known preparative electrochemistry by reproducing the known electrochemical oxidation of the proline derivative 3a[14c] to give 3b as well as the acyclic amide 4a to give 4b (Table 1, entries 1 and 2, respectively). Our initial experiments were conducted using a 6 V, 30 mA power source with later experiments conducted with a 5.2 V, 800 mA power source. Note that while the rate of electron flow (current) varied, both power sources had a voltage output significantly greater than the typical 1.95 to 2.10 V (vs. Ag/AgCl) oxidation potential of the amide or lactam.[16] Having validated the improvised device on model substrates we turned our attention to more complex amide substrates, readily available by azide methods developed in these laboratories.
Table 1.
Electrochemical Oxidation of Amidesa
| entry | substrate | product | yield, % (ratiob) |
|---|---|---|---|
| 1 |
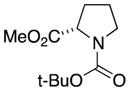 3a |
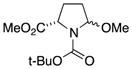 3b |
93 (1:1) |
| 2 |
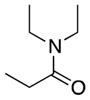 4a |
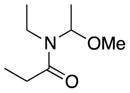 4b |
57 |
| 3 |
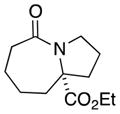 5a |
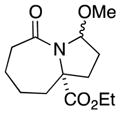 5b |
91 (4:1) |
| 4 |
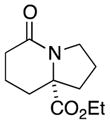 6a |
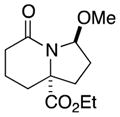 6b |
63 (single isomer) |
| 5 |
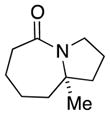 7a |
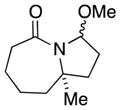 7b |
56 (1.7:1) |
| 6 |
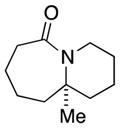 8a |
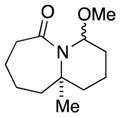 8b |
28 (1.5:1) |
| 7 |
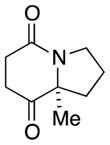 9a |
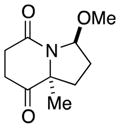 9b |
47 (single isomer) |
| 8 |
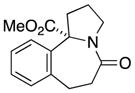 10a |
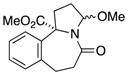 10b |
40 (single isomer) |
| 9 |
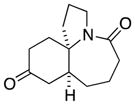 1a |
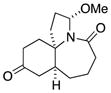 1b |
78 (single isomer) |
| 10 |
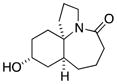 11a |
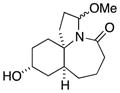 11b |
65 (2.8:1) |
| 11 |
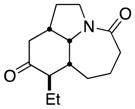 12a |
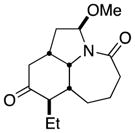 12b |
19 (single isomer) |
Conditions: MeOH, undivided cell, C anode/cathode, Et4NOTs or LiClO4.
Ratios approximated by 1H NMR; except where shown, diastereomeric structures were not determined.
In general, the results in Table 1 confirm the utility of this electrochemical oxidation across a range of ring systems. Entries 7 and 8 show that ketone or phenyl groups are tolerated in the electrochemical oxidation. Entries 9 and 10 extend the scope of the method to nonaromatic tricyclic lactam scaffolds. As expected, a diastereomeric mixture of methoxy amide products were obtained in all examples. In one case, reaction of a lactam containing more than one adjacent position with abstractable hydrogens belied a limitation of this method (Table 1, entry 11). Such substrates suffer from competing reactions between potential reactive sites and also the formation of overoxidized products, as previously observed for the electrochemical oxidation of lactams.[13] Thus, the product shown in entry 11 of Table 1 was isolated as a single isomer in 19% yield, while the mass balance was a complex and inseparable mixture of other isomers and side products. Moreover, we observed similar product mixtures when this reaction was performed using the fully-regulated constant current electrochemical setup traditionally used.
The versatility of these methoxyamides for the synthesis of an array of functionalized products was illustrated by the addition of various nucleophiles to the N-acyliminium ion generated in situ from either methoxy amide 5b or 7b (Scheme 2). Highlights from Scheme 2 include the Friedel-Crafts addition of aromatic compounds (derivative 16), butenolide or indole side chain introduction (derivatives 14 and 15), the use of boronic acids as nucleophiles (derivative 18) and addition of cuprate reagents such as the phenyl acetylide (derivative 20). The cuprate addition was unsuccessful under several different conditions using methoxy amide substrate 5b, but proceeded smoothly using the alkyl substrate 7b. We note here that the stereoselectivity of these substitutions depend on the intrinsic face selectivity of the particular scaffold. Thus, while 5b and 7b do not exhibit strong bias, the literature is replete with examples of highly selective additions to acyliminium ions[17] (for another example, see Scheme 4 below). As proposed in Scheme 1, the more complex methoxy amide 1b was readily converted to the N-acyliminium ion 2 and converted to the allyl derivative 21.
Scheme 2.

Diversification pathways for methoxy amides 5b or 7b (a) dimethyl malonate, Et3N, TiCl4; (b) 2-trimethylsilyloxyfuran, TiCl4; (c) N-methylindole, TiCl4; (d) 1,3,5-trimethoxybenzene, SnCl4; (e) TiCl4; Et3N; (f) thiopheneboronic acid; BF3•OEt2 (g) TiCl4; trimethylsilyl allyl silane (h) TiCl4; PhCCMgCuBr.
Scheme 4.

Synthesis and functionalization of lactam 27.
This methodology is attractive for target-oriented synthesis as well, isofar as amide or lactam intermediates are stable species able to survive numerous chemical conditions likely to be encountered in multistep synthesis. To demonstrate these, we targeted the derivatization of tricyclic lactam 27, a core skeleton we explored for the synthesis of pinnaic acid and related natural products (Figure 3).[18] Moreover, the recent disclosure that the related derivative 24 possessed potential anti-cancer properties[19] suggests that the tricyclic motif itself could serve as a scaffold for biologically relevant analogs. En route to the formal synthesis of pinnaic acid, we previously reported the selective synthesis of 22 in a 10:1 ratio over 23.[20] The trivially accessible and previously unreported lactam 27 would provide a blank canvas for the introduction of diversity as exemplified in Scheme 4.
Figure 3.
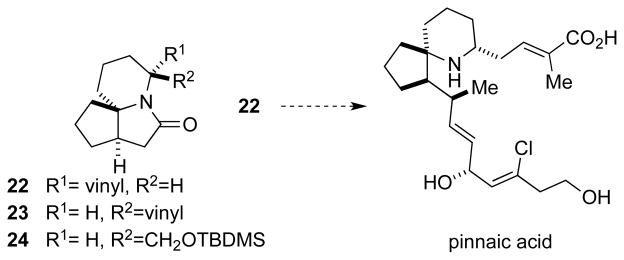
Tricyclic lactam pinnaic acid precursor and analogs.
Thus, the known acid 25, derived in a single step from commercially-available hept-6-enoic acid, underwent a ketene-mediated [2+2] cycloaddition to afford cyclobutanone 26 in 85% yield.[21] Subsequent azide displacement and intramolecular Schmidt reaction achieved the synthesis of tricyclic lactam 27 in 93% yield. The N-acyliminium ion intermediate was generated utilizing our electrochemistry apparatus and trapped by methanol as the methoxy amide 28. Subsequent allylation of the crude product gave lactam 29 as the sole observed product in 56% yield over two steps. The high stereoselectivity most likely arises from top attack of the nucleophile to the more stable conformation A (as opposed to the more strained B) in the N-acyliminium ion chair-like transition state (Scheme 4). The allyl derivative could be utilized directly as a handle to introduce functionality or additional nucleophiles could be introduced via the methoxyamide intermediate as illustrated in Scheme 1.
In summary, we have constructed a simple, improvised device for undivided cell electrochemistry. We have demonstrated how this device can enable the synthesis of novel lactam derivatives via N-acyliminium ion diversification and extended this chemistry to lactams of unprecedented complexity. We believe that this apparatus would be a useful addition to the standard methods available to synthetic organic chemists by providing a simple, reliable power source of sufficient voltage to carry out a variety of electrochemical transformations.[22]
Supplementary Material
Scheme 3.
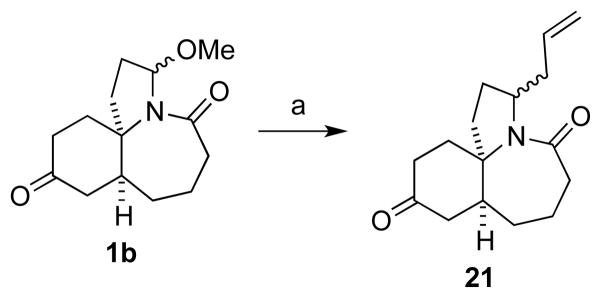
Substitution of methoxy amide 1b. (a) TiCl4; trimethylsilyl allyl silane.
Footnotes
We acknowledge the National Institute of General Medical Sciences (PO50-GM069663) for financial support, Victor Day for X-ray crystallography (NSF-MRI grant CHE-0923449), Hai-Chao Xu of the Moeller research group for instruction in electrochemistry techniques and Cady Bush for photographic assistance.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Kevin J. Frankowski, Department of Medicinal Chemistry, University of Kansas, 2034 Becker Drive, Lawrence, Kansas, 66047 (USA), Fax: (+1)785-864-8179
Dr. Ruzhang Liu, Department of Medicinal Chemistry, University of Kansas, 2034 Becker Drive, Lawrence, Kansas, 66047 (USA), Fax: (+1)785-864-8179
Prof. Dr. Gregory L. Milligan, Department of Chemistry, Saint Martin’s University, 5000 Abbey Way, Lacey, WA 98503
Prof. Dr. Kevin D. Moeller, Department of Chemistry, Washington University in St. Louis, St. Louis, MO 63130 (USA)
Prof. Dr. Jeffrey Aubé, Email: jaube@ku.edu, Department of Medicinal Chemistry, University of Kansas, 2034 Becker Drive, Lawrence, Kansas, 66047 (USA), Fax: (+1)785-864-8179
References
- 1.a) Shimokawa J. Tetrahedron Lett. 2014;55:6156–6162. [Google Scholar]; b) Sears JE, Boger DL, Dale L. Acct Chem Res. 2015 doi: 10.1021/ar500400w. [DOI] [Google Scholar]
- 2.a) O’Connor CJ, Beckmann HSG, Spring DR. Chem Soc Rev. 2012;41:4444–4456. doi: 10.1039/c2cs35023h. [DOI] [PubMed] [Google Scholar]; b) Serba C, Winssinger N. Eur J Org Chem. 2013:4195–4214. [Google Scholar]; c) Rizzo S, Waldmann H. Chem Rev. 2014;114:4621–4639. doi: 10.1021/cr400442v. [DOI] [PubMed] [Google Scholar]
- 3.a) Wencel-Delord J, Droege T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; b) Chen DYK, Youn SW. Chem Eur J. 2012;18:9452–9474. doi: 10.1002/chem.201201329. [DOI] [PubMed] [Google Scholar]; c) Schranck J, Tlili A, Beller M. Angew Chem Int Ed Engl. 2014;53:9426–9428. doi: 10.1002/anie.201405714. [DOI] [PubMed] [Google Scholar]
- 4.a) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; b) Prier CK, Rankic DA, MacMillan DWC. Angew Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vorontsov AV, Arsentyev AV. Curr Org Chem. 2013;17:2459–2481. [Google Scholar]
- 5.Reviews: Moeller KD. Tetrahedron. 2000;56:9527–9554.Sperry JB, Wright DL. Chem Soc Rev. 2006;35:605–621. doi: 10.1039/b512308a.Yoshida J-i, Kataoka K, Horcajada R, Nagaki A. Chem Rev. 2008;108:2265–2299. doi: 10.1021/cr0680843.Francke R. Beil J Org Chem. 2014;10:2858–2873. doi: 10.3762/bjoc.10.303.For some notable recent examples of oxidative electrochemistry in organic synthesis, see: Nokami T, Hayashi R, Saigusa Y, Shimizu A, Liu C-Y, Mong K-KT, Yoshida J-i. Org Lett. 2013;15:4520–4523. doi: 10.1021/ol402034g.Gao WJ, Li WC, Zeng CC, Tian HY, Hu LM, Little RD. J Org Chem. 2014;79:9613–9618. doi: 10.1021/jo501736w.Kabeshov MA, Musio B, Murray PRD, Browne DL, Ley SV. Org Lett. 2014;16:4618–4621. doi: 10.1021/ol502201d.Rosen BR, Werner EW, O’Brien AG, Baran PS. J Am Chem Soc. 2014;136:5571–5574. doi: 10.1021/ja5013323.Shoji T, Kim S, Yamamoto K, Kawai T, Okada Y, Chiba K. Org Lett. 2014;16:6404–6407. doi: 10.1021/ol503198p.Ding H, DeRoy PL, Perreault C, Larivée A, Siddiqui A, Caldwell CG, Harran S, Harran PG. Angewandte Chemie. 2015;127:4900–4904. doi: 10.1002/anie.201411663.Waldvogel SR, Möhle S. Angew Chem Int Ed. 2015;54:6398–6399. doi: 10.1002/anie.201502638.
- 6.Yoshida J-i, Ashikari Y, Matsumoto K, Nokami T. J Synth Org Chem, Jpn. 2013;71:1136–1144. [Google Scholar]
- 7.a) Watts K, Baker A, Wirth T. J Flow Chem. 2014;4:2–11. [Google Scholar]; b) Kabeshov MA, Musio B, Murray PRD, Browne DL, Ley SV. Org Lett. 2014;16:4618–4621. doi: 10.1021/ol502201d. [DOI] [PubMed] [Google Scholar]
- 8.Aubé J, Milligan GL. J Am Chem Soc. 1991;113:8965–8966.Milligan GL, Mossman CJ, Aubé J. J Am Chem Soc. 1995;117:10449–10459.For reviews, see: Lang S, Murphy JA. Chem Soc Rev. 2006;35:146–156. doi: 10.1039/b505080d.Nyfeler E, Renaud P. Chimia. 2006;60:276–284.Grecian S, Aubé J. In: Organic Azides: Syntheses and Applications. Bräse S, Banert K, editors. John Wiley & Sons Ltd; 2010. pp. 191–237.Wrobleski A, Aaron, Coombs TC, Huh CW, Li S-W, Aubé J. Org React. 2012;17:1–320.
- 9.a) Frankowski KJ, Neuenswander B, Aubé J. J Comb Chem. 2008;10:721–725. doi: 10.1021/cc800078h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Frankowski KJ, Setola V, Evans JM, Neuenswander B, Roth BL, Aubé J. Proc Natl Acad Sci USA. 2011;108:6727–6732. doi: 10.1073/pnas.1016558108. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McLeod MC, Singh G, Plampin JN, III, Rane D, Wang JL, Day VW, Aubé J. Nat Chem. 2014;6:133–140. doi: 10.1038/nchem.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Murahashi S, Naota T, Kuwabara T, Saito T, Kumobayashi H, Akutagawa S. J Am Chem Soc. 1990;112:7820–7822. [Google Scholar]; b) Zhang Y, Li CJ. Angew Chem Int Ed. 2006;45:1949–1952. doi: 10.1002/anie.200503255. [DOI] [PubMed] [Google Scholar]
- 11.Jones AM, Banks CE. Beil J Org Chem. 2014;10:3056–3072. doi: 10.3762/bjoc.10.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Shono T, Hamaguchi H, Matsumura Y. J Am Chem Soc. 1975;97:4264–4268. [Google Scholar]; b) Shono T, Matsumura Y, Tsubata K. J Am Chem Soc. 1981;103:1172–1176. [Google Scholar]; c) Asada S, Kato M, Asai K, Ineyama T, Nishi S, Izawa K, Shono T. J Chem Soc, Chem Commun. 1989:486–488. [Google Scholar]; d) Barrett AGM, Pilipauskas D. J Org Chem. 1991;56:2787–2800. [Google Scholar]; e) Moeller KD. Tetrahedron. 2000;56:9527–9554. [Google Scholar]; f) D’Oca MGM, Pilli RA, Vencato I. Tetrahedron Lett. 2000;41:9709–9712. [Google Scholar]
- 13.a) Mori M, Higuchi Y, Kagechika K, Shibasaki M. Heterocycles. 1989;29:853–856. [Google Scholar]; (b) Mori M, Uozumi Y, Ban Y. J Chem Soc, Chem Commun. 1986:841–842. [Google Scholar]; (c) Mori M, Kimura M, Uozumi Y, Ban Y. Tetrahedron Lett. 1985;26:5947–5950. [Google Scholar]
- 14.a) Frey DA, Wu N, Moeller KD. Tetrahedron Lett. 1996;37:8317–8320. [Google Scholar]; b) Anderson LA, Redden A, Moeller KD. Green Chem. 2011;13:1652–1654. [Google Scholar]; c) Duan S, Moeller KD. Tetrahedron. 2001;57:6407–6415. [Google Scholar]
- 15.Finney EE, Ogawa KA, Boydston AJ. J Am Chem Soc. 2012;134:12374–12377. doi: 10.1021/ja304716r. [DOI] [PubMed] [Google Scholar]
- 16.Sun H, Martin C, Kesselring D, Keller R, Moeller KD. J Am Chem Soc. 2006;128:13761–13771. doi: 10.1021/ja064737l. [DOI] [PubMed] [Google Scholar]
- 17.a) Martin SF. Acc Chem Res. 2002;35:895–904. doi: 10.1021/ar950230w. [DOI] [PubMed] [Google Scholar]; b) Speckamp WN, Moolenaar MJ. Tetrahedron. 2000;56:3817–3856. [Google Scholar]; c) Mizuta S, Onomura O. RSC Adv. 2012;2:2266–2269. [Google Scholar]
- 18.Clive DLJ, Yu M, Wang J, Yeh VSC, Kang S. Chem Rev. 2005;105:4483–4514. doi: 10.1021/cr0406209. [DOI] [PubMed] [Google Scholar]
- 19.Robbins D, Newton AF, Gignoux C, Legeay JC, Sinclair A, Rejzek M, Laxon CA, Yalamanchili SK, Lewis W, O’Connell MA, Stockman RA. Chem Sci. 2011;2:2232–2235. [Google Scholar]
- 20.Liu R, Gutierrez O, Tantillo DJ, Aubé J. J Am Chem Soc. 2012;134:6528–6531. doi: 10.1021/ja300369c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Collins MR, Mahmood K, Dowd P. Tetrahedron Lett. 1995;36:2729–2732. [Google Scholar]
- 22.CCDC 1403517 and CCDC 1403516 contain the supplementary crystallographic data for compounds 14 and 16. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.