

Abstract
A series of chemically modified 7-phenylpyrrolo[3,2-f] quinolinones was synthesized and evaluated as anticancer agents. Among them, the most cytotoxic (subnanomolar GI50 values) amidic derivative 5f was shown to act as an inhibitor of tubulin polymerization (IC50, 0.99 μM) by binding to the colchicine site with high affinity. Moreover, 5f induced cell cycle arrest in the G2/M phase of the cell cycle in a concentration dependent manner, followed by caspase-dependent apoptotic cell death. Compound 5f also showed lower toxicity in nontumoral cells, suggesting selectivity toward cancer cells. Additional experiments revealed that 5f inhibited the enzymatic activity of multiple kinases, including AURKA, FLT3, GSK3A, MAP3K, MEK, RSK2, RSK4, PLK4, ULK1, and JAK1. Computational studies showed that 5f can be properly accommodated in the colchicine binding site of tubulin as well as in the ATP binding clefts of all examined kinases. Our data indicate that the excellent antiproliferative profile of 5f may be derived from its interactions with multiple cellular targets.
INTRODUCTION
Cancer is a leading cause of disease worldwide, accounting for 12.7 million new cases every year, and this number is expected to rise to 26 million by 2030. Considering the impact on human health and economics, cancer presents a major challenge to the scientific world, and there is a necessity to discover new agents for the treatment of this disease. Most of the drugs in preclinical development are represented by small molecules. In 2013, for example, 9 out of 10 new cancer drugs launched on the market were small molecules.1
Currently, combined anticancer therapies or multitargeted drugs are preferred over traditional cytotoxic treatment, with the aim to overcome resistance and toxicity drawbacks. These events often prevent successful treatment and are responsible for reduced survival times.2 In the field of chemotherapeutics, the widely used antitubulin agents exhibit significant cytotoxicity by inhibiting microtubule dynamics. There are several structurally dissimilar small molecules with high affinity for the colchicine site on tubulin able to inhibit the proliferation of a wide variety of human cancer cells. Moreover, these agents also affect the tumor endothelial vasculature required for the growth of tumor mass. These types of tubulin inhibitors might provide new therapeutic approaches to treat cancers and overcome limitations of existing tubulin interactive drugs.3
Our previously described 2- and 7-phenylpyrrolo-quinolinones (2- and 7-PPyQs) (Figure 1) are a class of antiproliferative agents acting as tubulin polymerization inhibitors by binding at the colchicine site of β-tubulin.4,5 Members of the less cytotoxic 2-PPyQ family were also found to exhibit interesting in vitro and in vivo antiangiogenic properties.6
Figure 1.
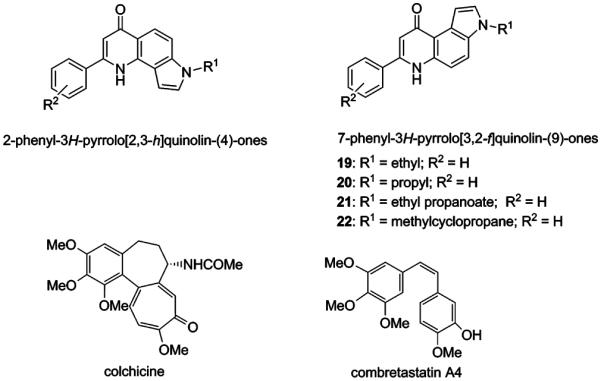
General chemical structures of 2-PPyQs, 7-PPyQs, the previous 7-PPyQs 19–22, and the known antitubulin agents colchicine and combretastatin A4.
The first generation of 7-PPyQs showed interesting in vitro biological properties. Some derivatives had micromolar GI50 values and good antitumor activity in vivo.5 The second generation, characterized by alkyl substitutions at the pyrrolic nitrogen, showed increased cytotoxicity with nanomolar GI50 values, and these compounds overcame cross resistance observed with the clinically used agents vincristine and paclitaxel.7 In the latter series, the 3N-cyclopropylmethyl 7-PPyQ derivative (22, MG 2477) was taken as lead compound due to its very strong cytotoxicity (nanomolar range GI50 values) and its potent interaction with tubulin. Its activities as an inhibitor of tubulin polymerization and of colchicine binding to tubulin were similar to those of the reference compound combretastatin A-4 (CA-4, Figure 1) (0.90 μM assembly IC50 and 83% inhibition of colchicine binding for 22 versus values of 1.1 μM and 99%, respectively, for CA-4).7
In an effort to produce additional highly active compounds, numerous related analogues were synthesized, and the effects of a variety of substitutions at diverse positions of the 3H-pyrrolo[3,2-f] quinolin-9-one nucleus (PyQ) were evaluated.8 In the present study, a number of new 7-PPyQ derivatives were designed, synthesized, and evaluated in cellular cytotoxicity and tubulin inhibition assays, resulting in the discovery of potent amidic derivatives. Furthermore, our recent study indicated that 22 is highly effective in reducing cell viability, and the reduced survival of A549 cells is associated with an initial autophagy that may be mediated by inhibition of the PI3K/Akt/mTOR pathway.9 This pathway plays a variety of physiological roles, including regulation of cell growth and proliferation, and is perhaps the most frequently disregulated pathway in human cancers conferring aggressiveness and poor prognosis.10,11 These findings prompted us to determine whether the new 7-PPyQ derivatives also interfere with the PI3K/Akt pathway and exert kinase inhibitory activity.
RESULTS AND DISCUSSION
Chemistry
The general method leading to the novel 3-substituted 7-PPyQ compounds was previously reported7 and consists of four steps as described in Scheme 1A. First, commercially available 5-nitroindole (1) was subjected to an N-alkylation or acylation reaction using appropriate halogenated compounds to obtain the nitroindole derivatives 2a–f. The same reaction conditions were used with all chloro and bromo compounds and with ethyl acrylate in anhydrous DMF in the presence of NaH at room temperature, resulting in high yields of the reaction products. The catalytic reduction (Pd/C 10%, H2 at atmospheric pressure, ethyl acetate) of intermediates 2a–d gave aminoindoles 3a–d in almost quantitative yields. In the case of 2f (and for 2e not shown) the catalytic procedure furnished the partially reduced derivative 6f as described in Scheme 1B. To obtain fully aromatic aminoindoles 3e and 3f, a chemical reduction with SnCl2 in methanol at reflux was necessary but resulted in lower final yields. The condensation reactions of aminoindoles 3a and 3c–f with ethyl benzoylacetate and of 3b with ethyl dimethoxybenzoylacetate were carried out in absolute ethanol at reflux and yielded the acrylate derivatives 4a–f as crude material, which first had to be purified by silica gel column chromatography and then submitted to thermal cyclization in boiling diphenyl ether (250 °C) to obtain 5a–f. Cyclization products 5a–f were further purified by recrystallization or column chromatography, with their purity verified by HPLC (>95%). In Scheme 1B, 6f obtained by means of catalytic reduction of the nitro compound 2f was reacted with ethyl benzoylacetate (7f) and thermally cyclized to the partially hydrogenated 7-PPyQ 8f.
Scheme 1. Synthetic Routes to PPyQs 5a–f (A) and 8f (B)a.

aReagents and conditions: (a) NaH 60%, bromoethyl alcohol, cyclopropylmethyl bromide, 3-bromopropionitrile, ethyl chloroacetate, cyclopropylcarbonyl chloride, n-propionyl chloride, anhydrous DMF, rt, 3 h, 47–99%; (b) H2, Pd/C 10%, ethyl acetate, 50 °C, 4–14 h, 90–98%; (c) SnCl2/HCl, CH3OH, reflux, 1.5 h, 47–80%; (d) ethyl benzoylacetate or ethyl dimethoxybenzoylacetate, absolute ethanol, reflux 15 h, 37–87%; (e) diphenyl ether, 250 °C, 10–15 min, 30–76%.
As shown in Scheme 2, the chemical transformations of the two previously reported 3-ethyl 7-PPyQs (19),6 ethyl 3N-propanoate (21)7 and of some 7-PPyQs described in Scheme 1 are shown. In Scheme 2A, 19, when reacted with CH3I, C4H9Br or NO2C6H5CH2Br in anhydrous DMF and in the presence of NaH, yielded the respective mixed ethers 9–11 as the only reaction products, bearing the alkoxylic function in the 9 position. Similarly, 19 reacted with benzylchloroformate and NaH in methanol at 0 °C to furnish the 9-substituted carbonate ester 12 as the only reaction product. In Scheme 2B, in order to obtain compound 13, the 6N-methyl derivative of 19, important for our SAR analysis, the 9-methoxy-PPyQ 9 was refluxed with excess CH3I for 2 h. This reaction provided the intermediate quinolinium iodide, which was not isolated but yielded 13 by treatment of the reaction mixture with 1 N NaOH at reflux.12 The exact structures with the precise alkylation or acylation sites in 9–13 were obtained by 1D and 2D NMR HMBC experiments (Figures 2–4 in Supporting Information).
Scheme 2. Synthesis of 7-PPyQs 9–18a.

a Reagents and conditions: (a) CH3 I or Br(CH2)3CH3, BrCH2C6H5NO2, NaH, DMF, rt, 2 h, 64–74%; (b) C6H5CH2OCOCl, NaH, DMF, 0 °C, 2 h, 96%; (c) CH3I, reflux; (d) NaOH 1 N, reflux, 18%; (e) LiAlH4, anhydrous THF, rt, 3 h, 70%; (f) NaOH 1 N, methanol, reflux, 2 h, 85%; (g) NaBH4, CoCl2·6H2O, di-tert-butyl dicarbonate, methanol, 10 h, 77%; (h) HCl dry gas, ethyl acetate, 58%.
As shown in Scheme 2C, the ethyl 3N-propanoate 218 was reduced with LiAlH4 in anhydrous THF to yield the corresponding alcohol 14. Alternatively, 21 underwent alkaline hydrolysis in 1 N NaOH in methanol to provide the corresponding carboxylic acid 15. Similarly, 5d yielded acid 16 (Scheme 2D).
As shown in Scheme 2E, the 3N-propanenitrile 5c, prepared as described in Scheme 1, was reduced with NaBH4 in the presence CoCl2 catalyst in methanol and in the presence of an equimolar quantity of di-tert-butyl dicarbonate. This somewhat laborious procedure13 was superior to others14,15 because the tBoc protected 7-PPyQ amine 17 was obtained as a solid product, which was more easily isolated from the reaction mixture than the free amine. Compound 17 was dissolved in an ethyl acetate/methanol mixture, and excess HCl dry gas was bubbled through the mixture to yield the hydrochloride compound 18 as a beige powder. 1D and 2D NMR spectrometry and absorption spectroscopy indicated that compound 18 was in the form of 9-hydroxypyrroloquinoline (Figures 7–10 in Supporting Information). We speculate that the hydroxylic form was obtained because of the treatment with HCl gas (see Experimental Section). This was confirmed through an investigation of the behavior of compound 18 in aqueous solution as a function of pH. On the basis of 1H NMR and UV–vis spectra, it was observed that the two quinolinonic and hydroxyquinolinic tautomers interconvert in a pH dependent way: above pH 4 the keto tautomer predominates, while below pH 2 the enolic one is dominant (Figure 28 in Supporting Information). In the pH range 4–9, the spectra did not change, indicating that the compound was in the keto tautomer. Thus, at physiologic pH all 7-PPyQs, including compound 18, are in the keto tautomer, with a carbonyl group at position 9 as previously proposed from the SAR findings.8
Biological Characterization
Antiproliferative Activity in Cellular Assays and SAR Analysis
On the basis of previous biological activity data on 7-PPyQs7,10 and docking simulations of 22 in the colchicine site of tubulin,9 the new compounds were designed to obtain additional SAR information by modifying the nature and size of substituents at the 3, 6, 7, and 9 positions of the PyQ tricycle. Evaluation of antiproliferative activities of 5a–f, 8–16, and 18 was performed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay16 against a panel of five solid tumor (HeLa, A549, HT-29, MCF-7, OVCAR-3) and nine leukemia cell lines (MOLT-3, CCRF-CEM, HL-60, K562, RS4;11, Jurkat, SEM, MV4;11, THP-1). GI50 values, the concentrations that inhibit cell growth by 50%, are presented in Table 1. Most of the novel 7-PPyQs possessed antiproliferative activity, inhibiting cell growth with nanomolar to micromolar GI50 values), except for the ethers 10 and 11 and the acids 15 and 16. Of particular note were the subnanomolar GI50 values obtained with 5f, but also with 5e and 12, in selected leukemic cell lines.
Table 1.
Growth Inhibitory Activity of PPyQ derivatives 5a–f, 8f, 9–16, and 18–22 for Human Cancer Solid Tumor (A) and Leukemic Cell Lines (B)
| (A) GI50 (nM)a |
|||||||||
| compd | HeLa | A549 | HT-29 | MCF-7 | OVCAR-3 | ||||
|
| |||||||||
| 5a | 1563 ± 135 | 7756 ± 1126 | 236 ± 16 | 256 ± 31 | 9863 ± 1236 | ||||
| 5b | 2856 ± 736 | 7456 ± 3601 | >10000 | >10000 | >10000 | ||||
| 5c | 191 ± 72 | 928 ± 125 | 356 ± 61 | 82 ± 21 | 326 ± 17 | ||||
| 5d | 2456 ± 231 | 4785 ± 432 | 34589 ± 119 | 2863 ± 61 | >10000 | ||||
| 5e | 5 ± 2 | 78 ± 4 | 12 ± 8 | 3 ± 1 | 12 ± 5 | ||||
| 5f | 1 ± 0.6 | 9 ± 0.4 | 1 ± 0.5 | 5 ± 1 | 71 ± 12 | ||||
| 8f | 38 ± 0.5 | 145 ± 78 | 15 ± 8 | 19 ± 8 | 243 ± 51 | ||||
| 9 | 111 ± 51 | 45 ± 9 | 1456 ± 623 | 1863 ± 921 | 32 ± 9 | ||||
| 10 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
| 11 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
| 12 | 15 ± 1 | 21 ± 5 | 18 ± 8 | 211 ± 20 | 9 ± 2 | ||||
| 13 | 936 ± 41 | 463 ± 65 | 923 ± 221 | 1656 ± 632 | 1236 ± 258 | ||||
| 14 | 175 ± 12 | 311 ± 125 | 48 ± 7 | 95 ± 16 | 38 ± 8 | ||||
| 15 | 5563 ± 55 | >10000 | >10000 | >10000 | >10000 | ||||
| 16 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
| 18 | 551 ± 5 | 1072 ± 119 | 1569 ± 62 | 389 ± 112 | 2136 ± 369 | ||||
| 19 b | 11 ± 8 | 32 ± 15 | 32 ± 12 | 45 ± 11 | 32 ± 11 | ||||
| 20 b | 2 ± 0.9 | nd | 5±1 | 2 ± 1 | nd | ||||
| 21 c | 452 ± 102 | nd | 492 ± 85 | 589 ± 189 | nd | ||||
| 22 b | 21 ± 12 | 21 ± 2 | 61 ± 1 | 42 ± 15 | 14 ± 8 | ||||
|
| |||||||||
| (B) GI50 (nM)a |
|||||||||
| compd | MOLT-3 | CCRF-CEM | HL-60 | K562 | RS4;11 | Jurkat | SEM | MV4;11 | THP-1 |
|
| |||||||||
| 5a | 252 ± 53 | 4705 ± 2122 | 254 ± 11 | 9±1 | 74 ± 32 | 845 ± 19 | 161 ± 33 | 311 ± 42 | 2850 ± 918 |
| 5b | 5850 ± 2301 | >10000 | 1324 ± 351 | 6936 ± 432 | 275 ± 171 | 2450 ± 323 | 452 ± 61 | 1986 ± 232 | 4632 ± 126 |
| 5c | 131 ± 33 | 392 ± 161 | 274 ± 42 | nd | 83 ± 42 | 1905 ± 832 | 92 ± 12 | 323 ± 6 | 49 ± 9 |
| 5d | 201 ± 67 | >10000 | 2415 ± 152 | 5213 ± 1265 | 263 ± 105 | 2211 ± 453 | 1650 ± 221 | 2305 ± 1123 | >10000 |
| 5e | 5.1 ± 1.3 | 2.1 ± 0.9 | 2.5 ± 0.5 | nd | 1 ± 0.5 | 0.4 ± 0.2 | 9 ± 2 | 36 ± 2 | 125 ± 32 |
| 5f | 2.3 ± 1.4 | 17 ± 4 | 2.0 ± 0.6 | 6 ± 2 | 0.1 ± 0.05 | 0.3 ± 0.03 | 0.4 ± 0.1 | 19 ± 8 | 74 ± 25 |
| 8f | 145 ± 52 | 232 ± 35 | 172 ± 5 | 82 ± 10 | 5 ± 1 | 14 ± 3 | 19 ± 2 | 513 ± 112 | 355 ± 72 |
| 9 | 163 ± 44 | 115 ± 42 | 27 ± 3 | 21 ± 9 | 3 ± 0.1 | 18 ± 2 | 23 ± 6 | 37 ± 13 | 34 ± 12 |
| 10 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 11 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 12 | 8.5 ± 3.2 | 18 ± 5 | 3 ± 0.5 | 3 ± 0.8 | 0.5 ± 0.2 | 0.4 ± 0.06 | 1 ± 0.1 | 4 ± 0.8 | 1 ± 0.6 |
| 13 | 145 ± 21.2 | 526 ± 123 | 145 ± 21 | 1142 ± 532 | 231 ± 61 | 30 ± 2 | 623 ± 212 | 1569 ± 521 | 821 ± 236 |
| 14 | 21.5 ± 11.6 | 405 ± 78 | 29 ± 6 | 78 ± 12 | 8 ± 2 | 8 ± 3 | 14 ± 3 | 78 ± 12 | 408 ± 51 |
| 15 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 16 | >10000 | >10000 | 2375 ± 372 | >10000 | 7756 ± 1231 | >10000 | >10000 | >10000 | >10000 |
| 18 | 515 ± 45 | 2423 ± 581 | 951 ± 51 | nd | 82 ± 11 | 482 ± 75 | 158 ± 51 | 492 ± 35 | 271 ± 18 |
| 19 | nd | nd | 0.5 ± 0.02 | 1 ± 0.1 | 2 ± 0.3 | 0.5 ± 0.2 | nd | nd | nd |
| 20 | nd | nd | nd | nd | 0.6 ± 0.4 | 0.9 ± 0.3 | nd | nd | nd |
| 21 | nd | nd | 2.0 ± 0.1 | 196 ± 61 | 2 ± 0.4 | 0.5 ± 0.01 | nd | nd | nd |
| 22 | nd | nd | 6.1 ± 0. 5 | 1 ± 0.4 | 2 ± 0.5 | 0.5 ± 0.03 | nd | nd | nd |
Overall, the most active of the new compounds was 5f (average GI50, 15 nM). It frequently had lower GI50 values than the previously described compounds 19–22. Three additional compounds also had average GI50 values of <150 nM: 5e (22 nM), 12 (22 nM), and 14 (120 nM). GI50 values below 100 nM in at least three cells lines were also observed with compounds 5c, 8f, and 9. In an attempt to razionalize structure–antiproliferative activity relationships, the 7-PPyQs mentioned here and the corresponding previously described active 3-alkyl derivatives were ordered into four subsets on the bases of length, chemical nature, and size of the various substitutions, as depicted in Scheme 3. The first three subsets were selected by taking in consideration the substitutions at position 3, in comparison with previously described potent 3N-alkyl substituted 7-PPyQs, compounds 19 (3-ethyl), 20 (3-propyl), and 22 (3-cyclopropylmethyl).7 The fourth subset includes compounds obtained by modifying the 9 position of compound 19 (9-C=O) to confirm the requirement for the carbonyl as a hydrogen bond acceptor in the active site on tubulin. Furthermore, the 6 position of 19 was methylated to gain SAR information on the 6 position and the phenyl at the 7 position was replaced by a dimethoxyphenyl moiety to determine the importance of the size and nature of the aryl substituent at that position.
Scheme 3. Subsets of 7-PPyQ Derivatives Prearranged for the Structure–Antiproliferative Activity Relationships Discussion.
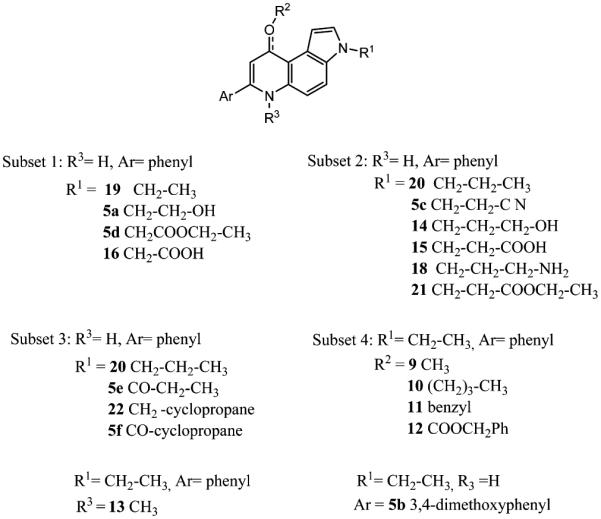
From a comparison of the first two subsets and their GI50 values (Table 1), cytotoxicity changes with the lipophilic character of the chain at the 3 position: the shorter and more polar is the chain, the weaker is the cytotoxicity, with the acids 15 and 16 being completely inactive.
In the third subset, an important chemical modification was introduced, consisting of an amidic connection between the indole ring and the side chain, to yield the amidic derivatives 5e and 5f. This chemical modification provided compounds with potent antiproliferative activities, comparable with the activity obtained with parent 22. As noted above, these two compounds, along with compound 12, had the strongest antiproliferative activity. The fourth subset includes analogues of compound 19 designed and synthesized in order to evaluate substitutions at the 9 position. Previously, we had stressed the essential role of the carbonyl moiety for the antitubulin activity of 7-PPyQs in comparison with the corresponding phenylpyrroloquinoline lacking this moiety,7 but we had not evaluated the effect of other substituents at position 9. The substituents in 9, 10, and 11, lacking a carbonyl moiety, had reduced or minimal antiproliferative activity as compared with 19. Moreover, as expected, the GI50 progressively decreased with steric hindrance in comparing the methyl derivative 9 with 10 and 11 with still bulkier groups. However, the bulkiest 9-benzyl carbonate derivative 12 was found to be a highly cytotoxic agent because it probably decomposed to 19 in the intracellular environment. Indeed, it is stable as a solid, while in dilute aqueous solution it quickly decomposes to the corresponding phenylpyrroloquinolinone (as proved by HPLC). Moreover, this degradation is in agreement with the data obtained with tubulin (Table 3) that show compound 12 to be as strong an inhibitor as its parent compound 19.
Table 3.
Inhibition of Tubulin Polymerization and Colchicine Binding by Compounds 5a–f, 8f, 9, 12–22, and CA–4
| compd | inhibition of tubulin assembly IC50 ± SD (μM)a,c |
inhibition of colchicine binding inhibition ± SD (%)b,c |
|---|---|---|
| 5a | 2.9 ± 0.1 | 11 ± 4 |
| 5b | >20 | nd |
| 5c | 1.3 ± 0.05 | 20 ± 1 |
| 5d | 6.6 ± 0.3 | nd |
| 5e | 0.99 ± 0.1 | 52 ± 5 |
| 5f | 0.99 ± 0.1 | 69 ± 3 |
| 8f | 2.6 ± 0.2 | 30 ± 4 |
| 9 | 5.7 ± 0.7 | nd |
| 12 | 0.74 ± 0.07 | 77 ± 2 |
| 13 | 1.8 ± 0.3 | 61 ± 1 |
| 14 | 1.2 ± 0.1 | 40 ± 0.9 |
| 15 | 8.0 ± 0.1 | nd |
| 16 | >20 | nd |
| 18 | >20 | nd |
| 19 d | 0.57 ± 0.02 | 73 ± 0.7 |
| 20 d | 0.58 ± 0.05 | 77 ± 3 |
| 21 e | 0.75 ± 0.04 | 75 ± 4 |
| 22 d | 0.90 ± 003 | 83 ± 0.5 |
| CA-4 | 1.2 ± 0.1 | 99 ± 0.06 |
To obtain data regarding the 6 position of the 7-PPyQs, we synthesized and evaluated compound 13 by attaching a methylic group at the quinolinic nitrogen of 19. This led to a compound with reduced antiproliferative activity with respect to the unsubstituted parent compound 19. Finally, in order to get further information about the size and nature of the pocket in tubulin with which the phenyl at position 7 interacts, 5b was designed and synthesized. In this compound the unsubstituted 7-phenyl group of 19 was replaced by a dimethoxyphenyl group. This compound was relatively inactive, leading us to conclude that the bulkier substituent led to a compound with a weaker interaction with tubulin. This is in agreement with what was previously observed with the series of 3-unsubstituted 7-PPyQs.5
We therefore conclude that the 7-PPyQ molecule tolerates very few chemical modifications and that for significant cytotoxicity only the 3-N position allows for a limited variety of substitutions. Substituents at this position require appropriate characteristics of size and polarity, and 3N-acylation yielded the highly active amidic compounds 5e and 5f. Due to its broad spectrum of potent antiproliferative activity, 5f was selected for further biological investigations on mechanism of action and for comparison with 22.
Evaluation of Antiproliferative Activity of 5f in Nontumor Cells
We investigated the effect of 5f and of 22 in human lymphocytes and human umbilical vein endothelial cells (HUVECs), isolated from healthy donors. As shown in Table 2, in both unstimulated and mitogen-activated lymphocytes, the two compounds showed low toxicity as compared with their potent activity in cancer cells. Somewhat greater cytotoxicity was observed in HUVECs when incubated with both compounds. In both cases the GI50 values of 7.8 (22) and 22.1 (5f) μM are significantly higher than the values obtained in the panel of tumor cells. These results suggest that both compounds may have a preferential selectivity toward cancer cells, especially 5f.
Table 2.
Cytotoxicity of 5f in Human Noncancer Cells
| GI50 (μM)a |
||
|---|---|---|
| cell line | 5f | 22 |
| PBLrestingb | >100 | 77.6 ± 20.0 |
| PBLPHAc | 95.7 ± 8.9 | 5.0 ± 0.9 |
| HUVEC | 22.1±10.9 | 7.8 ± 0.55 |
Compound concentration required to reduce cell growth by 50%. Values are the mean ± SEM for three separate experiments.
PBL not stimulated with PHA.
PBL stimulated with PHA.
5f Binds to the Colchicine Site of Tubulin and Inhibits Tubulin Assembly
In previous studies we found that the antiproliferative activity of other 7-PPyQs resulted from an interaction with tubulin at the colchicine site.17–20 Thus, the compounds of this new series were evaluated for their inhibition of tubulin polymerization and for their ability to inhibit [3H]colchicine binding to tubulin. For comparison, CA-4, a drug candidate in clinical trials in its phosphorylated form, was evaluated as a reference compound in simultaneous experiments (Table 3). CA-4 is a well described, highly potent competitive inhibitor of the binding of colchicine to tubulin.17 In the assembly assay,18 compound 5f was found to be the most active of the new agents as an inhibitor of tubulin polymerization, with an IC50 (0.99 μM) comparable with that of both 22 (0.90 μM) and CA-4 (1.20 μM). In the [3H]colchichine binding assay,19 compound 5f was the most active new derivative (69% inhibition at 5 μM), although this activity was somewhat less than that observed with both 22 and CA-4 (83% and 99% inhibition, respectively, at 5 μM). There was good correlation between inhibition of tubulin polymerization, inhibition of colchicine binding, and antiproliferative activity of the new derivatives, in particular since 5f had the highest activity in all three assays. Note that compound 12 behaved similarly to 19, which is in a good correlation with antiproliferative activity data. Of the compounds examined with tubulin, somewhat decreased activity was observed with 13 and 14 with respect to 5f. These compounds inhibited assembly with IC50 values of 1.8 and 1.2 μM, respectively. These compounds were also less active as inhibitors of colchichine binding than 5f and CA-4, 61% and 40% inhibition, respectively.
As previously described for 22, to further examine the competitive binding of 5f with colchicine in the active site of the 1SA0 structure, computer-based docking of 5f was performed using the MOE-Dock program.21,22 Figure 2 depicts the binding mode of 5f in the presumptive colchicine site. In particular, the colchicine site is mostly buried in the intermediate domain of the β-subunit, even if colchicine can also interact with loop T5 of the neighboring α-subunit (Figure 2), consistent with the observation that colchicine stabilizes the tubulin heterodimer. Not surprisingly, the accommodation of 5f in the colchicine cleft is similar to that previously described for 22.9 Also in this case, docking simulations showed that, similar to colchicine and 22, 5f also can be accommodated in the same hydrophobic cleft, adopting an energetically stable conformation. Moreover, the most stable conformation of 5f reproduced the scheme of chemical interactions (predominantly through hydrophobic interactions with Val181, Ala250, Cys241, Ala316, Val318, and Ile378 of the β-subunit) observed for colchicine, together with a similar interaction with the bordering T5 loop of α-tubulin, demonstrating a plausible competitive mechanism of action at the colchicine site.
Figure 2.
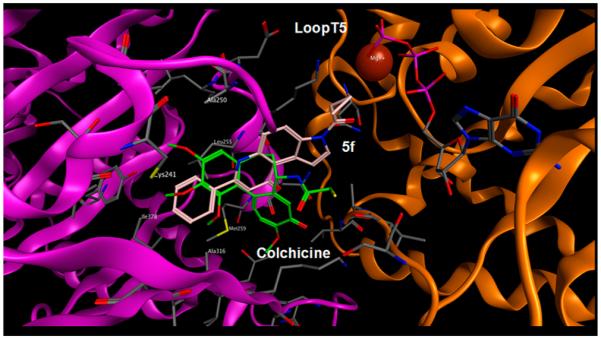
Comparison of the crystallographic structure of colchicine (in green) in complex with tubulin (Protein Data Bank code 1SA0) and the energetically most favorable pose of 5f (in pink) obtained by molecular docking simulation. Hydrogen atoms are omitted.
5f Induces Cell Cycle Arrest at the G2/M Phase of the Cell Cycle
The effect of 5f on cell cycle progression was examined by flow cytometry in the HeLa, Jurkat, and RS4;11 cell lines after a 24 h treatment with difierent concentrations of the agent (Figure 3). For all cell lines, the treatment resulted in the accumulation of cells in the G2/M phase of the cell cycle. In particular, the increase of G2/M cells began at 30 nM and was concentration dependent until a plateau was reached at 60 nM. In the three cell lines, G2/M arrest was accompanied by a concomitant reduction in the proportion of cells in the G1 phase. Only in HeLa cells was a significant decrease of S phase cells observed.
Figure 3.
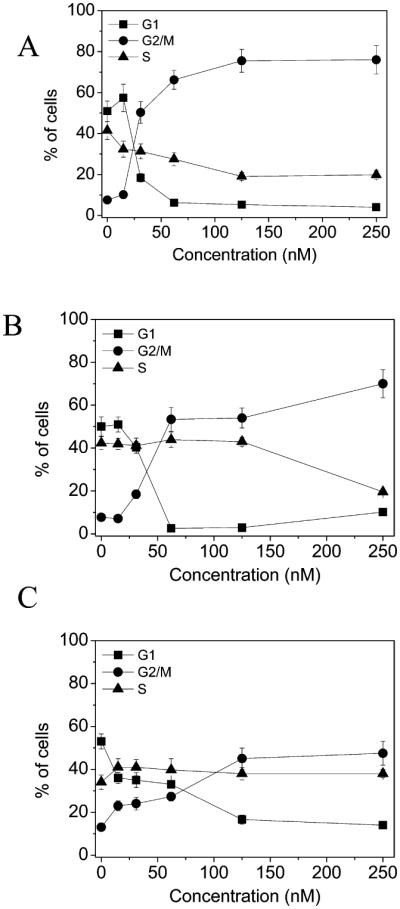
Percentage of cells in each phase of the cell cycle in HeLa (A), RS4;11 (B), and Jurkat (C) cells treated with 5f at the indicated concentrations for 24 h. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the Experimental Section. Data are represented as the mean ± SEM of three independent experiments.
We next studied the association between 5f-induced G2/M arrest and alterations in the expression of proteins that regulate cell division. The cdc2/cyclin B complex controls both entry into and exit from mitosis. Phosphorylation of cdc2 on Tyr15 and phosphorylation of cdc25C phosphatase on Ser216 negatively regulate the activation of the cdc2/cyclin B complex. Thus, dephosphorylation of these proteins is needed to activate the cdc2/cyclin B complex. Cdc25C is a major phosphatase that dephosphorylates the site on cdc2 and autodephosphorylates itself. Phosphorylation of cdc25C directly stimulates both its phosphatase and autophosphatase activities, a condition necessary to activate cdc2/cyclin B on entry of cells into mitosis.23–25 As shown in Figure 4, in Jurkat cells treatment with 5f at either 0.1 or 1 μM caused no significant variations in cyclin B expression after a 24 h treatment. However, we observed a marked decrease in the expression of the phosphorylated form of cdc2 (Tyr15) and that of cdc25C, in particular with 1 μM 5f. These data, along with accumulation of cells in the G2/M phase, suggest that 5f-induced G2/M arrest was not due to defects in G2/M regulatory proteins but rather is closely linked with acceleration of entry into mitosis.
Figure 4.
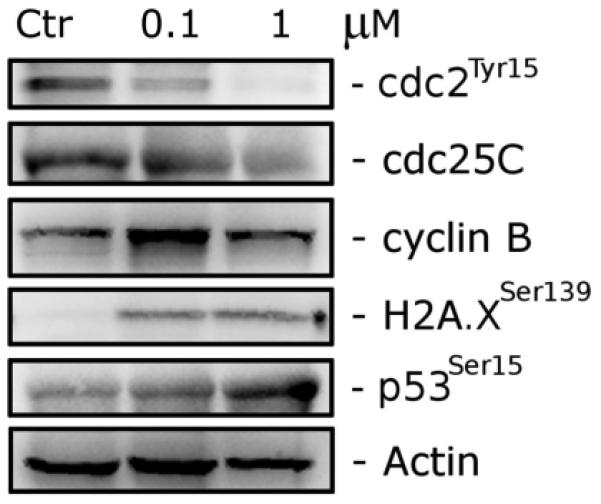
Effect of 5f on G2/M regulatory proteins and DNA-damage marker proteins. Jurkat cells were treated for 24 h with the indicated concentration of 5f. The cells were harvested and lysed for the detection of cyclin B, p-cdc2Tyr15, cdc25C, H2A.XSer139, and p53Ser15 expression by Western blot analysis. To confirm equal protein loading, each membrane was stripped and reprobed with an anti-β-actin antibody.
In addition to the analysis of proteins that control cell cycle checkpoints, we also examined the phosphorylation of the tumor suppressor p53 at Ser15 after treating Jurkat cells with 5f. It is well-known that prolonged mitotic arrest induces DNA damage and, consequently, the phosphorylation of p53 at Ser15 that leads to p53 stabilization and accumulation.26,27 As shown in Figure 4, we detected a concentration-dependent marked increase of p53-Ser15 that is particularly evident with 1 μM 5f. At the same time, we also observed a marked increase in the phosphorylation of histone γH2A.X at Ser139, which is an early sensitive indicator of DNA damage.28
5f Induces Apoptotic Cell Death
To characterize the mode of cell death induced by 5f, biparametric flow cytometric analysis with annexin-V/propidium iodide (PI) was performed on difierent cell lines. PI binds to DNA and is permeable only in dead, nonapoptotic cells, while annexin-V has high affinity for phosphatidylserine (PS) that is exposed only on the outer membrane leaffet of apoptotic cells. Thus, the dual staining permits quantitation of live cells (annexin-V−/PI−), early apoptotic cells (annexin-V+/PI−), late apoptotic cells (annexin-V+/PI+), and necrotic cells (annexin-V−/PI+). As shown in Figure 5, 5f induced an increase in the annexin V+ fraction, and thus of apoptosis, in a concentration dependent manner in HeLa cells (Figure 5A) and in leukemic cell lines such as Jurkat (Figure 5B), RS4;11 (Figure 5C), and K562 (Figure 5D).
Figure 5.

Flow cytometric analysis of apoptotic cells after treatment of HeLa (A), Jurkat (B), RS4;11 (C), and K562 (D) cells with 5f at the indicated concentrations after incubation for 24 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are represented as the mean ± SEM of three independent experiments.
To evaluate which caspases were involved in the apoptotic cell death induced by 5f, we analyzed Jurkat cell protein extracts by immunoblot. After a 24 h treatment with 5f, we observed the cleavage of activator caspases 2, 8, and 9 (Figure 6A). Moreover, we observed also the activation of the effector caspase-3 and the subsequent cleavage of its substrate PARP.
Figure 6.
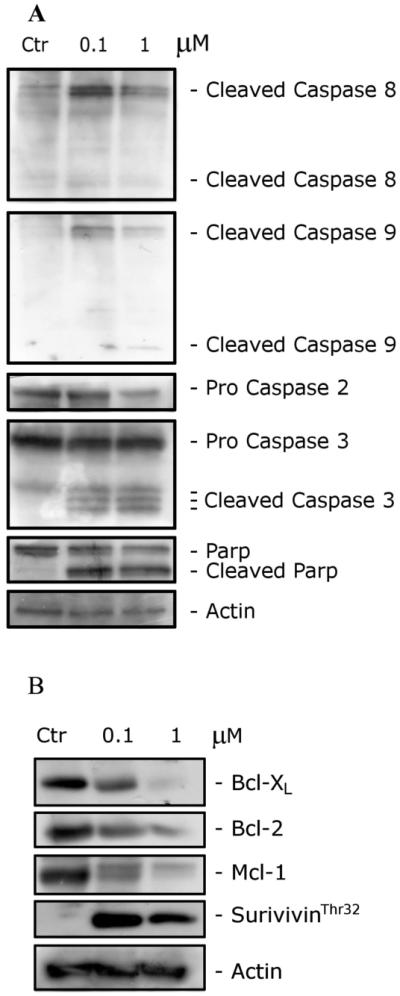
(A). Western blot analysis of caspase-8, caspase-9, caspase-2, caspase-3, and PARP after a 24 h treatment of Jurkat cells with 5f at the indicated concentrations. (B) Western blot analysis of Bcl-XL, Bcl-2, survivinThr32, and Mcl-1 after treatment of Jurkat cells with 5f at the indicated concentrations for 24 h. To confirm equal protein loading, each membrane was stripped and reprobed with an anti-β-actin antibody.
The intracellular apoptotic program can be modulated by the expression of Bcl-2 family proteins that include both proapoptotic and antiapoptotic members. Bcl-2 family proteins are the main regulators of the mitochondrial apoptotic pathway through the control of mitochondrial membrane permeability and release of cytochrome c.29 It is well-known that cancer cells express high levels of Bcl antiapoptotic proteins such as Bcl-2, Bcl-XL, Mcl-1, Bcl-w, Boo/Diva, Bcl-B, and NR-13 and that microtubule targeting agents induce the downregulation of the Bcl-2 antiapoptotic protein, thus promoting apoptosis.30,31 As shown in Figure 6B in Jurkat cells, after a 24 h incubation, compound 5f induced a significant decrease in Bcl-2, Bcl-XL, and Mcl-1 expression at the lower concentration (0.1 μM) examined. These results are in agreement with recent studies that point out the importance of Mcl-1 in cell death induced by antitubulin agents.32 In addition, Mcl-1 appears to play a major role in cancer cell survival, especially in leukemia cells. Thus, this observation with 5f suggestes that the agent could be an attractive pharmacological tool for induction of Mcl-1 depletion.33,34
Survivin is a member of IAP family (inhibitor of apoptosis protein).35 In general, the IAPs function through direct interactions to inhibit the activity of several caspases, including caspase-3, caspase-7, and caspase-9, and IAPs thereby inhibit the processing and activation of these enzymes.36 We found that survivin was phosphorylated on Thr32 following treatment with 5f. This effect is consistent with cell cycle arrest in mitosis and is shared by various antimitotic drugs.37,38
5f Induces Inhibition of the PI3K/Akt/mTOR Pathway
Recently, we identified 22, a structural analogue of 5f, as a potent antimitotic compound that might interfere with the PI3K/Akt/mTOR pathway.9 Therefore, we evaluated the effect of compound 5f on this pathway in Jurkat cells, which are a PTEN-null, T-cell acute lymphoblastic leukemia cell line. As shown in Figure 7, 5f induced the reduction of the p85 regulatory subunit of PI3K and caused the decrease of the phosphorylated (active) forms of mTOR (Ser2448) and AKT (Ser473, Thr308). 5f treatment also induced a decrease in the phosphorylation of mTOR downstream targets such as S6 kinase (Ser240/244), 4E-BP1 (Ser65), and GSK3 (Ser21).
Figure 7.
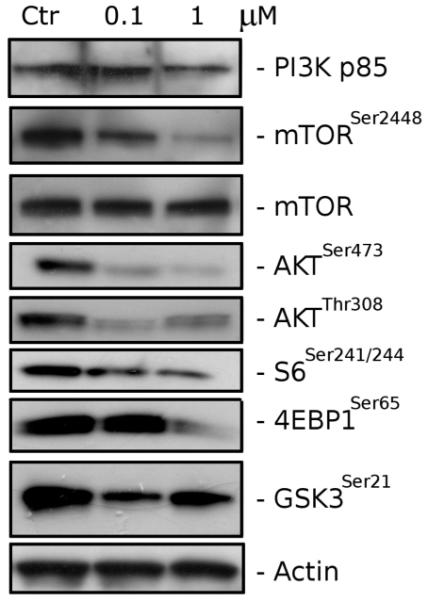
Western blot analysis of PI3K/AKT/mTOR after treatment of Jurkat cells with 5f at the indicated concentrations for 24 h. To confirm equal protein loading, each membrane was stripped and reprobed with an anti-β-actin antibody.
5f Inhibits Enzymatic Activity of Difierent Protein Kinases
Because of the significant effect of 5f on the PI3K/Akt/mTOR pathway, we performed DiscoveRx KINOMEscan39 profiles on a panel of 386 distinct human protein kinases at 1 μM 5f (Figure 8). The percent inhibition at this concentration is represented with red dots (Figure 8A) that, depending on their size, indicate the residual activity of the kinase.
Figure 8.

(A) TREEspot interaction maps: kinases found to bind 5f are marked with red circles, where larger circles indicate higher affinity binding and the side percentages indicate residual kinase activity. (B) Histogram of the inhibitory activity of 5f on kinases inhibited more than 50%.
With this screening platform, we found that 5f at 1 μM induced over 50% inhibition of 23 protein kinases (Figure 8B). We found that 5f did not directly inhibit PI3K, Akt, and mTOR. However, among the inhibited kinases were AURKA, FLT3, GSK3A, MAP3K, MEK4, RSK2, RSK4, PLK4, ULK1, and JAK2 that in cells could interact with the PI3K/Akt/mTOR pathway. These results suggest that suppression of these kinases could contribute to the high cytotoxicity caused by 5f.
To better understand the apparently unpredictable behavior of 22 and 5f as protein kinase inhibitors, a computer-based automated docking of both compounds was performed as described in detail in the Experimental Section. For this computational study, we selected from among the kinases that were inhibited more than 50% (Figure 8B) and whose crystallographic structures were deposited in the Protein Data Bank. As reported in detail in the Experimental Section, the ATP binding sites of five protein kinases (RSK2, PLK4, FLT3, JAK1, and GSK3)40–42 were used to explore the binding of both 22 and 5f. We found that these 7-phenylpyrrolo-quinolinones could be readily accommodated in the ATP binding clefts of all selected protein kinases but that the ligand–kinase stabilization energy associated with the binding of 5f was much higher (around 4–9 kcal/mol higher) as compared with 22. In particular, the experimental inhibitory activity shown by 5f can be attributed to the presence of the amide moiety at the 3-position through the formation of a strong H-bonding interaction with the side chain of amino acids located inside the ATP binding cavity (Figure 9). In Table 4 are summarized the most relevant binding features of 5f with the five selected protein kinases.
Figure 9.
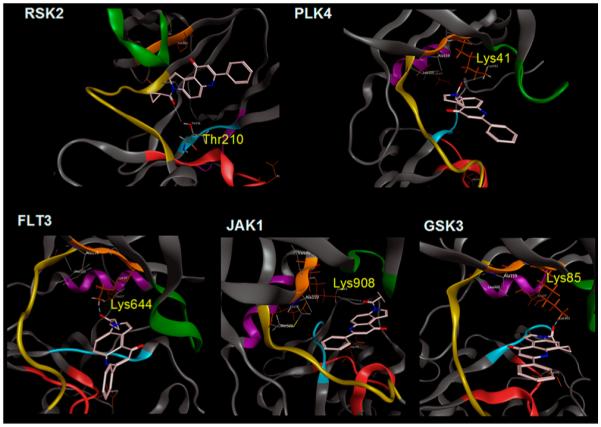
Energetically most favorable pose of 5f (in pink) in the ATP binding side of selected protein kinases.
Table 4.
Relevant Binding Features of 5f against Five Selected Protein Kinases
| protein kinase |
crucial H-bonding interaction with amide moiety in 3-position of 5f |
Δ(ΔEinter)IV→5fa
(kcal/mol) |
|---|---|---|
| RSK2 | Thr210 (dC=O/HO ≅ 2.8 Å) | −6.5 |
| PLK4 | Lys41 (dC=O/HN ≅ 3.1 Å) | −4.5 |
| FLT3 | Lys644 (dC=O/hn ≅ 2.1 Å) | −9.0 |
| JAK1 | Lys908 (dC=O/HN ≅ 3.0 Å) | −5.0 |
| GSK3 | Lys85 (dC=O/HN ≅ 2.2 Å) | −6.0 |
Δ(ΔEinter)IV→5f = [(ΔEinter)IV] – [(ΔEinter)5f].
5f Synergizes with Conventional Chemotherapeutic Agents in Inhibiting Leukemia Cell Proliferation
Since 5f induced a strong decrease in cancer cell proliferation, we evaluated whether 5f had promise when used in combination with commonly used chemotherapeutics in leukemia treatment. To this end, three difierent leukemia cell lines, two glucocorticoid-resistant (Jurkat, THP-1) and one glucocorticoid-sensitive (MV4;11) were treated for 48 h with 5f in combination with selected chemotherapeutic agents (i.e., dexamethasone, daunorubicin, and Ara-C) used frequently to treat leukemic patients. Compound 5f was combined with difierent drugs at a fixed molar combination ratio, and cell viability was analyzed by the MTT assay (Figure 10 and Supporting Information Figure 29).
Figure 10.
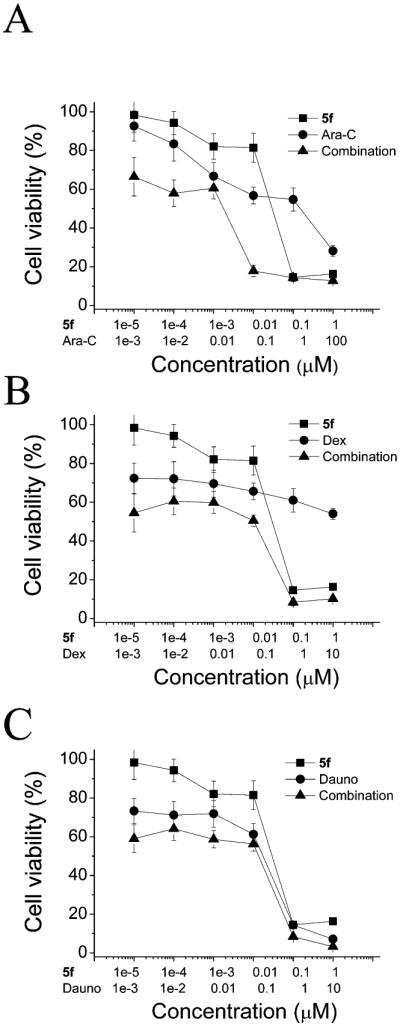
Effect of 5f treatment alone and in combination with difierent chemotherapic drugs in MV4;11 cells. Cells were treated at the indicated concentrations and at a fixed combination ratio, and viability was assessed by the MTT test after a 48 h incubation. Data are expressed as the mean ± SEM of three independent experiments.
As described above, 5f alone had significant cytotoxicity when used as a single agent (Table 1). When used in combination with chemotherapeutic drugs, we observed a synergistic increase in cytotoxicity, as demonstrated by combination index (CI) values obtained by the analytic method of Chou and Talalay.43–45 As summarized in Table 5, in which CI values calculated at the GI50, GI75, and GI90 values are shown, in almost all cases, but not with Jurkat cells for Ara-C treatment, 5f and the selected chemotherapeutic drugs acted in a synergistic fashion (CI < 1). Moreover, in the glucocorticoid resistant cells, Jurkat and THP-1, 5f was able to restore glucocorticoid sensitivity, suggesting that 5f might be able to optimize the efficacy of existing therapies for leukemia.
Table 5.
Compound 5f Synergizes with Drugs Used in Leukemia Therapy
| CIa | Dex + 5f (10:1)b | Dauno + 5f (10:1)b | AraC + 5f (100:1)b |
|---|---|---|---|
| Jurkat Cell Line | |||
| CI (GI50) | 0.67 | 0.21 | 11.9 |
| CI (GI75) | 0.81 | 0.45 | 0.79 |
| CI (GI90) | 0.97 | 1.0 | 14.7 |
| MV4;11 Cell Line | |||
| CI (GI50) | 0.016 | 0.22 | 0.09 |
| CI (GI75) | 0.06 | 0.17 | 0.38 |
| CI (GI90) | 0.25 | 0.14 | 1.8 |
| THP-1 Cell Line | |||
| CI (GI50) | 0.22 | 0.17 | 0.14 |
| CI (GI75) | 0.17 | 0.17 | 0.15 |
| CI (GI90) | 0.13 | 0.17 | 0.15 |
Combination indexes evaluated at different points.
Molar combination ratios.
CONCLUSION
A series of new 7-PPyQs was efficiently synthesized and biologically evaluated as anticancer agents. With respect to the previously reported analogues, the new compounds were further modified at the 3, 6, 7, and 9 positions. Our findings reported here show that the only chemical modifications tolerated by the 7-PPyQ pharmacophore without a major loss in antiproliferative potency occur at the 3N position. However, substituents at this position must show suitable size and polarity properties in order to preserve the strong cytotoxicity of the most active compounds. This was observed, for example, with compounds 20, 5a, and 14 (Table 1), in which the potency decreased in the following order: 20 (3N-propyl, low nanomolar GI50) > 14 (3N-propyl alcohol, high nanomolar GI50) > 5a (3N-ethyl alcohol, high micromolar GI50). Compound 15 (N-propanoic acid) and compound 16 (N-ethanoic acid) were both inactive. Among all compounds reported here, the amidic derivatives 5e and 5f, obtained by acylation of the pyrrolic N, showed very high cytotoxicity.
Importantly, when tested in human noncancer cell lines, 22 and especially 5f showed very low toxicity compared with tumor cell lines (Table 2).
All 7-PPyQs examined share the same primary mechanism of action, inhibition of tubulin polymerization by binding in the colchicine site (Table 3). In general, their potency as antitubulin agents correlated well with their potency as antiproliferative agents. The effect of 5f on cell cycle progression was examined with three cell lines (Figure 2), and the agent caused G2/M arrest. We showed alterations in expression of proteins that regulate cell division (Figure 3). These studies indicated that 5f at 1 μM induced G2/M arrest not because of defects in G2/M regulatory proteins but rather because there was an acceleration of entry into mitosis (Figure 4). As observed in some cancer cell lines, in a concentration dependent manner, 5f, like 22, caused cell death by apoptosis involving activator caspases 2, 8, and 9, activation of the effector caspase-3, and subsequent cleavage of PARP. Moreover, as with other microtubule targeting agents, at 0.1 μM in Jurkat cells, 5f induced downregulation of the antiapoptotic proteins Bcl-2, Bcl-XL, and particularly Mcl-1, which is a key factor governing cell survival and often overexpressed in cancer cells. Also, as noted with various antimitotic drugs, survivin was found to be phosphorylated upon treatment with 5f, additional evidence of cell cycle arrest in mitosis (Figure 5).
Another interesting finding of our work is that 5f modulates the PI3K/Akt/mTOR pathway which plays a major role in tumorigenesis and progression, since it is often hyperactivated in many types of tumors. Although this observation was already described by our group in A549 cell lines with compound 22, in this work we show this effect was maintained also in PTEN null cell line Jurkat. In this context it is worthwhile to note that PTEN mutations are associated with bad prognosis and that PTEN null tumors are particularly aggressive due to the strong activation of PI3K/Akt/mTOR pathway. Thus, the therapeutic potential of 5f with respect to other antimitotic compounds could be strongly enhanced and extended to difierent kinds of tumor.
By a KINOMEscan analysis with 1 μM 5f, we found greater than 50% inhibition of 23 kinases (Figure 8B), but there was no direct inhibitory effect on the enzymatic activity on PI3K, Akt, and mTOR. Therefore, our results indicated that a simple chemical modification made by linking an acyl side chain to the pyrrolic nitrogen endowed this compound class with a kinase inhibiting activity and that suppression of the activity of selected kinases might contribute to the high cytotoxicity of 5f.
Finally, 5f sensitized leukemia cell lines to the action of frequently used chemotherapeutic drugs, leading to cell death in a synergistic way. This effect was observed with agents with difierent mechanisms of action. In particular, we observed that 5f was able to restore the activity of dexamethasone in glucocorticoid resistant cells (Jurkat and THP-1). If confirmed in planned in vivo studies, this would be a valuable property of 5f.
EXPERIMENTAL SECTION
Materials and Methods
Melting points were determined on a Buchi M-560 capillary melting point apparatus and are uncorrected. Infrared spectra were recorded on a PerkinElmer 1760 FTIR spectrometer with potassium bromide pressed disks; all values are expressed in cm−1. UV–vis spectra were recorded on a Thermo Helyos α spectrometer. 1H NMR spectra were determined on Bruker 300 and 400 MHz spectrometers, with the solvents indicated; chemical shifts are reported in δ (ppm) downfield from tetramethylsilane as internal reference. Coupling constants are given in hertz. In the case of multiplets, chemical shifts were measured starting from the approximate center. Integrals were satisfactorily in line with those expected on the basis of compound structure. Elemental analyses were performed in the Microanalytical Laboratory, Department of Pharmaceutical Sciences, University of Padova, on a PerkinElmer C, H, N elemental analyzer model 240B, and analyses indicated by the symbols of the elements were within ±0.4% of the theoretical values. Analytical data are presented in detail for each final compound in the Supporting Information. Mass spectra were obtained on a Mat 112 Varian Mat Bremen (70 eV) mass spectrometer and Applied Biosystems Mariner System 5220 LC/MS (nozzle potential 140 eV). Column flash chromatography was performed on Merck silica gel (250–400 mesh ASTM); chemical reactions were monitored by analytical thin-layer chromatography (TLC) on Merck silica gel 60 F-254 glass plates. Solutions were concentrated on a rotary evaporator under reduced pressure. Starting materials were purchased from Sigma-Aldrich and Alfa Aesar, and solvents were from Carlo Erba, Fluka and Lab-Scan. DMSO was obtained anhydrous by distillation under vacuum and stored on molecular sieves.
The purity of new tested compounds was checked by HPLC using the instrument HPLC VARIAN ProStar model 210, with detector DAD VARIAN ProStar 335. The analysis was performed with a flow of 1 mL/min, a C-18 column of dimensions 250 mm × 4.6 mm, a particle size of 5 μm, and a loop of 10 μL. The detector was set at 300 nm. The mobile phase consisted of phase A (Milli-Q H2O, 18.0 MΩ, TFA 0.05%) and phase B (95% MeCN, 5% phase A). The gradient elution was performed as reported: 0 min, % B = 10; 0–20 min, % B = 90; 20–25 min, % B = 90; 25–26 min, % B = 10; 26–31 min, % B = 10.
General Procedure for the Synthesis of 1N-Substituted Nitroindoles (2a–f)
As a typical procedure, the synthesis of 2-(5-nitro-1H-indol-1-yl)ethanol 2a is described in detail. Into a two-necked 50 mL round-bottomed flask, 0.888 g (37 mmol) of NaH, 60% dispersion in mineral oil, was placed and washed with toluene (3 × 10 mL). With stirring, a solution of commercial 5-nitroindole 1, 1.50 g (9.25 mmol) in 5 mL of anhydrous DMF, was dropped into the flask, and the initial yellow color changed to red with the formation of H2 gas. After 30 min at room temperature, a solution of 2-bromoethanol, 0.981 mL (13.87 mmol; d = 1.763 g/mL) in 1 mL dry DMF, was added, and the reaction mixture was left to stir for 24 h. The reaction was monitored by TLC analysis (eluent toluene/n-hexane/ethyl acetate, 1:1:0.3). At the end of the reaction, 25 mL of water was added, and the solvent was evaporated under reduced pressure, leaving a residue, which was extracted with ethyl acetate (3 × 50 mL). The organic phase, washed with water and dried over anhydrous Na2SO4, was concentrated under vacuum giving a crude yellow solid (1.898 g). This crude product was purified with a silica gel chromatographic column (d 3 cm, l 35 cm, 230–400 mesh, eluent ethyl acetate/n-hexane, 7:3), yielding 1.587 g of a pure yellow solid.
2-(5-Nitro-1H-indol-1-yl)ethanol (2a)
Yield 82.2%; Rf = 0.31 (ethyl acetate/n-hexane 7:3); mp = 82 °C; 1H NMR (400 MHz, DMSO-d6) δ = 3.86 (q, 2H, J = 5.23 Hz, CH2-CH2-OH), 4.44 (t, 2H, J = 5.23 Hz, CH2-CH2-OH), 5.07 (t, 1H, J = 5.23 Hz, OH), 6.87 (dd, 1H, J = 3.18 and J = 0.59 Hz, 3-H), 7.76 (d, 1H, J = 3.18 Hz, 2-H), 7.82 (d, 1H, J = 9.15 Hz, 7-H), 8.15 (dd, 1H, J = 9.15 and J = 2.30 Hz, 6-H), 8.70 (d, 1H, J = 2.30 Hz, 4-H); 13C NMR (101 MHz, DMSO-d6) δ = 49.08 (NCH2CH2OH), 61.07 (NCH2CH2OH), 103.24 (3-C), 112.36 (7-C), 116.93 (6-C), 119.24 (4-C), 134.12 (2-C), 135.22 (3a-C), 138.24 (7a-C), 143.16 ppm (5-C). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C10H11N2O3+, 207.2054; found, 207.1987.
1-(Cyclopropylmethyl)-4-nitro-1H-indole (2b)
Compound 2b was prepared as for compound 2a, and the analytical data are reported in ref 7.
3-(5-Nitro-1H-indol-1-yl)propanenitrile (2c)
Compound 2c was prepared as for compound 2a by reacting 0.720 g of NaH 60% (30 mmol) and 1.621 g (10.0 mmol) of 5-nitroindole 1 dissolved in 5 mL of DMF and 1.24 mL of 3-bromopropionitrile (15 mmol, d = 1.615 g/mL). Reaction time 5 h (TLC ethyl acetate/n-hexane/toluene, 1:1:1). A solid product (2.564 g) was obtained. Yield 99%; Rf = 0.27 (toluene/n-hexane/ethyl acetate, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 3.10 (t, 2H, J = 6.60 Hz, NCH2CH2CN), 4.61 (t, 2H, J = 6.52 Hz, NCH2CH2CN), 6.81 (dd, 1H, J = 3.24 Hz and J = 0.8 Hz, 3-H), 7.72 (d, 1H, J = 3.31 Hz, 2-H), 7.83 (dt, 1H, J = 9.13 Hz and J = 0.7 Hz, 7-H), 8.06 (ddd, 1H, J = 9.08 Hz, J = 2.29 Hz and J = 0.25 Hz, 6-H), 8.59 ppm (dd, 1H, J = 2.24 Hz and J = 0.25 Hz, 4-H); 13C NMR (101 MHz, DMSO-d6) δ = 19.08 (NCH2CH2CN), 42.07 (NCH2CH2CN), 104.76 (3-C), 111.06 (7-C), 117.03 (6-C), 118.04 (4-C), 119.10 (CN), 132.76 (2-C), 132.76 (3a-C), 139.07 (7a-C), 141,48 ppm (5-C). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C11H10N3O+, 216.2155; found, 216.1290.
Ethyl 2-(5-Nitro-1H-indol-1-yl)acetate (2d)
Compound 2d was prepared as for compound 2a by reacting 0.445 g of NaH 60% (18.51 mmol) and 1.00 g (6.17 mmol) of 5-nitroindole 1 dissolved in 10 mL of toluene and 1.5456 g (9.25 mmol, d = 1.506 g/L) of bromoethyl acetate in 5 mL toluene. Reaction time was 4 h by TLC analysis (ethyl acetate/n-hexane/toluene, 1:1:1). After extraction, 1.400 g of the crude material was obtained, which was chromatographed on a silica gel column (d = 3 cm, l = 30 cm, 230–400 mesh, ethyl acetate/n-hexane/toluene, 1:1:1) giving 0.665 g of a pure bright white solid. Yield 47.5%; mp = 52–53 °C; Rf = 0.63 (eluent ethyl acetate/n-hexane/toluene, 1:1:1); 1H NMR (300 MHz, DMSO-d6) δ = 1.21 (t, 3H, J = 7.05 Hz, -OCH2CH3), 4.17 (q, 2H, J = 7.05 Hz, -OCH2CH3), 5.26 (s, 2H, NCH2), 6.78 (dd, 1H, J = 3.24 Hz and J = 0.76 Hz, 3-H), 7.61 (d, 1H, J = 3.24 Hz, 2-H), 7.64 (d, 1H, J = 9.15 Hz and J = 0.76 Hz, 7-H), 8.04 (dd, 1H, J = 9.15 Hz and J = 2.28 Hz, 6-H), 8.58 ppm (d, 1H, J = 2.09 Hz, 4-H); 13C NMR (75 MHz, DMSO-d6) δ = 16.83 (NCH2COOCH2CH3), 47.15 (NCH2COOCH2CH3), 64.34 (NCH2COOCH2CH3), 102.02 (3-C), 111.66 (7-C), 117.74 (6-C), 120.00 (4-C), 132.64 (2-C), 134.73 (3a-C), 137.98 (7a-C), 145.05 (5-C), 167.72 ppm (NCH2COOCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C12H13N2O4+, 249.2421; found, 249.1497.
1-(5-Nitro-1H-indol-1-yl)propan-1-one (2e)
Compound 2e was prepared as for compound 2a by reacting 0.444 g of NaH 60% (18.50 mmol) and 1.00 g (6.17 mmol) of commercial 5-nitroindole (1) dissolved in 5 mL of anhydrous DMF and 0.808 mL (9.25 mmol, d = 1.059 g/mL) of propionyl chloride dissolved in 1 mL of anhydrous DMF. On addition of the propionyl chloride solution, a white precipitate formed. After about 1 h, the solution was cooled (ice bath) and treated with 15 mL of water to quench the excess NaH. The precipitate formed was collected by filtration under vacuum, washed several times with water, and desiccated under vacuum, yielding 0.920 g of a white crystalline compound. Yield 68%; Rf = 0.63 (ethyl acetate/n-hexane/toluene, 1:1:1); mp = 177 °C; 1H NMR (400 MHz, DMSO-d6) δ = 1.19 (t, 3H, J = 7.25 Hz, C(O)CH2CH3), 3.13 (q, 2H, J = 7.25 Hz, C(O)CH2CH3), 6.97 (d, 1H, J = 3.34 Hz, 3-H), 8.14 (d, 1H, J = 3.34 Hz, 2-H), 8.20 (dd, 1H, J = 9.29 Hz and J = 2.25 Hz, 6-H), 8.52 (dt, 1H, J = 9.29 Hz, 7-H), 8.58 ppm (d, 1H, J = 2.25 Hz, 4-H); 13C NMR (101 MHz, DMSO-d6) δ = 8.75 (C(O)CH2CH3), 28.97 (C(O) CH2CH3), 109.15 (3-C), 116.63 (7-C), 117.50 (4-C), 120.11 (6-C), 130.27 (2-C), 130.41 (3a-C), 138.41 (7a-C), 143.80 (5-C), 173.73 ppm (NC(O)). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C11H11N2O +, 219.1261; found, 219.1291.
Cyclopropyl(5-nitro-1H-indol-1-yl)methanone (2f)
Compound 2f was prepared as for compound 2a by reacting 0.445 g of NaH 60% (18.51 mmol) and 1.012 g (6.24 mmol) of 5-nitroindole 1 dissolved in 10 mL DMF and 1.10 mL of cylopropanecarbonyl chloride (12.20 mmol, d = 1.152 g/mL). Reaction time was 3 h, monitoring by TLC (ethyl acetate/n-hexane/toluene, 1:1:1). After extraction, 0.860 g of crude reaction product, a golden solid (70%), was obtained, which was chromatographed on a silica gel column (d = 3 cm, l = 35 cm, 230–400 mesh, ethyl acetate/n-hexane/toluene, 1:1:1) to yield 0.752 g of pure compound 2f. Yield 52%; Rf = 0.62 (ethyl acetate/n-hexane/toluene, 1:1:1); mp = 45–48 °C; 1H NMR (400 MHz, DMSO-d6) δ = 1.17 (m, 4H, CH2-CH2), 2.77 (m, 1H, CH), 7.05 (dd, 1H, J = 3.81 Hz and J = 0.76 Hz, 3-H), 8.20 (dd, 1H, J = 9.15 Hz and J = 2.48 Hz, 6-H), 8.47 (d, 1H, J = 3.18 Hz, 2-H), 8.50 (d, 1H, J = 9.15 Hz, 7-H), 8.61 ppm (d, 1H, J = 2.48 Hz, 4-H); 13C NMR (101 MHz, DMSO-d6) δ = 15.14 (CH2CH2), 26.34 (CH), 101.99 (3-C), 113.14 (7-C), 115.82 (6-C), 118.34 (4-C), 133.84 (2-C), 134.72 (3a-C), 137.46 (7a-C), 144.04 (5-C), 172.01 ppm (NC(O)CHCH2CH2). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C12H11N2O3+, 231.2268; found, 231.1382.
General Procedure for the Synthesis of 1N-Substituted Aminoindoles 3a–d and 6f
As a typical procedure, the synthesis of 5-aminoindole 3a is described in detail. Into a three-necked flask of 500 mL, previously dried in an oven, about 0.300 g of C/Pd 10% and approximately 60 mL of ethyl acetate were placed. After connecting the flask to an elastomer balloon containing hydrogen gas, the mixture was stirred at room temperature for 1 h in order to saturate the suspension of C/Pd with hydrogen. Then, 7.70 mmol (1.587 g) of nitroindole derivative 2a in 15 mL of ethyl acetate was added dropwise to the suspension, and the mixture was stirred under hydrogen at atmospheric pressure and heated by means of an oil bath at 50–60 °C for 15 h, monitoring the progress of the reaction by TLC analysis (ethyl acetate/n-hexane, 7:3). At the end of the reaction, the mixture was filtered, and the solution was concentrated to dryness on a rotavapor to give 1.234 g of semisolid dark purple product.
2-(5-Amino-1H-indol-1-yl)ethanol (3a)
Yield 91%; mp = 124 °C; Rf = 0.20 (ethyl acetate/n-hexane, 7:3). 1H NMR (400 MHz, DMSO-d6) δ = 4.55 (q, 2H, J = 5.67 Hz, CH2CH2OH), 4.92 (bs, 2H, NH2), 4.96 (t, 2H, J = 5.67 Hz, CH2CH2OH), 5.80 (t, 1H, J = 5.67 Hz, OH), 7.06 (dd, 1H, J = 3.25 Hz and J = 0.41 Hz, 3-H), 7.62 (dd, 1H, J = 8.79 Hz and J = 2.13 Hz, 6-H), 7.87 (d, 1H, J = 2.13 Hz, 4-H), 7.93 (d, 1H, J = 3.25 Hz, 2-H), 8.04 ppm (d, 1H, J = 8.79 Hz, 7-H); 13C NMR (101 MHz, DMSO-d6) δ = 47.34 (NCH2CH2OH), 60.77 (NCH2CH2OH), 100.34 (3-C), 105.72 (4-C), 111.34 (7-C), 113.75 (6-C), 127.96 (2-C), 128.14 (3a-C), 129.94 (7a-C), 142.90 ppm (5-C). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C10H13N2O+, 177.2225, found:177.1565.
1-(Cyclopropylmethyl)-4-amino-1H-indole (3b)
Compound 3b was prepared as for compound 3a, and the analytical data are reported in ref 7.
3-(5-Amino-1H-indol-1-yl)propanenitrile (3c)
Compound 3c was prepared as for compound 3a by reacting 2.853 g (13.26 mmol) of nitroindole derivative 2c, obtaining 1.842 g of a brown oil. Yield 97%; Rf = 0.11 (ethyl acetate/n-hexane/toluene, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 2.95 (t, 2H, J = 6.88 Hz, NCH2CH2CN), 4.36 (t, 2H, J = 6.88 Hz, NCH2CH2CN), 4.52 (s, 2H, NH2), 6.17 (dd, 1H, J = 3.12 Hz and J = 0.85 Hz, 3-H), 6.55 (ddd, 1H, J = 8.73 Hz, J = 2.13 Hz and J = 0.33 Hz, 6-H), 6.70 (dd, 1H, J = 2.12 Hz and J = 0.61 Hz, 4-H), 7.21 (d, 1H, J = 3.12 Hz, 2-H), 7.23 ppm (dt, 1H, J = 8.74 Hz and J = 0.73 Hz, 7-H); 13C NMR (101 MHz, DMSO-d6) δ = 19.05 (NCH2CH2CN), 41.70 (NCH2CH2CN), 100.29 (3-C), 104.03 (4-C), 110.32 (7-C), 112.33 (6-C), 119.44 (CN), 128.45 (2-C), 129.70 (3a-C), 129.75 (7a-C), 142.19 ppm (5-C). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C11H12N3+, 186.1031; found, 186.1012.
Ethyl 2-(5-Amino-1H-indol-1-yl)acetate (3d)
Compound 3d was prepared as for compound 3c by reacting 0.500 g of nitroindole derivative 2d (2.01 mmol), obtaining 0.396 g of a violet solid. Yield 90%; Rf = 0.43 (n-hexane/ethyl acetate, 1:1); 1H NMR (300 MHz, DMSO-d6) δ = 1.21 (t, 3H, J = 7.05 Hz, CH2CH3), 4.13 (q, 2H, J = 7.05 Hz, CH2CH3), 4.50 (bs, 2H, NH2), 4.95 (s, 2H, CH2), 6.15 (dd, 1H, J = 3.05 Hz and J = 0.76 Hz, 3-H), 6.50 (dd, 1H, J = 8.58 Hz and J = 2.09 Hz, 6-H), 6.67 (d, 1H, J = 2.09 Hz and J = 0.52 Hz, 4-H), 7.02 (d, 1H, J = 8.77 Hz and J = 0.76 Hz, 7-H), 7.11 ppm (d, 1H, J = 3.05 Hz, 2-H); 13 C NMR (75 MHz, DMSO-d6) δ = 15.22 (NCH2COOCH2CH3), 45.53 (NCH2COOCH2CH3), 64.22 (NCH2COOCH2CH3), 101.20 (3-C), 104.73 (4-C), 112.74 (7-C), 114.01 (6-C), 126.06 (2-C), 127.94 (3a-C), 130.02 (7a-C), 143.06 (5-C), 168.25 ppm (NCH2COOCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C12H15N2O2+, 219.2592; found, 219.2537.
(5-Aminoindolin-1-yl)(cyclopropyl)methanone (6f)
Compound 6f was prepared as for compound 3a by reacting 3.27 mmol (0.759 g) of 5-nitroindole 2f at room temperature, obtaining 0.653 g of a slightly pink solid. Yield 98%; Rf = 0.37 (ethyl acetate/n-hexane, 8:2); mp = 159 °C; 1H NMR (300 MHz, DMSO-d6) δ = 0.80 (m, 4H, CH2-CH2), 1.85 (m, 1H, CH), 3.04 (t, 2H, J = 8.01 Hz, 2-H2), 4.18 (t, 2H, J = 8.01 Hz, 3-H2), 4.81 (bs, 2H, NH2), 6.30 (dd, 1H, J = 8.58 Hz and J = 2.09 Hz, 6-H), 6.45 (s, 1H, 4-H), 6.71 ppm (d, 1H, J = 8.58 Hz, 7-H); 13C NMR (75 MHz, DMSO-d6) δ = 8.26 (CH2CH2), 14.02 (CH), 28.66 (3-C), 48.96 (2-C), 103.76 (4-C), 111.22 (7-C), 115.92 (6-C), 125.76 (3a-C), 127.42 (7a-C), 141.01 (5-C), 170.44 ppm (NC(O)CHCH2CH2). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C12H15N2O+, 203.2598; found, 203.2524.
General Procedure for the Synthesis of 5-Aminoindoles 3e and 3f
As a typical procedure, the synthesis of 5-aminoindole 3e is described in detail. Into a two-necked 50 mL round-bottomed flask, 0.350 g of 1-(5-nitro-1H-indol-1-yl)propan-1-one (2e) (1.60 mmol), 1.809 g of SnCl2·2H2O (8.02 mmol), and 25 mL of methanol were added. The reaction mixture was refluxed for 3 h, and the reaction progress was monitored by TLC (n-hexane/ethyl acetate, 2:8). At the end, the solvent was evaporated, the residue was taken up with aqueous NaOH 20% (20 mL), and the resulting suspension was extracted with diethyl ether (4 × 50 mL). The combined extracts, washed with anhydrous Na2SO4, were evaporated to dryness on a rotary evaporator to yield 0.240 g of a semisolid yellow product.
1-(5-Amino-1H-indol-1-yl)propan-1-one (3e)
Yield 80%; mp = 132 °C; Rf = 0.27 (ethyl acetate/n-hexane/toluene, 1:1:1). 1H NMR (400 MHz, DMSO-d6) δ = 1.16 (t, 3H, J = 7.33 Hz, C(O)CH2CH3), 2.96 (q, 2H, J = 7.33 Hz, C(O)CH2CH3), 4.91 (bs, 2H, NH2), 6.17 (dd, 1H, J = 3.12 Hz and J = 0.85 Hz, 3-H), 6.55 (ddd, 1H, J = 8.74 Hz, J = 2.13 Hz and J = 0.33 Hz, 6-H), 6.70 (dd, 1H, J = 2.13 Hz and J = 0.61 Hz, 4-H), 7.21 (d, 1H, J = 3.12 Hz, 2-H), 7.23 ppm (dt, 1H, J = 8.74 Hz and J = 0.85 Hz, 7-H); 13C NMR (101 MHz, DMSO-d6) δ = 9.36 (C(O)CH2CH3), 28.45 (C(O)CH2CH3), 100.29 (3-C), 104.03 (4-C), 110.31 (7-C), 112.33 (6-C), 128.44 (2-C), 129.70 (3a-C), 129.75 (7a-C), 142.19 (5-C), 174.80 ppm (NC(O)). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C11H13N2O+, 189.2332; found, 189.2388.
Cyclopropyl(5-amino-1H-indol-1-yl)methanone (3f)
Compound 3f was prepared as for compound 3e by reacting 0.500 g of of 5-nitroindole 2f (2.15 mmol), 2.9 g of SnCl2·2H2O (12.85 mmol), and 30 mL of methanol to yield 0.205 g of a pearly white solid. Yield 47%. Rf = 0.70 (n-hexane/ethyl acetate, 2:8); 1H NMR (300 MHz, DMSO-d6) δ = 1.03 (m, 4H, CH2-CH2), 2.60 (m, 1H, CH), 4.91 (s, 2H, NH2), 6.54 (dd, 1H, J = 3.62 Hz and J = 0.57 Hz, 3-H), 6.57 (dd, 1H, J = 8.77 Hz and J = 2.09 Hz, 6-H), 6.71 (d, 1H, J = 2.09 Hz, 4-H), 7.98 (d, 1H, J = 3.24 Hz, 2-H), 8.00 ppm (dd, 1H, J = 8.77 Hz and J = 0.57 Hz, 7-H); 13C NMR (75 MHz, DMSO-d6) δ = 15.95 (CH2CH2), 28.44 (CH), 101.15 (3-C), 104.33 (4-C), 112.77 (7-C), 113.24 (6-C), 128.06 (2-C), 128.94 (3a-C), 129.74 (7a-C), 142.52 (5-C), 171.61 ppm (NC(O)CHCH2CH2). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C12H13N2O+, 201.2439; found, 201.2482.
General Procedure for the Synthesis of Acrylate Derivatives 4a–f and 7f
As a typical procedure, the synthesis of acrylate derivative 4a is described in detail. In a 100 mL round-bottomed flask, 1.234 g (7.00 mmol) of 3-substituted aminoindole 3a in 25 mL of absolute ethanol was condensed with 1.82 mL (10.5 mmol; d = 1.11 g/mL) of commercial ethyl benzoylacetate and 0.5 mL of glacial acetic acid in the presence of 100 mg of Drierite. The mixture was refluxed for about 24 h, the reaction being monitored by TLC analysis (n-hexane/ethyl acetate, 3:7). Even though the reaction was not complete after 24 h, the mixture was cooled and filtered to remove the Drierite; the resulting solution was evaporated to dryness under vacuum and the residue (2.420 g) purified by silica gel chromatography (d = 3 cm, l = 35 cm, 230–400 mesh, eluent n-hexane/ethyl acetate, 3:7) to yield 1.47 g of a semisolid yellow product.
(E,Z)-Ethyl 3-(1-(2-Hydroxyethyl)-1H-indol-5-ylamino)-3-phenylacrylate (4a)
Yield 60%; Rf = 0.58 (n-hexane/ethyl acetate, 3:7); 1H NMR (300 MHz, DMSO-d6) δ = 1.11 (t, 3H, J = 7.05 Hz, CH2CH3), 3.89 (q, 2H, J = 7.05 Hz, CH2CH2OH), 4.13 (q, 2H, J = 7.05 Hz, CH2CH3), 4.61 (t, 2H, J = 7.05 Hz, CH2CH2OH), 4.83 (s, 1H, CH), 5.15 (t, 1H, J = 7.05 Hz, OH), 7.16 (d, 1H, J = 3.05 Hz, 3-H), 7.67 (m, 5H, 2′-H, 3′-H, 4′-H, 5′-H, 6′-H), 7.71 (dd, 1H, J = 8.77 and J = 2.10 Hz, 6-H), 8.02 (d, 1H, J = 2.10 Hz, 4-H), 8.07 (d, 1H, J = 3.05 Hz, 2-H), 8.13 (d, 1H, J = 8.77 Hz, 7-H), 10.25 ppm (s, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 14.75 (CHCOOCH2CH3), 46.94 (NCH2 CH2 OH), 60.24 (CHCOOCH2 CH3), 61.56 (NCH2CH2OH), 89.44 (CHCOOCH2CH3), 102.62 (3-C), 110.75 (7-C), 114.73 (4-C), 118.12 (6-C), 127.22 (3a-C), 128.94 (2′-C and 6′-C), 129.44 (5′-C and 3′-C), 129.87 (4′-C), 130.44 (2-C), 132.03 (7a-C), 133.76 (1′-C), 137.42 (5-C), 159.92 (NHCCH), 169.84 ppm (CHCOOCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C21H23N2O3+, 351.4184; found, 351.4194.
(E,Z)-Ethyl 3-(1-(Cyclopropylmethyl)-1H-indol-5-ylamino)-3-(3,4-dimethoxyphenyl)acrylate (4b)
Compound 4b was prepared as for compound 4a by reacting 0.500 g (2.68 mmol) of 5-aminoindole 3b, obtaining after column chromatography 0.387 g of a semisolid product. Yield 87%; Rf = 0.76 and Rf = 0.70 (ethyl acetate); 1H NMR (300 MHz, DMSO-d6) including NMR signals of both isomers δ = 0.32 (dd, 2H, J = 4.95 Hz and J = 1.14 Hz, CH2-CH2), 0.46 (dd, 2H, J = 8.20 Hz and J = 1.90 Hz, CH2-CH2), 1.17 (t, 3H, J = 7.05 Hz, CH2CH3), 1.23 (t, 3H, J = 7.05 Hz, CH2CH3), 1.29 (m, 1H, CH), 3.54 and 3.68 (s, 6H, -OCH3), 3.81 and 3.85 (s, 6H, -OCH3), 4.09 (m, 2H, J = 6.29 Hz, CH2CH3), 4.13 (m, 2H, J = 7.05 Hz, CH2CH3), 4.88 (s, 1H, CH), 6.10 and 6.25 (dd, 2H, J = 2.86 Hz and J = 0.76 Hz, J = 3.05 Hz, J = 0.57 Hz, 3-H), 6.51 (dd, 2H, J = 9.15 Hz and J = 2.09 Hz, 6-H), 6.64 (dd, 2H, J = 8.39 and J = 1.90 Hz, 6′-H), 6.82 (d, 1H, J = 8.96 Hz, 5′-H), 7.02 (d, 1H, J = 0.90 Hz, 2′-H), 7.12 (d, 1H, J = 8.58 Hz, 7-H), 7.18 (dd, 2H, J = 6.29 Hz and J = 3.05 Hz, 4-H), 7.31 and 7.36 (d, 2H, J = 8.77 Hz and J = 3.05 Hz, 5-H), 7.44 (d, 1H, J = 2.09 Hz, 2-H), 7.61 (dd, 1H, J = 8.58 Hz and J = 2.28 Hz, 6-H), 10.19 ppm (s, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 8.24 (CH2CH2), 13.07 (CH), 14.34 (CHCOOCH2CH3), 60.24 (CHCOOCH2CH3), 62.04 (OCH3), 62.61 (OCH3), 88.78 (CHCOOCH2CH3), 101.91 (3-C), 111.77 (7-C), 114.97 (4-C), 115.85 (2′-C), 117.24 (6-C), 119.82 (5′-C), 122.45 (6′-C), 126.92 (3a-C), 130.44 (2-C), 132.03 (7a-C), 135.62 (1′-C), 137.23 (5-C), 160.44 (NHCCH), 163.03 (3′-C), 164.24 (4′-C), 169.84 ppm (CHCOOCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C25H29N2O4+, 421.5082; found, 421.5095.
(E,Z)-Ethyl 3-(1-(2-Cyanoethyl)-1H-indol-5-ylamino)-3-phenylacrylate (4c)
Compound 4c was prepared as for compound 4a by reacting 2.148 g (11.60 mmol) of 5-aminoindole derivative 3c and 3.01 mL (17.7 mmol) of ethyl benzoylacetate, obtaining after column chromatography (d = 3 cm, l = 30 cm, 230–400 mesh, eluent n-hexane/ethyl acetate, 1:1) 2.54 g of a dark red solid. Yield 71%; Rf = 0.72 (n-hexane/ethyl acetate, 1:1); 1H NMR (400 MHz, DMSO-d6) δ = 1.24 (t, 3H, J = 6.92 Hz, CHCOOCH2CH3), 2.97 (t, 2H, J = 6.34 Hz, NCH2CH2CN), 4.14 (q, 2H, J = 6.92 Hz,, CHCOOCH2CH3), 4.39 (t, 2H, J = 6.34 Hz, NCH2CH2CN), 4.85 (s, 1H, CHCOOCH2CH3), 6.28 (dd, 1H, J = 3.19 Hz and J = 0.69 Hz, 3-H), 6.66 (dd, 1H, J = 8.71 Hz and J = 2.08 Hz, 6-H), 6.98 (m, 1H, J = 2.08 Hz, 4-H), 7.29 (m, 1H, 2-H), 7.29 (m, 1H, 4′-H), 7.35 (m, 1H, 7-H), 7.37 (m, 2H, 2′-H and 6′-H), 7.35 (m, 2H, 3′-H and 5′-H), 10.27 ppm (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 14.94 (CHCOOCH2CH3), 19.01 (NCH2CH2CN), 41.71 (NCH2CH2CN), 59.14 (CHCOOCH2CH3), 89.12 (CHCOOCH2CH3), 101.68 (3-C), 110.44 (7-C), 115.25 (4-C), 118.75 (6-C), 119.29 (CN), 128.64 (3a-C), 128.89 (2′-C and 6′-C), 128.59 (5′-C and 3′-C), 129.85 (4′-C), 129.87 (2-C), 132.93 (7a-C), 133.06 (1′-C), 136.31 (5-C), 160.50 (NHCCH), 169.79 ppm (CHCOOCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C22H22N3O2+, 360.4284; found, 360.4242.
(E,Z)-Ethyl 3-(1-((Ethoxycarbonyl)methyl)-1H-indol-5-ylamino)-3-phenylacrylate (4d)
Compound 4d was prepared as for compound 4a by reacting 0.800 g (3.66 mmol) of 5-aminoindole 3d, to yield after silica gel column chromatography (d = 2.5 cm, l = 30 cm, 230–400 mesh, eluent n-hexane/ethyl acetate, 1:1) 1.024 g of a semisolid red product. Yield 70%; Rf = 0.54 (n-hexane/ethyl acetate, 1:1); 1H NMR (300 MHz, DMSO-d6) δ = 1.11 (t, 3H, J = 7.05 Hz, CH2CH3), 1.16 (t, 3H, J = 7.05 Hz, CH2CH3), 4.01 (q, 2H, J = 7.05 Hz, CH2CH3), 4.13 (q, 2H, J = 7.05 Hz, CH2CH3), 4.83 (s, 1H, CH), 4.95 (s, 2H, CH2) 6.20 (d, 1H, J = 3.05 Hz, 3-H), 6.60 (dd, 1H, J = 8.77 Hz and J = 2.10 Hz, 6-H), 6.97 (d, 1H, J = 2.10 Hz, 4-H), 7.30 (m, 7H, 2-H, 7-H, 2′-H, 3′-H, 4′-H, 5′-H, and 6′-H), 10.25 ppm (s, 1H, NH); 13 C NMR (75 MHz, DMSO-d6) δ = 14.33 (CHCOOCH2CH3), 15.64 (NCH2COOCH2CH3), 46.55 (NCH2COOCH2CH3), 60.24 (CHCOOCH2CH3), 61.15 (NCH2COOCH2CH3), 88.95(CHCOOCH2CH3), 102.36 (3-C), 110.82 (7-C), 114.46 (4-C), 118.25 (6-C), 127.16 (3a-C), 128.77(2′-C and 6′-C), 129.41 (5′-C and 3′-C), 129.82 (4′-C), 130.11 (2-C), 132.09 (7a-C), 133.24 (1′-C), 137.77 (5-C), 160.14 (NH C CH), 168.88 (CHCOOCH2CH3), 169.76 ppm (NCH2COOCH2CH3);HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C23H25N2O4, 393.4551; found, 393.4577.
(E,Z)-Ethyl 3-(1-Propionyl-1H-indol-5-ylamino)-3-phenylacrylate (4e)
Compound 4e was prepared as for compound 4a by reacting 0.240 g (1.28 mmol) of 5-aminoindole 3e and 0.332 mL of ethyl benzoyl acetate (1.92 mmol, d = 1.11 g/mL). An amount of 0.525 g of a crude product was obtained, and this was purified by silica gel column chromatography (d = 2 cm, l = 15 cm, 230–400 mesh, eluent n-hexane/ethyl acetate/toluene, 1:1:1), giving 0.250 g of a semisolid yellow product. Yield 53%; Rf = 0.72 (ethyl acetate/n-hexane/toluene, 1:1:1); 1H NMR (400 MHz, DMSO-d6) δ = 1.16 (t, 3H, J = 7.33 Hz, C(O)CH2CH3), 1.24 (t, 3H, J = 6.92 Hz, COOCH2CH3), 2.96 (q, 2H, J = 7.33 Hz, C(O)CH2CH3), 4.14 (q, 2H, J = 6.92 Hz, COOCH2CH3), 4.85 (s, 1H, CH), 6.28 (dd, 1H, J = 3.19 Hz and J = 0.69 Hz, 3-H), 6.66 (dd, 1H, J = 8.70 Hz and J = 2.08 Hz, 6-H), 6.97 (m, 1H, J = 2.08 Hz, 4-H), 7.28 (d, 1H, J = 3.19 Hz, 2-H), 7.29 (m, 1H, 4′-H), 7.34 (m, 2H, 3′-H and 5′-H), 7.35 (dd, 1H, J = 8.70 Hz and J = 0.69 Hz, 7-H), 7.37 (m, 2H, 2′-H and 6′-H), 10.26 ppm (bs, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 9.36 (C(O)CH2CH3), 14.94 (COOCH2CH3), 28.45 (C(O)CH2CH3), 59.14 (COOCH2CH3), 89.11 (CH), 101.68 (3-C), 110.44 (7-C), 115.25 (4-C), 118.76 (6-C), 128.59 (3′-C and 5′-C), 128.64 (3a-C), 128.89 (2′-C and 6′-C), 129.84 (4′-C), 129.87 (2-C), 132.93 (7a-C), 133.06 (1′-C), 136.31 (5-C), 160.51 (C-CH), 169.79 (COOCH2CH3), 174.80 ppm (COCH2CH3). HRMS (ESI-MS, 140 eV): m/z [M + H ] calculated for C22H23N2O3+, 363.4291; found, 363.4273.
(E,Z)-Ethyl 3-(1-Cyclopropylmethanone-1H-indol-5-ylamino)-3-phenylacrylate (4f)
Compound 4f was prepared as for compound 4a by reacting 0.750 g (3.74 mmol) of 5-aminoindole derivative 3f with ethyl benzoylacetate (1.72 mL, 5.61 mmol). An amount of 1.150 g of crude product was obtained, and this was chromatographed on a silica gel column (d = 2.5 cm, l = 30 cm, 230–400 mesh, eluent ethyl acetate/n-hexane, 4:6) yielding 0.694 g of a yellow oil. Yield 49%; Rf = 0.76 (ethyl acetate/n-hexane, 4:6); 1H NMR (300 MHz, DMSO-d6) δ = 1.05 (m, 4H, CH2CH2), 1.17 (t, 3H, J = 7.05 Hz, CH2CH3), 2.64 (m, 1H, CH), 4.13 (q, 2H, J = 7.05 Hz, CH2CH3), 4.92 (s, 1H, CH), 6.58 (d, 1H, J = 3.62 Hz, 3-H), 6.73 (dd, 1H, J = 8.84 Hz and J = 2.01 Hz, 6-H), 6.99 (d, 1H, J = 2.01 Hz, 4-H), 7.32 (m, 3H, 3′-H, 5′-H and 4′-H), 7.94 (m, 2H, J = 7.43 Hz, 2′-H and 6′-H), 8.01 (d, 1H, J = 8.77 Hz, 7-H), 8.11 (d, 1H, J = 3.81 Hz, 2-H), 10.24 ppm (s, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 14.75 (CHCOOCH2CH3), 15.36 (CH2CH2), 28.44 (CH), 61.04 (CHCOOCH2CH3), 89.27 (CHCOOCH2CH3), 102.94 (3-C), 110.35 (7-C), 114.68 (4-C), 118.24 (6-C), 127.36 (3a-C), 128.83 (2′-C and 6′-C), 129.26 (5′-C and 3′-C), 129.65 (4′-C), 130.25 (2-C), 132.25 (7a-C), 133.78 (1′-C), 137.89 (5-C), 160.44 (NHCCH), 169.44 (CHCOOCH2CH3), 171.91 ppm (NC(O)CHCH2CH2). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C23H23N2O3+, 375.4398; found, 375.4356.
(E,Z)-Ethyl 3-(1-Cyclopropylmethanone-1H-indolin-5-ylamino)-3-phenylacrylate (7f)
Compound 7 was prepared as for compound 4a by reacting 0.653 g (3.22 mmol) of 5-aminoindole derivative 2f and 0.836 mL diethyl benzoylacetate (4.83 mmol). An amount of 0.956 g of crude product was obtained, and this was purified by silica gel column chromatography (d = 2.5 cm, l = 35 cm, 230–400 mesh, eluent ethyl acetate/n-hexane, 3:7), giving 0.459 g of a yellow solid. Yield 37%. Rf = 0.79 (ethyl acetate/n-hexane, 8:2); mp = 133 °C; 1H NMR (300 MHz, DMSO-d6) δ = 0.80 (m, 4H, CH2CH2), 1.17 (t, 3H, J = 6.95 Hz, CH2CH3), 2.64 (m, 1H, CH-COOCH2CH3), 3.18 (t, 2H, J = 7.84 Hz, 3-H2), 4.13 (q, 2H, J = 6.95 Hz, CH2CH3), 4.29 (t, 2H, J = 7.84 Hz, 2-H2), 6.73 (dd, 2H, J = 8.84 Hz and J = 2.01 Hz, 6-H), 6.99 (d, 1H, J = 2.01 Hz, 4-H), 7.32 (m, 3H, 3′-H, 5′-H and 4′-H), 7.94 (m, 2H, 2′-H and 6′-H), 8.01 (d, 1H, J = 8.84 Hz, 7-H), 10.25 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 8.44 (CH2CH2), 13.96 (CH), 14.26 (CHCOOCH2CH3), 28.02 (3-C), 48.47 (2-C), 60.84 (CHCOOCH2CH3), 89.72 (CHCOOCH2CH3), 110.26 (7-C), 114.75 (4-C), 118.23 (6-C), 127.75 (3a-C), 128.34 (2′-C and 6′-C), 129.34 (5′-C and 3′-C), 129.75 (4′-C), 132.11 (7a-C), 133.87 (1′-C), 137.23 (5-C), 160.86 (NHCCH), 170.07 (CHCOOCH2CH3), 172.51 ppm (NC(O)CHCH2CH2). HRMS (ESI-MS, 140 eV): m/z [M + H] + calculated for C23H25N2O3+, 377.4557; found, 377.4547.
General Procedure for the Synthesis of Phenylpyrrolo-quinolinones 5a–f and 8f
As a typical procedure, the synthesis of the phenylpyrroloquinolinone derivative 5a is described in detail. In a two-necked round-bottomed flask, 50 mL of diphenyl ether was heated to boiling. An amount of 1.47 g (4.2 mmol) of acrylate derivative 4a was then added portionwise, and the resulting mixture was refluxed for 15 min. After cooling to room temperature, an amount of 25 mL of diethyl ether was added, and the mixture was left for 12 h. Then the separated precipitate was collected by filtration and washed many times with diethyl ether. The product (0.945 g) was purified by silica gel column chromatography (d = 2.5, l = 30, 230–400 mesh, eluent ethyl acetate/methanol, 9:1), obtaining 0.511 g of a slightly brown product.
3-(2-Hydroxyethyl)-7-phenyl-3H-pyrrolo[3,2-f] quinolin-9(6H)-one (5a)
Yield 40%; Rf = 0.32 (blue fluorescent spot, dichloromethane/methanol, 9:1); mp = 161 °C; 1H NMR (300 MHz, DMSO-d6) δ = 3.76 (m, 2H, CH2CH2OH), 4.34 (t, 2H, J = 5.62 Hz, CH2CH2OH), 4.94 (bs, 1H, OH), 6.47 (s, 1H, 8-H), 7.49 (d, 1H, J = 2.94 Hz, 2-H), 7.53 (m, 1H, 1-H), 7.58 (m, 4H, 5-H, 3′-H, 4′-H and 5′-H), 7.78 (m, 2H, 2′-H and 6′-H), 7.90 (dd, 1H, J = 9.02 Hz and J = 0.46 Hz, 4-H), 11.74 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 49.00 (CH2CH2OH), 61.22 (CH2CH2OH), 103.98 (1-C), 108.22 (8-C), 112.83 (5-C), 116.38 (4-C), 117.84 (9a-C), 123.38 (9b-C), 127.80 (6′-C and 2′-C), 129.42 (3′-C and 5′-C), 129.72 (2-C), 130.42 (4′-C), 132.16 (1′-C), 132.18 (3a-C), 137.11 (5a-C), 149.63 (7-C), 178.86 ppm (9-C). IR (ATR ZnSe): ν = 3150, 2900, 2850, 1610, 1470, 1055 cm−1. UV–vis (MeOH): λmax (ε) = 206 (1.435), 270 (1.341), 345 nm (0.642); λmin (ε) = 195 (0.453), 247 (0.600), 314 nm (0.344). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C19H17N2O2+, 305.1290; found, 305.1298. RP-C18-HPLC: 97.24%, tR = 10.02 min.
3-(Cyclopropylmethyl)-7-(3,4-dimethoxyphenyl)-3H-pyrrolo[3,2-f] quinolin-9(6H)-one (5b)
Compound 5b was prepared as for compound 5a by reacting 1.5 g (3.56 mmol) of dimethoxyphenylacrylate derivative 4b. An amount of 0.801 g of a rough dark green solid was obtained, and this was chromatographed on a silica gel column (d = 2.5 cm, l = 30 cm, 230–400 mesh, eluent ethyl acetate) yielding 0.450 g of a green solid. Yield 35%; mp = 291 °C; Rf = 0.83 (blue fluorescent spot, ethyl acetate); 1H NMR (300 MHz, DMSO-d6) δ = 0.41 (m, 2H, CH2-CH2), 0.53 (m, 2H, CH2-CH2), 1.26 (m, 1H, CH), 3.85 (s, 3H, -OCH3), 3.90 (s, 3H, -OCH3), 4.15 (d, 2H, CH2), 6.37 (bs, 1H, 8-H), 7.14 (d, 1H, J = 8.21 Hz, 5-H), 7.56 (m, 2H, 1-H and 5′-H), 7.86 (m, 3H, 2-H, 2′-H and 6′-H), 7.91 (d, 1H, J = 8.92 Hz, 4-H), 11.50 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 4.87 (CH2CH2), 5.28 (CH2CH2), 8.83 (CHCH2CH2), 56.16 (-OCH3), 61.52 (-OCH3), 63.51 (–CH2-) 102.74 (1-C), 106.44 (8-C), 108.21 (5-C), 112.11 (4-C), 116.81 (9a-C), 124.14 (9b-C), 124.86 (4′-C), 125.43 (3′-C), 125.98 (2′-C), 126.14 (2-C), 130.42 (1′-C), 134.21 (3a-C), 135.15 (5a-C), 147.13 (6′-C), 148.56 (7-C), 153.31 (5′-C), 175.23 ppm (9-C). IR (ATR ZnSe): = 3180, 2950, 2880, 1615, 1490 cm−1. UV–vis (MeOH): λmax (ε) = 221 (0.717), 293 (0.670), 343 nm (0.320); λmin (ε) = 258 (0.226), 318 nm (0.172); fluorescence (MeOH), λexc = 300 nm, λems = 451 nm. HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C23H23N2O3+, 375.1709; found, 375.1715. RP-C18-HPLC: 99.82%, tR = 12.53 min.
3-(9-Oxo-7-phenyl-6H-pyrrolo[3,2-f] quinolin-3(9H)-yl)-propanenitrile (5c)
Compound 5c was prepared as for compound 5a by reacting 2.54 g (7.06 mmol) of phenylacrylate derivative 4c. An amount of 1.680 g of a yellow solid product was obtained. Yield 76%; Rf = 0.72 (blue fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 3.10 (t, 2H, J = 6.51 Hz, NCH2CH2CN), 4.63 (t, 2H, J = 6.51 Hz, NCH2CH2CN), 6.40 (s, 1H, 8-H), 7.50 (m, 1H, 4′-H), 7.56 (m, 1H, 2-H), 7.57 (m, 2H, 3′-H and 5′-H), 7.60 (m, 1H, 5-H), 7.61 (m, 1H, 1-H), 7.86 (m, 2H, 2′-H and 6′-H), 8.00 (d, 1H, J = 8.64 Hz, 4-H), 11.69 ppm (bs, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 19.55 (NCH2CH2CN), 41.85 (NCH2CH2CN), 104.81 (1-C), 108.72 (8-C), 113.06 (5-C), 116.04 (4-C), 118.26 (9a-C), 119.27 (CN), 123.90 (9b-C), 127.83 (2′-C and 6′-C), 129.16 (4′-C), 129.44 (3′-C and 5′-C), 130.51 (2-C), 131.63 (5a-C), 135.00 (7-C), 137.04 (3a-C), 137.04 (1′-C), 178.38 ppm (9-C). IR (KBr): υ = 3424, 2247, 1506 cm−1. UV–vis (MeOH): λmax (ε) = 204 (0.608), 269 (0.378), 336 nm (0.179); λmin (ε) = 245 (0.183), 311 nm (0.101). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C20H16N3O+, 314.1293; found, 314.1303. RP-C18 HPLC: tR = 11.09 min, 98.18%.
Ethyl 2-(9-Oxo-7-phenyl-6H-pyrrolo[3,2-f] quinolin-3(9H)-yl)acetate (5d)
Compound 5d was prepared as for compound 5a by reacting 1.024 g (3.15 mmol) of phenylacrylate derivative 4d. An amount of 0.634 g of a beige solid was obtained, and this was purified by silica gel column chromatography (d = 2.5, l = 35, 230–400 mesh, eluent ethyl acetate) to yield 0.380 g of a golden beige solid. Yield 30%; mp = 107 °C; Rf = 0.27 (fluorescent blue spot, ethyl acetate); 1H NMR (300 MHz, DMSO-d6) δ = 1.63 (t, 3H, J = 7.02 Hz, CH2CH3), 4.20 (q, 2H, J = 7.02 Hz, CH2CH3), 4.95 (s, 2H, CH2), 6.79 (d, 1H, J = 1.38 Hz, 8-H), 7.87 (d, 1H, J = 3.01 Hz, 1-H), 7.99 (m, 5H, 4′-H, 5′-H, 3′-H, 4-H and 2-H), 8.21 (d, 1H, J = 9.06 Hz, 5-H), 8.26 (m, 2H, 2′-H and 6′-H), 11.66 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 14.15 (CH2CH3), 61.85 (CH2CH3), 68.34 (–CH2-), 102.71 (1-C), 107.62 (8-C), 111.06 (5-C), 114.04 (4-C), 117.26 (9a-C), 124.17 (9b-C), 128.33 (2′-C and 6′-C), 130.15 (4′-C), 130.84 (3′-C and 5′-C), 134.56 (2-C), 135.67 (5a-C), 138.00 (7-C), 138.54 (3a-C), 139.94 (1′-C), 178.68 ppm (9-C). IR (KBr): ν = 3500, 1650, 1500 cm−1. UV–vis (MeOH): λmax (ε) = 203 (0.700), 265 (0.840), 339 nm (0.360), λmin (ε) = 246 (0.650), 314 nm (0.250). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C21H19N2O3+, 347.1396; found, 347.1372. RP-C18-HPLC: 98.84%, tR = 10.07 min.
7-Phenyl-3-propionyl-3H-pyrrolo[3,2-f] quinolin-9(6H)-one (5e)
Compound 5e was prepared as for compound 5a by reacting 0.250 g (0.689 mmol) of phenylacrylate derivative 4e. An amount of 0.130 g of a solid product was obtained, and this was recrystallized from methanol giving 0.065 g of a slightly brown solid. Yield 30%; mp = 289 °C (with decomposition); Rf = 0.73 (blue fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 1.21 (t, 3H, J = 7.23 Hz, C(O)CH2CH3), 3.15 (q, 2H, J = 7.23 Hz, CH2), 6.42 (s, 1H, 8-H), 7.59 (m, 3H, 3′-H, 4′-H and 5′-H), 7.75 (d, 1H, J = 9.06 Hz, 5-H), 7.84 (m, 1H, 1-H), 7.86 (m, 2H, 2′-Hand6′-H), 8.03 (d, 1H, J = 3.61 Hz, 2-H), 8.69 (d, 1H, J = 9.06 Hz, 4-H), 11.84 ppm (bs, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 9.03 (CH2CH3), 28.87 (CH2CH3), 109.01 (8-C), 109.93 (1-C), 116.03 (5-C), 117.78 (9a-C), 120.77 (4-C), 126.75 (9b-C), 127.43 (2-C), 127.91 (2′-C and 6′-C), 129.45 (3′-C and 5′-C), 130.73 (4′-C), 131.03 (3a-C), 134.77 (1′-C), 138.60 (5a-C), 149.10 (7-C), 173.03 (C(O)CH2CH3), 178.51 ppm (9-C). IR (ATR ZnSe): ν = 2937, 1701, 1627, 1461 cm−1. UV–vis (MeOH): λmax (ε) = 203 (0.374), 272 (0.445), 326 nm (0.165); λmin (ε)= 243 (0.183), 306 nm (0.112); fluorescence (MeOH), λexc = 272 nm, λems = 413 nm. HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C20H17N2O2+, 317.1290; found, 317.1301. RP-C18-HPLC: 97.23%, tR = 12.97 min.
3-(Cyclopropylmethanone)-7-phenyl-H-pyrrolo[3,2-f]-quinolin-9(6H)-one (5f)
Compound 5f was prepared as for compound 5a by reacting 0.650 g (2.13 mmol) of phenylacrylate derivative 4f. An amount of 0.430 g of a solid product was obtained, and this was recrystallized from ethyl acetate giving 0.266 g of a gray solid. Yield 46%; mp = 298 °C; Rf = 0.62 (chloroform/methanol, 9:1); 1H NMR (300 MHz, DMSO-d6) δ = 1.15 (m, 4H, CH2CH2), 2.79 (m, 1H, CH), 6.41 (s, 1H, 8-H), 7.71 (m, 3H, 3′-H, 4′-H and 5′-H), 7.83 (d, 1H, J = 3.46 Hz, 1-H), 7.97 (m, 3H, 5-H, 2′-H, 6′-H), 8.44 (d, 1H, J = 3.46 Hz, 2-H), 8.65 (d, 1H, J = 8.82 Hz, 4-H), 11.94 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 12.45 (CH2CH2), 13.05 (CH2CH2), 107.94 (8-C), 108.75 (1-C), 115.54 (5-C), 116.40 (9a-C), 119.65 (4-C), 125.65 (9b-C), 126.71 (2-C), 127.15 (2′-C and 6′-C), 128.32 (3′-C and 5′-C), 129.50 (4′-C), 130.83 (3a-C), 132.91 (1′-C), 137.56 (5a-C), 148.40 (7-C), 172.44 (C(O)CHCH2CH2), 177.31 ppm (9-C). IR (ATR ZnSe): υ = 3350, 3085, 2940, 1724, 1654, 1455 cm−1. UV–vis (MeOH): λmax (ε) = 203 (0.748), 274 (0.880), 325 nm (0.340); λmin (ε)= 243 (0.122), 306 nm (0.168); fluorescence (MeOH), λexc = 300 nm, λems = 412 nm. HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C21H17N2O2+, 329.1290; found, 329.1297. RP-C18 HPLC: tR = 12.93 min, 98.13%.
3-(Cyclopropylmethanone)-2,3-dihydro-7-phenyl-1H-pyrrolo[3,2-f] quinolin-9(6H)-one (8f)
Compound 8f was prepared as for compound 5a by reacting 0.350 g (0.94 mmol) of phenylacrylate derivative 7f. An amount of 0.202 g of a yellow solid product was obtained, and this was purified by silica gel column chromatography (d = 2.5, l = 30, 230–400 mesh, eluent ethyl acetate/methanol, 8:2) yielding 0.120 g of a slightly yellow solid. Yield 30%; mp = 317 °C (with decomposition); Rf = 0.67 (blue fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 0.88 (m, 4H, CH2CH2), 1.95 (m, 1H, CH), 3.77 (t, 2H, J = 8.34 Hz, 1-H2), 4.36 (t, 2H, J = 8.34 Hz, 2-H2), 6.22 (d, 1H, J = 1.82 Hz, 8-H), 7.58 (m, 4H, 5-H, 3′-H, 4′-H, and 5′-H), 7.81 (m, 2H, 2′-H and 6′-H), 8.41 (d, 1H, J = 8.49 Hz, 4-H), 11.75 ppm (bs, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 8.25 (CH2CH2), 13.68 (CH), 29.69 (1-C), 49.06 (2-C), 107.58 (8-C), 118.08 (5-C), 120.78 (4-C), 122.38 (9a-C), 127.68 (2′-C and 6′-C), 129.43 (3′-C and 5′-C), 130.27 (9b-C), 130.83 (4-C), 134.57 (1′-C), 138.00 (3a-C), 139.96 (5a-C), 149.63 (7-C), 171.43 (C(O)CHCH2CH2), 178.86 (9-C). IR (ATR ZnSe): υ = 2960, 2920, 1630, 1590, 1440 cm−1. UV–vis (MeOH): λmax (ε) = 204 (0.992), 279 (1.557), 352 nm (0.281); λmin (ε)= 195 (0.322), 237 (0.443), 336 nm (0.225); fluorescence (MeOH), λexc = 277 nm, λems = 460 nm. HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C21H19N2O2+, 331.1441; found, 331.1423. RP-C18 HPLC: tR = 11.56 min, 96.33%.
General Procedure for Synthesis of 9-Alkoxyphenylpyrrolo-quinolinones 9–11
As a typical procedure, the synthesis of 9-methoxyl derivative 9 is described in detail. Into a two-necked 50 mL flask 0.0625 g (2.60 mmol) of NaH 60% was placed and washed three times with toluene and finally suspended in 15 mL of anhydrous THF under a nitrogen atmosphere. Then, to the cooled suspension, 0.150 g (0.520 mmol) of starting material 19 (MG 2474)7 was added. The reaction mixture changed from yellow to slightly brown while developing white fumes (hydrogen) and was stirred for 45 min. Next, 0.01 mL (1.56 mmol, d = 2.28 g/mL) of CH3I in 1 mL of anhydrous THF was introduced, and the resulting mixture was stirred at room temperature. The progress of the reaction was monitored by TLC analysis (ethyl acetate/methanol, 9:1). After 12 h, the reaction mixture was treated with 20 mL of water, neutralized with 3 N HCl and extracted with ethyl acetate (3 × 50 mL). The combined extracts were dried with anhydrous Na2SO4, and the solvent was removed on a rotavapor to yield 0.156 g of a dark dense liquid. The crude reaction product was purified by silica gel column chromatography (d = 2.5, l = 25 cm, 230–400 mesh, eluent ethyl acetate/methanol, 9:1) to yield 0.100 g of a white solid.
3-Ethyl-9-methoxy-7-phenyl-3H-pyrrolo[3,2-f] quinoline (9)
Yield 64%; mp = 248 °C; Rf = 0.88 (green fluorescent spot, ethyl acetate/methanol, 9:1); 1H NMR (400 MHz, DMSO-d6) δ = 1.42 (t, 3H, J = 7.21 Hz, CH2CH3), 4.23 (s, 3H, OCH3), 4.37 (q, 2H, J = 7.21 Hz, CH2CH3), 7.20 (dd, 1H, J = 3.00 and J = 0.69 Hz), 7.44 (m, 1H, 4′-H), 7.54 (m, 3H, 2-H, 3′-H, 5′-H), 7.60 (s, 1H, 8-H), 7.74 (d, 1H, J = 9.08 Hz, 5-H), 7.96 (dd, 1H, J = 9.08 Hz and J = 0.69 Hz, 4-H), 8.29 ppm (dd, 2H, J = 8.34 Hz and J = 1.31 Hz, 6′-H, 2′-H); 13C NMR (75 MHz, DMSO-d6) δ = 15.48 (CH2CH3), 40.14 (CH2CH3), 55.39 (OCH3), 104.38 (C-1), 113.35 (C-8), 114.75 (C-9a), 118.09 (C-4), 119.08 (C-5), 122.11 (C-9b), 126.28 (C-2), 126.48 (C-2′ and C-6′), 128.28 (C-3′ and C-5′), 130.93 (C-4′), 139.16 (C-3a), 139.45 (C-1′), 145.33 (C-5a), 153.17 (C-7), 162.66 ppm (C-9). UV–vis (MeOH): λmax= 218 (0.984), 265 (0.525), 351 nm (0.367); λmin= 247 (0.634), 310 nm (0.145). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C20H19N2O+, 303.1497; found, 303.1491. RP-C18-HPLC: 99.02%, tR = 15.93 min.
3-Ethyl-7-phenyl-9-butoxy-3H-pyrrolo[3,2-f] quinoline (10)
Compound 10 was prepared as for compound 9 by reacting 0.300 g (1.04 mmol) of compound 22 dissolved in 5 mL of anhydrous DMF and 0.17 mL (1.56 mmol; d = 1.27 g/mL) of BrC4H9 in 1 mL of di-DMF. An amount of 0.348 g of a dark brown solid was obtained, and this was recrystallized from methanol giving 0.259 g of a slightly brown solid. Yield 72%; mp = 120 °C; Rf = 0.87 (green fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (300 MHz, DMSO-d6) δ = 1.03 (t, 3H, J = 7.52 Hz, CH2CH2CH2CH3), 1.41 (t, 3H, J = 7.23 Hz, CH2CH3), 1.63 (sextet, 2H, J = 7.52 Hz, CH2CH2CH2CH3), 1.98 (quintuplet, 2H, J = 7.52 Hz and J = 6.15 Hz, CH2CH2CH2CH3), 4.34 (q, 2H, J = 7.23 Hz, CH2CH3), 4.44 (t, 2H, J = 6.15 Hz, CH2CH2CH2CH3), 7.23 (d, 1H, J = 2.81 Hz, 1-H), 7.46 (m, 1H, 4′-H), 7.54 (d, 1H, J = 2.81 Hz, 2-H), 7.54 (m, 3H, 2′-H, 3′-H, 5′-H), 7.60 (s, 1H, 8-H), 7.75 (d, 1H, J = 9.09 Hz, 5-H), 7.94 (d, 1H, J = 9.09 Hz, 4-H), 8.30 ppm (m, 1H, 6′-H); 13C NMR (75 MHz, DMSO-d6) δ = 14.2 2 (CH2CH2CH2CH3), 16. 46 (CH2CH3), 19.54 (CH2CH2CH2CH3), 31.26 (CH2CH2CH2CH3), 41.10 (CH2CH3), 68.45 (CH2CH2CH2CH3), 99.25 (C-8), 105.16 (C-1), 114.36 (C-9a), 115.64 (C-4), 120.17 (C-9b), 123.17 (C-4 and C-5), 127.29 (C-2′ and C-6′), 129.01 (C-3′ and C-5′), 129.21 (C-4′), 131.94 (C-3a), 140.19 (C-1′), 146.41 (C-5a), 154.14 (C-7), 162.96 ppm (C-9). UV–vis (MeOH): λmax (ε) = 210 (1.011), 239 (0.743), 284 nm (1.610); λmin (ε)= 229 (0.663), 248 nm (0.591). IR (ATR ZnSe): 2953–2869 (CH2, CH3), 1524–1469 (C=C), 1230 (C–O) cm−1; fluorescence (MeOH), λexc = 284 nm, λem = 514.92 nm. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C23H25N2O+, 345.1967; found, 345.1997. RP-C18 HPLC: 99.84%, tR = 17.60 min.
9-(Benzyloxy)-3-ethyl-7-phenyl-3H-pyrrolo[3,2-f] quinoline (11)
Compound 11 was prepared as for compound 9 by reacting 0.150 g (0.520 mmol) of compound 22 dissolved in 5 mL of anhydrous DMF and 0.360 g (1.560 mmol) of 4-nitrobenzylbromide dissolved in 2–3 mL of anhydrous DMF. An amount of 0.164 g of a dark brown solid product was obtained, and this was purified by silica gel column chromatography (d = 2.5, l = 25 cm, 230–400 mesh, eluent ethyl acetate/methanol, 9:1) giving 0.132 g of a brown solid. Yield 74%; mp = 131–134 °C; Rf = 0.76 (green fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (300 MHz, CDCl3) δ = 1.50 (t, 3H, J = 7.25 Hz, CH2CH3), 4.25 (q, 2H, J = 7.31 Hz, CH2CH3), 5.96 (s, 2H, -CH2-), 7.15 (d, 1H, J = 3.01 Hz, 1-H), 7.23 (d, 1H, J = 3.02 Hz, 2-H), 7.51 (m, 3H, 3′-H, 4′-H, and 5′-H), 7.61 (d, 2H, J = 8.55 Hz, 2″-H and 6″-H), 7.84 (d, 1H, J = 9.09 Hz, 5-H), 7.93 (s, 1H, 8-H), 8.15 (d, 1H, J = 9.03 Hz, 4-H), 8.27 (dd, 2H, J = 7.17 Hz and J = 3.23 Hz, 2′-H and 6′-H), 8.35 ppm (m, 3H, J = 8.77 Hz, 3″-H, 4″-H, and 5″-H); 13C NMR (75 MHz, CDCl3) δ = 15.31 (CH2CH3), 41.04 (CH2CH3), 45.96 (–CH2-), 104.05 (C-1), 110.02 (C-8), 115.18 (C-4), 115.52 (C-5), 118.45 (C-9a), 126.10 (C-9b), 127.96 (C-2), 128.39 (C-2″ and C-6″), 128.43 (C-2′ and C-6′), 128.53 (C-3″ and C-5″), 128.75 (C-3′ and C-5′), 129.23 (C-4″), 129.54 (C-4′), 131.97 (C-3a), 134.28 (C-1″), 134.81 (C-1′), 152.33 (C-5a), 154.26 (C-7), 164.72 (C-9), 170.94 ppm (–COO). UV–vis (MeOH): λmax (ε) = 205 (0.925), 239 (0.524), 281 nm (0.295); λmin (ε) = 231 (0.628), 247 nm (0.263). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H23N2O+ = 379.1810; found, 379.1805. RP-C18-HPLC: 96.54%, tR = 16.74 min.
Synthesis of Benzyl 3-Ethyl-7-phenyl-3H-pyrrolo[3,2-f]-quinolin-9-ylcarbonate (12)
Into a two-necked 50 mL flask 0.0625 g (2.60 mmol) of NaH 60% was placed and washed at least three times with toluene and finally suspended in 15 mL of anhydrous THF under a nitrogen atmosphere. Then, to the ice-bath cooled suspension, 0.150 g (0.520 mmol) of the starting material 197 was added. The reaction mixture changed from yellow to slightly brown while developing white fumes (hydrogen) and was stirred for 45 min. Next, 0.15 mL (1.04 mmol, d = 1.195 g/(mL) of benzylchloroformate in 2 mL of anhydrous THF was introduced, and the mixture was stirred at 0 °C. The progress of the reaction was monitored by TLC analysis (ethyl acetate/methanol, 9:1). After 3 h, the reaction mixture was treated with 15 mL of water, neutralized with 3 N HCl, and extracted with ethyl acetate (3 × 50 mL). The combined extracts were dried with anhydrous Na2SO4, and the solvent was removed on a rotavapor to yield a dark yellow semisolid residue that was recrystallized from methanol giving 0.212 g of a yellow solid. Yield 96%; mp = 215–218 °C; Rf = 0.89 (blue fluorescent spot, ethyl acetate/methanol, 9:1); 1H NMR (300 MHz, CDCl3) δ = 1.53 (t, 3H, J = 7.30 Hz, CH2CH3), 4.31 (q, 2H, J = 7.31 Hz, CH2CH3), 5.40 (s, 2H, CH2-Benz), 7.13 (d, 1H, J = 3.05 Hz, 1-H), 7.23 (d, 1H, J = 3.06 Hz, 2-H), 7.40 (m, 3H, 3″-H, 4″-H, and 5″-H), 7.46 (d, 2H, J = 8.57 Hz, 6″-H and 2″-H), 7.51 (m, 3H, 3′-H, 4′-H, and 5′-H), 7.80 (d, 1H, J = 8.02 Hz, 5-H), 7.94 (s, 1H, 8-H), 8.05 (d, 1H, J = 8.05 Hz, 4-H), 8.18 ppm (m, 2H, J = 7.19 Hz and J = 3.21 Hz, 6′-H and 2′-H); 13C NMR (75 MHz, CDCl3) δ = 15.62 (CH2CH3), 41.08 (CH2CH3), 69.37 (CH2-Benz), 104.05 (C-1), 110.02 (C-8), 115.18 (C-4), 115.52 (C-5), 118.45 (C-9a), 126.10 (C-9b), 127.09 (C-2), 127.96 (C-2″ and C-6″), 128.18 (C-2′ and C-6′), 128.22 (C-3″ and C-5″), 128.27 (C-3′ and C-5′), 129.39 (C-4′), 129.43 (C-4″), 131.97 (C-3a), 134.28 (C-1″), 134.81 (C-1′), 154.26 (C-5a), 154.37 (C-7), 164.72 (C-9), 178.24 ppm (-OCOO-). UV–vis (MeOH): λmax (ε) = 207 (0.458), 284 (0.273), 339 nm (0.101); λmin (ε) = 250 (0.317), 316 nm (0.082). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C27H23N2O3+, 423.1709; found, 423.1705. RP-C18-HPLC: 98.52%, tR = 17.04 min.
3-Ethyl-6-methyl-7-phenyl-3H-pyrrolo[3,2-f] quinolin-9(6H)-one (13)
In a 100 mL flask, 0.175 g (0.58 mmol) of 9-methoxy-3-ethyl-7-phenyl-3H-pyrrolo[3,2-f] quinoline (9) was dissolved in 3 mL of CH3I. The solution was maintained at reflux for 12 h, and the reaction was monitored by TLC (ethyl acetate/methanol, 8:2) until the starting material 9 disappeared and a new yellow fluorescent spot appeared. Then, 5 mL of 1.5 M NaOH in water was added to the reaction mixture and heating at reflux resumed for 2 h. After cooling, the mixture was extracted with ethyl acetate (4 × 50 mL), and the combined extracts were dried with sodium sulfate and filtered. The solvent was evaporated, giving 0.031 g of a pure yellow solid. Yield 18%; Rf = 0.86 (blue fluorescent spot, ethyl acetate/methanol, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 1.42 (t, 3H, J = 7.20 Hz, NCH2CH3), 3.67 (s, 3H, N(quinoline)-CH3), 4.37 (q, 2H, J = 7.20 Hz, NCH2CH3), 6.04 (s, 1H, H-8), 7.57 (m, 1H, H-2), 7.57 (m, 5H, H-2′, H-3′, H-4′, H-5′, H-6′), 7.59 (d, 1H, J = 9.35 Hz, H-5), 7.67 (dd, 1H, J = 2.92 Hz and J = 0.72 Hz, H-1), 8.00 ppm (dd, 1H, J = 9.35 Hz and J = 0.70 Hz, H-4); 13C NMR (101 MHz, DMSO-d6) δ = 16.37 (NCH2CH3), 38.69 (N(quinoline)-CH3), 41.02 (NCH2CH3), 104.38 (C-1), 110.96 (C-5), 112.59 (C-8), 115.94 (C-4), 119.63 (C-9a), 124.30 (C-2), 124.30 (C-9b), 129.15 (C-3′ and C-5′), 129.24 (C-2′ and C-6′), 129.45 (C-4′), 131.65 (C-1′), 136.68 (C-5a), 138.05 (C-3a), 177.16 ppm (C-9). IR (KBr): 3447.02 (C=O), 2924.41 (CH3, CH2CH3), 1582.55 (C=C), 1450.86 (CH3) cm−1. UV–vis (MeOH): λmax (ε) = 351 (0.415), 264 (0.746), 219 nm (0.99); λmin (ε) = 306.35 (0.138), 246 nm (0.467). HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for C20H19N2O+, 303.1497; found, 303.1500. RP-C18-HPLC: 98.77%, tR = 11.49 min.
3-(3-Hydroxypropyl)-7-phenyl-3H-pyrrolo[3,2-f] quinolin-9(6H)-one (14)
Into a 100 mL dry two-necked flask, 0.084 g (2.22 mmol) of LiAlH4 and 15 mL of anhydrous THF were placed. After stirring the suspension for 2–3 min under a nitrogen atmosphere, 0.200 g (0.562 mmol) of pyrroloquinolinone derivative 21 dissolved in 15 mL of anhydrous THF was added and the mixture stirred for 2 h at room temperature under a nitrogen atmosphere. The reaction was monitored by TLC (ethyl acetate/n-hexane, 8:2). At the end of the reaction, excess LiAlH4 was deactivated by adding 10 mL of saturated NH4Cl in water. After filtering the suspension, the filtrate was concentrated to dryness on a rotavapor, yielding a dark brown residue. The latter was treated with water and extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed several times with water and dried with sodium sulfate. The solvent was removed in a rotavapor, giving a dark yellow solid (0.156 g). Finally, the reaction product was purified with a silica gel chromatography column (d = 3 cm, l = 30 cm, 230–400 mesh, eluent ethyl acetate/n-hexane, 8:2) to give 0.123 g of a pale-yellow solid. Yield 70%; Rf = 0.45 (blue fluorescent spot, ethyl acetate/n-hexane, 8:2); mp = 213-215 °C; 1H NMR (300 MHz, DMSO-d6) δ = 1.94 (m, 2H, J = 6.51 Hz, NCH2CH2CH2OH), 3.38 (t, 2H, J = 6.13 Hz, NCH2CH2CH2OH), 4.38 (t, 2H, J = 6.89 Hz, NCH2CH2CH2OH), 5.47 (bs, 1H, OH), 6.70 (s, 1H, 8-H), 7.53 (m, 2H, J = 2.92 Hz, 1-H and 2-H), 7.61 (m, 3H, 3′-H, 4′-H and 5′-H), 7.71 (d, 1H, J = 8.98 Hz, 5-H), 7.90 (m, 2H, J = 7.54 Hz and J = 2.31 Hz, 6′-H and 2′-H), 8.01 (d, 1H, J = 9.00 Hz, 4-H), 12.46 ppm (bs, 1H, NH); 13C NMR (75 MHz, DMSO-d6) δ = 33.41 (NCH2CH2CH2OH), 42.68 (NCH2CH2CH2OH), 57.60 (NCH2CH2CH2OH), 103.78 (C-1), 116.56 (C-8), 118.53 (C-4), 122.10 (C-5), 123.36 (C-9a), 127.51 (C-9b), 127.51 (C-2), 128.97 (C-2′ and C-6′), 129.16 (C-3′ and C-5′), 130.23 (C-4′), 131.49 (C-3a), 134.19 (C-1′), 148.29 (C-5a), 156.56 (C-7), 178.74 ppm (C-9). UV– vis (MeOH): λmax (ε) = 205 (0.925), 271 (0.714), 346 nm (0.451); λmin = 248 (0.613), 314 nm (0.514). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C20H19N2O2+ = 319.1447; found, 319.1472. RP-C18-HPLC: 98.78%, tR = 10.25 min.
3-(9-Oxo-7-phenyl-6H-pyrrolo[3,2-f] quinolin-3(9H)-yl)-propanoic Acid (15)
Into a 50 mL two-necked flask, a solution of 0.200 g (0562 mmol) of pyrroloquinolinone derivative II (MG 2540) in 15 mL of methanol was placed, and 3 mL of 20% NaOH in water was added. The mixture was stirred at 60–70 °C for about 2 h, and the reaction was monitored by TLC (chloroform/methanol, 9:1). At the end of the reaction, the solution was cooled to room temperature, and the solvent was removed on a rotavapor, yielding a dark yellow solid. The dry residue was treated with water and then extracted with ethyl acetate (2 × 50 mL) to remove unreacted compounds. The aqueous phase was then acidified to pH 3–4 with 37% HCl, and immediately a yellow precipitate formed. The precipitate was collected, washed several times with water, and dried under vacuum to yield 0.157 g of an intensely yellow solid. Yield 85%; Rf = 0.20 (blue fluorescent spot, chloroform/methanol, 9:1); mp = 280 °C; 1H NMR (300 MHz, DMSO-d6) δ = 2.80 (t, 2H, J = 6.76 Hz, NCH2CH2COOH), 4.52 (t, 2H, J = 6.76 Hz, NCH2CH2COOH), 6.54 (s, 1H, 8-H), 7.51 (dd, 2H, J = 2.98 Hz, 2-H and 1-H), 7.58 (m, 3H, 3′-H, 4′-H and 5′-H), 7.63 (d, 1H, J = 8.96 Hz, 5-H), 7.86 (dd, 2H, J = 7.71 Hz and J = 2.01 Hz, 6′-H and 2′-H), 7.96 (d, 1H, J = 9.00 Hz, 4-H), 12.46 ppm (bs, 1H, NH), (no signal for the carboxylic acid proton); 13C NMR (75 MHz, DMSO-d6) δ = 34.60 (NCH2CH2 COOH), 41.20 (NCH2CH2COOH), 103.43 (C-1), 112.40 (C-8), 115.44 (C-5), 122.10 (C-4), 123.24 (C-9a), 127.41 (C-9b), 127.47 (C-2), 128.77 (C-2′ and C-6′), 129.06 (C-3′ and C-5′), 129.97 (C-4′), 130.13 (C-3a), 131.35 (C-1′), 134.22 (C-5a), 147.44 (C-7), 167.35 (COOH), 171.87 ppm (C-9). UV–vis (MeOH): λmax (ε) = 206 (0.981), 271 (0.613), 347 nm (0.451); λmin (ε) = 247 (0.714), 314 nm (0.581). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C20H17N2O3+ = 333.1239; found, 333.1265. RP-C18-HPLC: 97.98%, tR = 9.17 min.
2-(9-Oxo-7-phenyl-6H-pyrrolo[3,2-f] quinolin-3(9H)-yl)acetic Acid (16)
Into a 50 mL two-necked flask, 0.150 g (0.43 mmol) of pyrroloquinolinone derivative 5d in 8 mL of methanol was place, and 3 mL of 20% NaOH in water was added. The mixture was stirred at 60–70 °C for about 2 h, and the reaction was monitored by TLC (chloroform/methanol, 9:1). At the end of the reaction, the solution was cooled to room temperature, and the solvent was removed on a rotavapor, yielding a greyish-white solid (0.120 g). The residue was treated with water and then extracted with ethyl acetate (2 × 50 mL) to remove unreacted compounds. The aqueous phase was then acidified to pH 3–4 with 37% HCl, and immediately a white precipitate formed. The precipitate was collected, washed several times with water, and dried under vacuum, giving 0.085 g of a white crystalline product. Yield 63%; Rf = 0.18 (chloroform/methanol, 7:3); mp = 273 °C; 1H NMR (300 MHz, DMSO-d6) δ = 5.67 (s, 2H, CH2COOH), 6.79 (d, 1H, J = 1.38 Hz, 8-H), 7.87 (d, 1H, J = 3.01 Hz, 1-H), 7.99 (m, 5H, 3′-H, 5′-H, 4′-H, 4-H and 2-H), 8.21 (d, 1H, J = 9.06 Hz, 5-H), 8.26 (m, 2H, 6′-H and 2′-H), 11.66 (bs, 1H, NH), 13.10 ppm (bs, 1H, COOH); 13C NMR (75 MHz, DMSO-d6) δ = 55.72 (NCH2COOH), 104.33 (C-1), 113.30 (C-5), 116.34 (C-8), 123.00 (C-4), 124.14 (C-9a), 126.31 (C-9b), 128.31 (C-2), 128.67 (C-2′ and C-6′), 129.67 (C-3′ and C-5′), 130.87 (C-3a), 131.03 (C-4′), 132.15 (C-1′), 135.12 (C-5a), 148.34 (C-7), 168.32 (COOH), 174.12 ppm (C-9). UV–vis (MeOH): λmax (ε) = 226 (0.815), 268 (0.613), 346 nm (0.481); λmin (ε) = 249 (0.724), 315 nm (0.523). HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C19H15N2O3+ 319.1083, found 319.1060. RP-C18-HPLC: 98.12%, tR = 8.74 min.
tert-Butyl 3-(9-Oxo-7-phenyl-6H-pyrrolo[3,2-f] quinolin-3(9H)-yl)propylcarbamate (17)
In a 100 mL flask, 0.626 g of pyrroloquinolinone derivative 5c was dissolved in hot MeOH. The solution was cooled to 0 °C, and 1.310 g (6 mmol) of di-tert-butyl dicarbonate and 0.110 g (0.4 mmol) of CoCl2·6H2O were added. To the resulting suspension 0.757 g (20 mmol), NaBH4 was added portionwise, with development of hydrogen and formation of a black precipitate. The mixture was stirred for 12 h at room temperature, and the formation of the reaction product was monitored by TLC (ethyl acetate/methanol, 8:2). Finally, 0.8 mL (2 mmol, d = 0.955 g/mL) of diethylenetriamine was added, and the mixture was dried on a rotavapor. The resulting clear solid was dissolved in 50 mL of ethyl acetate and the solution washed with a saturated solution of NaHCO3 (3 × 25 mL). The organic phase was dried with anhydrous Na2SO4 and evaporated to dryness, yielding 0.644 g of a light brown solid. Yield 77%; Rf = 0.79 (ethyl acetate/methanol, 8:2); 1H NMR (400 MHz, DMSO-d6) δ = 1.37 (s, 3H, OC(CH3)3), 1.39 (s, 6H, OC(CH3)3), 1.89 (quintuplet, 2H, J = 6.942 Hz, NCH2CH2CH2NHCO), 2.91 (m, 2H, J = 6.66 Hz and J = 5.88 Hz, NCH2CH2CH2NHCO), 4.29 (t, 2H, J = 6.93 Hz, NCH2CH2CH2NHCO), 6.38 (d, 1H, J = 1.78 Hz, H-8), 6.97 (t, 1H, J = 5.05 Hz, NHCO), 7.50 (d, 1H, J = 2.87 Hz, H-2), 7.55 (d, 1H, J = 2.77 Hz, H-1), 7.58 (m, 4H, H-3′, H-4′, H-5′, and H-5), 7.85 (m, 2H, H-2′ and H-6′), 7.87 (d, 1H, J = 9.04 Hz, H-4), 11.64 ppm (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ = 28.68 (OC(CH3)3), 28.72 (OC(CH3)3), 31.03 (NCH2CH2CH2NHCO), 37.96 (NCH2CH2CH2NHCO), 43.84 (NCH2CH2CH2NHCO), 104.02 (C-1), 108.63 (C-8), 112.71 (C-5), 118.31 (C-9a), 123.57 (C-9b), 127.81 (C-2′ and C-6′), 129.38 (C-2), 129.43 (C-3′ and C-5′), 130.46 (C-4′), 131.70 (C-3a), 135.04 (C-1′), 136.82 (C-5a), 147.71 (C-7), 156.10 (NHCOO), 178.37 ppm (C-9). IR (KBr): 3461.11 (C=O), 1653 (CONH) cm−1. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C25H28N3O3+ = 418.2131; found, 418.2137. RP-C18-HPLC: 96.55%, tR = 10.76 min.
3-(3-Aminopropyl)-7-phenyl-3H-pyrrolo[3,2-f] quinolin-9-ol hydrochloride (18)
In a 100 mL flask, 0.644 g (1.54 mmol) of pyrroloquinolinone derivative 17 was dissolved in 55 mL of an ethyl acetate/ethanol 2:1 mixture, and dry HCl gas was bubbled into the solution until a yellow precipitate formed. The mixture was left at 0–4 °C overnight, and the resulting precipitate was collected and dried under vacuum to yield 0.413 g of a powdery beige solid. Yield: 58%; Rf = 0.20 (blue fluorescent spot, chloroform/methanol, 8:2); mp = 194 °C; 1H NMR (400 MHz, DMSO-d6) δ = 2.16 (quintuplet, 2H, J = 6.84 Hz, NCH2CH2CH2NH3+Cl−), 2.77 (m, 2H, NCH2CH2CH2NH3+Cl−), 4.58 (t, 2H, J = 6.86 Hz, NCH2CH2CH2NH3+Cl−), 7.42 (dd, 1H, J = 2.99 Hz and J = 0.65 Hz, H-1), 7.67 (s, 1H, H-8), 7.70 (m, 1H, H-4′), 7.71 (m, 2H, H-3′ and H-5′), 7.86 (d, 1H, J = 3.21 Hz, H-2), 8.02 (m, 2H, H-2′ and H-6′), 8.17 (d, 1H, J = 9.19 Hz, H-5), 8.19 (m, 3H, NH3+), 8.46 (d, 1H, J = 6.86 Hz, H-4), 14.70 ppm (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6) δ = 28.94 (NCH2CH2CH2NH3+Cl−), 36.78 (NCH2CH2CH2NH3+Cl−), 43.82 (N-CH2CH2CH2NH3+Cl−), 105.19 (C-8), 105.40 (C-1), 113.22 (C-5), 114.10 (C-9a), 120.13 (C-4), 120.68 (C-9b), 129.05 (C-2′ and C-6′), 129.83 (C-3′ and C-5′), 130.95 (C-2), 132.12 (C-4′), 132.58 (C-3a), 132.61 (C-1′), 137.87 (C-5a), 151.15 (C-7), 169.98 ppm (CO). IR (KBr): 3411.56 (OH), 1618 (C=C) cm−1. UV–vis (MeOH): λmax (ε) = 338 (0.081), 270 (0.162), 204 nm (0.169); λmin (ε) = 312 nm (0.046), 246 nm (0.079); fluorescence (H2O), λexc = 215.00 nm, λem = 430.00 nm. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C20H20N3O+ = 318.1606; found, 318.1641, m/(2z) [M + 2H]2+ calculated for C20H21N3O2+ 159.5837; found, 159.5801. RP-C18-HPLC: 99.57%, tR = 8.96 min.
Biology. Cell Lines and Growth Inhibition Assays
The human cell lines were the following: cervix carcinoma (HeLa), adenocarcinomic alveolar basal epithelial cells (A549), colon adenocarcinoma (HT-29), breast adenocarcinoma (MCF-7), ovarian carcinoma (OVCAR-3), acute T-cell lymphoblastic leukemia (MOLT-3, Jurkat, and CCRF-CEM), chronic myelogenous leukemia cells (K562), B-cell leukemia (RS4;11and SEM), acute myelocytic leukemia cells (MV4;11), and acute monocytic leukemia cells (THP-1). All cell lines were purchased from the American Type Culture Collection. Leukemic cell lines were cultured in RPMI-1640 (Gibco, Milano, Italy), while solid tumor cell lines were cultured in DMEM (Gibco, Milano, Italy). Both media were supplemented with 10% fetal bovine serum (FBS), glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL; all media components were from Life Technologies, Italy) and maintained at 37 °C in a humidified atmosphere with 5% CO2. The cytotoxic activity of the compounds was determined using a standard MTT based colorimetric assay (Sigma-Aldrich, Milano, Italy). Briefly, cells were seeded at a density of 8 × 103 cells/well in 96-well microtiter plates. After 24 h, cells were exposed to various concentrations of the test compounds. After difierent times, cell survival was determined by the addition of an MTT solution as described previously.16 The GI50 value is defined as the compound concentration required to inhibit cell proliferation by 50%.
PBL from healthy donors were obtained by separation on a Lymphoprep (Fresenius KABI Norge AS) gradient. After extensive washing, cells were resuspended (1.0 × 106 cells/mL) in RPMI-1640 with 10% FBS and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, nonadherent cells were resuspended at 5 × 105 cells/mL in growth medium, containing 2.5 μg/mL PHA (Irvine Scientific). Difierent concentrations of the test compounds were added, and viability was determined 72 h later by the MTT test. For cytotoxicity evaluations in resting PBL cultures, nonadherent cells were resuspended (5 × 105 cells/mL) and treated for 72 h with the test compounds, as described above.
HUVECs were prepared from human umbilical cord veins, as previously described.29 The adherent cells were maintained in M200 medium supplemented with low serum growth supplement containing FBS, hydrocortisone, human epithelial growth factor (hEGF), basic fibroblast growth factor (bFGF), heparin, gentamycin/amphotericin (Life Technologies, Monza, Italy). Once confluent, the cells were detached by trypsin–EDTA solution and used in experiments from the first to sixth passages.
Effects on Tubulin Polymerization and on Colchicine Binding to Tubulin
To evaluate the effect of the compounds on tubulin assembly in vitro,17 varying concentrations of compounds were preincubated with 10 μM bovine brain tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After addition of 0.4 mM GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C. Tubulin assembly was followed turbidimetrically at 350 nm. The IC50 was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The ability of the test compounds to inhibit colchicine binding to tubulin was measured as described19 except that the reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine, and 5 μM test compound.
Analysis of Cell Cycle Distribution
5 ×105 cells in exponential growth were treated with difierent concentrations of test compound for difierent times. After the incubation, cells were collected, centrifuged, and fixed with ice-cold ethanol (70%) and stained with PI as described previously.45
Externalization of PS
Surface exposure of PS on apoptotic cells was measured by flow cytometry with a Coulter Cytomics FC500 (Beckmann Coulter, USA) instrument by adding annexin-V-FITC and PI to cells according to the manufacturer′s instructions (annexin-V Fluos, Roche Diagnostic, Monza, Italy).
Western Blot Analysis
Cells were treated with test compounds and, after difierent times, collected, centrifuged, and washed with ice cold phosphate buffered saline. The pellet was then resuspended in lysis buffer as described.25 The protein concentration in the supernatant was determined using the BCA protein assay (Pierce, Milano, Italy). Equal amounts of protein (10–20 μg) were resolved using SDS–PAGE gel electrophoresis (Criterion precast Tris-HCl gel, Biorad Laboratories, Milano, Italy) and transferred to PVDF Hybond-p membranes (GE Healthcare, Milano, Italy). Membranes were blocked with 2% ECL-blocking solution (GE Healthcare, Milano, Italy) for 2 h, with rotation at room temperature. Membranes were then incubated with primary antibodies against Bcl-2, p53, PARP, procaspase-9, procaspase-8, procaspase-2, Mcl-1, Bcl-XL (Cell Signaling, Milano, Italy), β-actin (Sigma-Aldrich, Milano, Italy), and caspase-3 (Novus Biologicals, Milano, Italy) overnight. Membranes were next incubated with peroxidase-conjugated secondary antibodies (Invitrogen, Milano, Italy) for 60 min. All membranes were visualized using ECL Select (GE Healthcare, Milano, Italy) and exposed to Hyperfilm MP (GE Healthcare, Milano, Italy). To ensure equal protein loading, each membrane was stripped and reprobed with anti-β-actin antibody.
Drug Combination Studies
Cell proliferation was assessed by the MTT assay after treatment. Equal concentrations of cells were plated in triplicate in a 96-well plate and treated for 48 h using scalar dilutions of 5f, combined with cytarabine (Aractyn, Pfizer), daunorubicin (Pfizer), and dexamethasone (Sigma-Aldrich) at fixed combination ratios. The effectiveness of various drug combinations was analyzed by the Calcusyn, version 2.1 software (Biosoft). The combination index (CI) was calculated according to the Chou–Talalay method. A combination index of 1 indicates an additive effect of the two drugs. Combination index values less than 1 indicate synergy, and combination index values greater than 1 indicate antagonism.
Molecular Docking Simulations
Target Structures
The three-dimensional structures of tubulin in complex with colchicine as well as all selected protein kinases were retrieved from the Protein Data Bank (www.rcsb.org).21 The collected PDB structures are summarized in Table 2 in Supporting Information.
The assessment of crystallographic structure quality has been performed with the Structure Preparation tool of the Molecular Operation Environment (MOE, version 2014.09) program.46 Critical structural issues (such as missing or poorly resolved atomic data, anomalous topological properties present in amino acids units as well as anomalous bonding patterns of non amino acids units) were fixed when necessary. Hydrogen atoms were added and their appropriate protonation state was fixed using the Protonate3D tool as implemented in the MOE program. To minimize contacts between hydrogen atoms, the structures were subjected to Amber99 force field47 minimization until the rms of the conjugate gradient was <0.1 kcal mol−1 Å−1, keeping the heavy atoms fixed at their crystallographic positions.
Molecular Docking Protocol
All 3-substituted 7-PPyQs were built using the “Builder” module of MOE, and each compound was docked into the presumptive binding sites (colchicine or ATP binding sites) using flexible MOE-Dock methodology. The purpose of MOE-Dock is to search for favorable binding configurations between a small, flexible ligand and a rigid macromolecular target. Searching is conducted within a user-specified 3D docking box, using the “Tabu search”48 protocol and the MMFF94 force field.49 Charges for ligands were imported from the MOPAC program50 output files. MOE-Dock performs a user-specified number of independent docking runs (50 in the present case) and writes the resulting conformations and their energies to a molecular database file. The resulting ligand/protein complexes were subjected to MMFF94 all-atom energy minimization until the rms of conjugate gradient was <0.1 kcal mol−1 Å−1. GB/SA approximation51 has been used to model the electrostatic contribution to the free energy of solvation in a continuum solvent model. The interaction energy values were calculated as the energy of the complex minus the energy of the ligand minus the energy of the protein:
ΔEinter = Ecomplex − (EL + Eprotein)
Supplementary Material
ACKNOWLEDGMENTS
The synthetic work coordinated by M.G.F. was carried out with financial support from the University of Padova, Italy, and the Italian Ministry for University and Research (MIUR), Rome, Italy. The molecular modeling work coordinated by S.M. was carried out with financial support from the University of Padova, Italy, and the Italian Ministry for University and Research (MIUR), Rome, Italy. S.M. is also very grateful to the Chemical Computing Group for its scientific and technical partnership. G.V., R.B. and G.B. thank the Fondazione Cariparo by the “Progetto Ricerca Pediatrica”. The content is solely the responsibility of the authors and does not necessarily reffect the official views of the National Institutes of Health.
ABBREVIATIONS USED
- PPyQ
phenylpyrroloquinolinone
- CA-4
combretastatin A-4
- PyQ
3H-pyrrolo[3,2-f] quinolin-9-one
- ATR
attenuated total reffectance
- MOE
Molecular Operating Environment
- FBS
fetal bovine serum
- PI
propidium iodide
- PS
phosphatidylserine
- HUVEC
human umbilical vein endothelial cell
- PBL
peripheral blood lymphocyte
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00805.
Complete 1H and 13C NMR signals assignment and structure elucidation for compound 10; 1H NMR spectrum (400 MHz, DMSO-d6) of compound 10; 13C{1H} NMR spectrum (101 MHz, DMSO-d6) of compound 10; 1H–1H 2D COSY and 1H–13C 2D HMBC NMR correlation tables (DMSO-d6) of compound 10; HRMS (ESI-MS, 140 eV) spectrum of compound 10; HPLC trace of compound 10; complete 1H and 13C NMR signals assignment and structure elucidation for compound 18; 1H NMR spectrum (400 MHz, DMSO-d6) of compound 18; 13C{1H} NMR spectrum (101 MHz, DMSO-d6) of compound 18; 1H–1H 2D COSY NMR correlation table (DMSO-d6) of compound 18; HRMS (ESI-MS, 140 eV) spectrum of compound 18; HPLC trace of compound 18; HRMS (ESI, 140 eV) spectra and HPLC traces of compounds 5a–f, 8f, 9, 11–17; behavior of compound 18 in aqueous solution at various pH values; elemental analysis of all final compounds 5a–f, 8f, and 9–18; combination cytotoxicity of 5f and daunorubucine (Dauno), dexamethasone (Dex), cytarabine (Ara-C); dose–response curves of Jurkat (A) and THP1 (B) to 5f; Table S2 of the collected PDB structures and the corresponding references; Table S3 of simplified molecular-input line-entry for all final compounds 5a–f, 8f, and 9–18 (PDF) Molecular formula strings (CSV)
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. Ca-Cancer J. Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Shuttleworth SJ, Silva FA, Cecil ARL, Tomassi CD, Hill TJ, Raynaud FI, Clarke PA, Workman P. Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinositide 3-kinase (PI3K) inhibitors. Curr. Med. Chem. 2011;18:2686–2714. doi: 10.2174/092986711796011229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferlin MG, Chiarelotto G, Gasparotto V, Dalla Via L, Pezzi V, Barzon L, Palu G, Castagliuolo I. Synthesis and in vitro and in vivo antitumor activity of 2-phenylpyrroloquinolin-4-ones. J. Med. Chem. 2005;48:3417–3427. doi: 10.1021/jm049387x. [DOI] [PubMed] [Google Scholar]
- 5.Gasparotto V, Castagliuolo I, Chiarelotto G, Pezzi V, Montanaro D, Brun P, Palù G, Viola G, Ferlin MG. Synthesis and biological activity of 7-phenyl-6,9-dihydro-3H-pyrrolo[3,2-f] quinolin-9-ones: A new class of antimitotic agents devoid of aromatase activity. J. Med. Chem. 2006;49:1910–1915. doi: 10.1021/jm0510676. [DOI] [PubMed] [Google Scholar]
- 6.Ferlin MG, Conconi MT, Urbani L, Oselladore B, Guidolin D, Di Liddo R, Parnigotto PP. Synthesis, in vitro and in vivo preliminary evaluation of anti-angiogenic properties of some pyrroloazaflavones. Bioorg. Med. Chem. 2011;19:448–457. doi: 10.1016/j.bmc.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 7.Gasparotto V, Castagliuolo I, Ferlin MG. 3-Substituted 7-phenyl-pyrroloquinolinones show potent cytotoxic activity in human cancer cell lines. J. Med. Chem. 2007;50:5509–5513. doi: 10.1021/jm070534b. [DOI] [PubMed] [Google Scholar]
- 8.Ferlin MG, Bortolozzi R, Brun P, Castagliuolo I, Hamel E, Basso G, Viola G. Synthesis and in vitro evaluation of 3H-pyrrolo[3,2-f]-quinolin-9-one derivatives that show potent and selective anti-leukemic activity. ChemMedChem. 2010;5:1373–1385. doi: 10.1002/cmdc.201000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viola G, Bortolozzi R, Hamel E, Moro S, Brun P, Castagliuolo I, Ferlin MG, Basso G. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem. Pharmacol. 2012;83:16–26. doi: 10.1016/j.bcp.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase-moving towards therapy. Biochim. Biophys. Acta, Proteins Proteomics. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Goodwin S, Smith AF, Horning EC. Alkaloids of Lunasia amara blanco. 4-methoxy-2-phenylquinoline. J. Am. Chem. Soc. 1957;79:2239–2241. [Google Scholar]
- 13.Caddick S, Judd DB, Lewis AKDK, Reich MT, Williams MRV. A generic approach for the catalytic reduction of nitriles. Tetrahedron. 2003;59:5417–5423. [Google Scholar]
- 14.Wu C, Wu F, Bai Y, Yi B, Zhang H. Cobalt boride catalysts for hydrogen generation from alkaline NaBH4 solution. Mater. Lett. 2005;59:1748–1751. [Google Scholar]
- 15.Sahakitpichan P, Ruchirawat S. Highly efficient synthesis of buflavine: A unique Amaryllidaceae alkaloid. Tetrahedron Lett. 2003;44:5239–5241. [Google Scholar]
- 16.van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol. Biol. 2011;731:237–245. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- 17.Lin CM, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 18.Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharyya B, Panda D, Gupta S, Banerjee M. Antimitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- 20.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 21.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 23.Clarke PR, Allan LA. Cell-cycle control in the face of damage - a matter of life or death. Trends Cell Biol. 2009;19:89–98. doi: 10.1016/j.tcb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Kiyokawa H, Ray D. In vivo roles of CDC25 phosphatases: biological insight into the anti-cancer therapeutic targets. Anti-Cancer Agents Med. Chem. 2008;8:832–836. doi: 10.2174/187152008786847693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donzelli M, Draetta GF. Regulating mammalian checkpoints through cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012;199:871–881. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell. 2012;23:567–576. doi: 10.1091/mbc.E11-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: The histone guardian of the genome. DNA Repair. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 29.García-Sáez AJ. The secrets of the Bcl-2 family. Cell Death Differ. 2012;19:1733–1740. doi: 10.1038/cdd.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- 31.Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997;57:229–233. [PubMed] [Google Scholar]
- 32.Romagnoli R, Baraldi PG, Brancale A, Ricci A, Hamel E, Bortolozzi R, Basso G, Viola G. Convergent synthesis and biological evaluation of 2-amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl thiazoles as microtubule targeting agents. J. Med. Chem. 2011;54:5144–5153. doi: 10.1021/jm200392p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matson DR, Stukenberg PT. Spindle poisons and cell fate: A tale of two pathways. Mol. Interventions. 2011;11:141–150. doi: 10.1124/mi.11.2.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJC, Leong KG, Dueber EC, Maecker H, Huang DCS, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 35.Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castedo M, Perfettini, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: A molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 37.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34(cdc2) phosphorylation of survivin. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Porcù E, Viola G, Bortolozzi R, Persano L, Mitola S, Ronca R, Presta M, Romagnoli R, Baraldi PG, Basso G. TR-644 a novel potent tubulin binding agent induces impairment of endothelial cells function and inhibits angiogenesis. Angiogenesis. 2013;16:647–662. doi: 10.1007/s10456-013-9343-z. [DOI] [PubMed] [Google Scholar]
- 39.Profiling Service LeadHunter . KINOMEscan. DiscoveRx; Birmingham, U.K.: [Google Scholar]
- 40.Utepbergenov D, Derewenda U, Olekhnovich N, Szukalska G, Banerjee B, Hilinski MK, Lannigan DA, Stukenberg PT, Derewenda ZS. Insights into the inhibition of the p90 ribosomal S6 kinase (RSK) by the flavonol glycoside SL0101 from the 1.5 Å crystal structure of the N-terminal domain of RSK2 with bound inhibitor. Biochemistry. 2012;51:6499–6510. doi: 10.1021/bi300620c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell. 2004;13:169–178. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 42.Kulagowski JJ, Blair W, Bull RJ, Chang C, Deshmukh G, Dyke HJ, Eigenbrot C, Ghilardi N, Gibbons P, Harrison TK, Hewitt PR, Liimatta M, Hurley CA, Johnson A, Johnson T, Kenny JR, Bir Kohli P, Maxey RJ, Mendonca R, Mortara K, Murray J, Narukulla R, Shia S, Steffek M, Ubhayakar S, Ultsch M, Van Abbema A, Ward SI, Waszkowycz B, Zak M. Identification of imidazo-pyrrolopyridines as novel and potent JAK1 inhibitors. J. Med. Chem. 2012;55:5901–5921. doi: 10.1021/jm300438j. [DOI] [PubMed] [Google Scholar]
- 43.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 44.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 45.Bortolozzi R, Viola G, Porcù E, Consolaro F, Marzano C, Pellei M, Gandin V, Basso G. A novel copper(I) complex induces ER-stress-mediated apoptosis and sensitizes B-acute lymphoblastic leukemia cells to chemotherapeutic agents. Oncotarget. 2014;5:5978–5991. doi: 10.18632/oncotarget.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molecular Operating Environment (MOE) Chemical Computing Group Inc.; 1010 Sherbooke St. West, Suite No. 910, Montreal, QC, H3A 2R7, Canada: 2015. version 2014.09. [Google Scholar]
- 47.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 48.Baxter CA, Murray CW, Clark DE, Westhead DR, Eldridge MD. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins: Struct., Funct., Genet. 1998;33:367–382. [PubMed] [Google Scholar]
- 49.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. [Google Scholar]
- 50.Stewart JJP. MOPAC 7. Fujitsu Limited; Tokyo, Japan: 1993. [Google Scholar]
- 51.Wojciechowski M, Lesyng B. Generalized Born model: Analysis, refinement, and applications to proteins. J. Phys. Chem. B. 2004;108:18368–18376. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.