

Abstract
The ability to rapidly and specifically modify the genome of mammalian cells has been a long-term goal of biomedical researchers. Recently, the clustered, regularly interspaced, short palindromic repeats (CRISPR)/Cas9 system from bacteria has been exploited for genome engineering in human cells. The CRISPR system directs the RNA-guided Cas9 nuclease to a specific genomic locus to induce a DNA double-strand break that may be subsequently repaired by homology-directed repair using an exogenous DNA repair template. Here we describe a protocol using CRISPR/Cas9 to achieve bi-allelic insertion of a point mutation in human cells. Using this method, homozygous clonal cell lines can be constructed in 5–6 weeks. This method can also be adapted to insert larger DNA elements, such as fluorescent proteins and degrons, at defined genomic locations. CRISPR/Cas9 genome engineering offers exciting applications in both basic science and translational research.
INTRODUCTION
Genome engineering is a term used to describe the process of making specific, targeted alterations in the genome of a living organism. Genome engineering exploits the repair of a DNA double-strand break (DSB) through the endogenous pathway of homologous recombination (HR). By providing an exogenous DNA repair template that contains homology to the targeted site, it is possible to exploit the HR machinery to create defined alterations close to the site of a DSB. However, mammalian genomes comprise billions of base pairs and there is a low probability of a spontaneous DSB occurring close to the region to be targeted; as a consequence, desired recombination events occur extremely infrequently (Capecchi, 1989). A major breakthrough came with the demonstration that targeted DSBs greatly increase the frequency of homology-directed repair (HDR) at a specific locus (Choulika, Perrin, Dujon, & Nicolas, 1995; Plessis, Perrin, Haber, & Dujon, 1992; Rouet, Smih, & Jasin, 1994; Rudin, Sugarman, & Haber, 1989). This discovery has spurred the development of programmable endonucleases that can be exploited to promote site-specific cleavage of the genome.
Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are artificial restriction enzymes produced by fusing customizable DNA binding domains to the sequence-independent nuclease domain of the restriction enzyme Fok1 (Boch et al., 2009; Christian et al., 2010; Miller et al., 2007, 2011; Moscou & Bogdanove, 2009; Urnov et al., 2005). Fok1 requires dimerization for its activity, and thus a pair of ZFNs or TALENs is required to bind to opposite strands of DNA on either side of a target site to allow Fok1 dimerization and DNA cleavage. While ZFNs and TALENs have been shown to be capable of creating targeted DNA breaks and introducing genomic sequence changes through HDR, difficulties in protein design and synthesis proved to be a barrier to their widespread use (Hsu, Lander, & Zhang, 2014).
1. CRISPR/Cas SYSTEM
Recently, a new tool based on clustered, regularly interspaced, short palindromic repeats (CRISPR) systems from bacteria have been exploited for genome engineering in human cells and have generated considerable excitement (Hsu et al., 2014). CRISPR systems have the distinct advantage of using RNA-guided nuclease activity to target cleavage of DNA and thereby eliminate the need for protein engineering and optimization.
CRISPR/Cas modules were identified in bacteria as part of an adaptive immune system that enables hosts to recognize and cleave foreign invading DNA (Horvath & Barrangou, 2010; Marraffini & Sontheimer, 2010). CRISPR modules comprise arrays of short nucleotide repeats interspersed with unique spacers that share homology with foreign phage or plasmid DNA. Of the three CRISPR/Cas systems that have evolved in bacteria, the type II system is the simplest and involves only three components: a processed RNA that is complementary to the spacers, known as a CRISPR-RNA (crRNA), a trans-activating tracrRNA that hybridizes to the crRNA, and the Cas9 nuclease. The crRNA and the tracrRNA form an RNA double-strand structure that directs Cas9 to generate DSBs at a site complementary to the targeting region of the crRNA (Brouns et al., 2008; Deltcheva et al., 2011; Garneau et al., 2010). The RNA components of the CRISPR/Cas9 system (the crRNA and the tracrRNA) can be combined into a singular guide RNA (gRNA) (Jinek et al., 2012). The gRNA directs Cas9 to induce DSBs in the genome of cells at sites complementary to a ~20 base pair targeting sequence in the gRNA. The simplicity of these RNA-guided nucleases has allowed scientists to repurpose the CRISPR/ Cas9 system to create site-specific DNA breaks in a variety of eukaryotic cells (Cong et al., 2013; Mali et al., 2013).
2. ANALOG-SENSITIVE KINASES
Nearly one-third of the proteome is subject to phosphorylation by protein kinases. Adenosine triphosphate (ATP)-competitive small molecule inhibitors are powerful tools for probing the function of kinases in living cells. However, many kinases possess a similar catalytic core, and thus achieving specificity in inhibiting kinase activity in cells is a major challenge. One method to overcome this limitation is to exploit a chemical genetic strategy in which a kinase is engineered to accept ATP analogs that are not efficiently utilized by wild-type kinases. These engineered kinases are referred to as analog-sensitive (AS) kinases (Bishop et al., 2000). This is achieved through the mutation of a bulky hydrophobic ‘gatekeeper’ amino acid in the ATP binding pocket to a smaller amino acid (alanine or glycine) (to identify the gatekeeper residue for a kinase see http://sequoia.ucsf.edu/ksd) (Liu et al., 1999). An AS kinase can be specifically inhibited with generic nonhydrolyzable bulky ATP analogs, allowing rapid and reversible control of kinase activity in cells. Despite broad utility, the use of the AS kinase approach in mammalian cells has been hampered by the difficulty of functionally replacing an endogenous kinase with appropriate levels of an AS kinase. The development of CRISPR/Cas9 offers a facile method for introducing AS mutations into endogenous mammalian kinases.
Here, we outline a method for using CRISPR/Cas9 genome engineering to introduce an AS point mutation in a single step into both alleles of Polo-like kinase 4 (Plk4). This protocol can be used to introduce point mutations into any target gene of choice and could also be adapted to insert larger DNA elements, such as fluorescent proteins and degrons, at defined genomic locations.
3. METHODS
3.1 DESIGNING A GUIDE RNA FOR SEQUENCE-SPECIFIC DNA CLEAVAGE BY SpCas9
The CRISPR/Cas9 system is capable of generating targeted DSBs in the genome of mammalian cells. The Streptococcus pyogenes (Sp) CRISPR/Cas9 system is the most widely used system and will be the focus of this protocol. The specificity of SpCas9 targeting is determined by a 20-nucleotide (nt) targeting sequence within the gRNA that is complementary to the genomic target sequence. The genomic target sequence must precede an “NGG” sequence known as the protospacer adjacent motif (PAM), which is necessary for target cleavage, but is not encoded within the gRNA (Figure 1) (Mojica, Diez-Villasenor, Garcia-Martinez, & Almendros, 2009; Shah, Erdmann, Mojica, & Garrett, 2013). The PAM sequence has evolved to ensure that the CRISPR/Cas9 system does not self-target the CRISPR modules in the bacterial genome (Shah et al., 2013). SpCas9 usually cleaves the DNA 3-nt upstream of (i.e., 5′ to) the PAM to produce a blunt-ended DSB (Figure 1). Breaks can either be repaired by HDR or through error-prone nonhomologous end-joining (NHEJ) pathway, which usually introduces insertions and deletions (InDels) of bases at the cut site.
FIGURE 1. Genome Editing Using the CRISPR/Cas9 System.

SpCas9 nuclease is directed to a specific locus through base-pairing of the targeting sequence (underlined) of its associated guide RNA (gRNA) with a genomic target sequence. The genomic target sequence is followed by a protospacer-adjacent motif (PAM, red (light gray in print versions)) that is required for SpCas9 recognition and cleavage. Double-stranded cleavage usually occurs three base pairs upstream of the PAM. The double-strand break (DSB) may be repaired through homology directed repair (HDR) using an exogenously supplied repair template that contains a mutation of interest (green (dark gray in print versions)). Inserting a second mutation (blue (dark gray in print versions)) in the PAM prevents additional rounds of SpCas9/ gRNA cutting at this locus. Asterisks represent locations of point mutations in the repair template.
Several plasmid constructs are available for SpCas9/gRNA expression in mammalian cells. In our experiments we have used the PX459 vector (available from Addgene, vector #62988), which enables expression of a gRNA, SpCas9, and a puromycin resistance gene from a single vector. In this section we describe how to design a gRNA to direct cleavage at a specific genomic site.
Download the genomic sequence of the target gene from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/gene).
Identify the codon for the amino acid that will be mutated and copy a sequence of 50 nucleotides on either side of the desired mutation.
Visit crispr.mit.edu and paste the copied sequence into the query box. Select “other region” as the sequence type and choose the correct target genome. Click “Submit Query” and then select “Guides & offtargets.” Results may take some time to appear as the program searches the genome database.
The results will show a rank-ordered list of the potential genomic targets for gRNA recognition. All of the 20-nt genomic target sites are followed at the 3′ end by a PAM shown in green. Choice of gRNA targeting sequence depends on two main parameters: (1) HDR efficiency decreases as the distance between the desired mutation and the site of the DSB increases; therefore, the site of genomic target cleavage should be as close as possible to the site of the introduced mutation. (2) gRNAs can direct SpCas9 cleavage of nonidentical target sequences in the genome, possibly resulting in the introduction of undesired mutations. For each selected gRNA, a list of “off-target” binding sites is shown along with the position of mismatches within the gRNA sequence. gRNAs with higher “quality scores” have a greater predicted target specificity. For a detailed analysis of the effect of mismatches on gRNA recognition, see Hsu et al. (2013). We recommend choosing a gRNA that has at least four base pair mismatches to any other sequence in the genome and promotes cutting at <20-nt from the site of the intended mutation. For some genomic target sequences, it is not possible to achieve these parameters. In this case we select the highest scoring gRNA that directs cutting within 20-nt on either side of the desired mutation site.
We recommend selecting two to three gRNAs and testing each for cleavage efficiency using the SURVEYOR® Mutation Detection Kit (Transgenomic), as described in Ran et al. (2013).
Copy the 20-nt sequence of the genomic target sequence. The PX459 vector uses a U6 promoter to transcribe the gRNA and this requires that a G be the first nucleotide in the transcript. In cases where the genomic target sequence does not begin with a G, append an extra G at the 5′ end of the gRNA.
Generate the reverse complement of the genomic target sequence (including the 5′ G if it was added) (Figure 2(A)).
Add the overhang sequence 5′-CACC-3′ to the 5′ end of genomic target and the sequence 5′-AAAC-3′ to the 5′ end of the reverse complement (Figure 2(A)). These sequences will produce the correct overhangs for cloning into the PX459 vector.
Order single-stranded DNA oligonucleotides for the two sequences generated in Step 8.
FIGURE 2. Guide RNA and Repair Template Design.

(A) A Plk4 genomic target sequence (underlined) is chosen due to the close proximity of the cut site to the desired mutation (gatekeeper residue L89G, green (gray in print versions)). The PAM site required for SpCas9/gRNA cleavage (red (light gray in print versions)) lies immediately downstream of the genomic target sequence. The 5′ end of the genomic target sequence must start in a G for efficient transcription from the U6 promoter on the PX459 vector. When the genomic target sequence does not begin with a G, this must be added to the gRNA targeting sequence (blue (dark gray in print versions)). To clone the gRNA targeting sequence into the BbsI-digested PX459 vector, the sequence 5′-CACC-3′ must be added onto the 5′ end of the genomic target sequence and the sequence 5′-AAAC-3′ must be added onto the reverse complement of the genomic target sequence. (B) The Plk4AS repair template contains the gatekeeper residue mutation (L89G), a PAM site mutation, and introduces an AflIII restriction site to facilitate identifying clones that have undergone HDR (all mutations are shown in green (light gray in print versions)). Note that the PAM mutation and restriction enzyme recognition site insertion do not alter the coding sequence.
3.2 CLONING OLIGONUCLEOTIDES INTO THE PX459 VECTOR
The PX459 vector contains two BbsI cleavage sites that allow for the insertion of annealed oligonucleotides containing the gRNA target sequence. BbsI cleaves DNA outside of its recognition site to produce overhangs complementary to those added in Step 8 above. Below we describe how to clone the gRNA into the PX459 expression vector.
3.2.1 Vector preparation
-
1
Digest 1 μg of the PX459 vector with BbsI for 2 h at 37 °C.
-
2
To remove terminal phosphates, add 0.1 μL of calf intestinal phosphatase (CIP) to the reaction and incubate at 37 °C for 30 min. Since the two overhangs produced following BbsI digestion are not complementary this step is not required, but can reduce background.
-
3
Purify the cut vector using a standard polymerase chain reaction (PCR) cleanup kit. Note that BbsI digestion of PX459 produces a small 22-nt fragment that will pass through the column, leaving a 9153-nt, linear piece of vector DNA. The purified linear vector can be stored at −20 °C until ready for use.
3.2.2 Oligonucleotide annealing
-
4
Combine:
43 μL of H2O of molecular biology grade.
1 μL of each oligonucleotide from a 100 μM stock.
5 μL of New England Biolabs Buffer 3 (www.neb.com/).
-
5
Anneal oligonucleotides in a thermocycler with the following protocol:
4 min at 95 °C.
10 min at 70 °C.
Cool to 4°C at 1 °C/min.
Annealed oligonucleotides can be stored at −20 °C until ready for use.
3.2.3 Oligonucleotide phosphorylation
We use T4 polynucleotide kinase (PNK) from New England Biolabs to add terminal phosphates to the annealed oligonucleotides.
-
6
Combine:
5 μL of H2O of molecular biology grade
2 μL of the annealed oligonucleotides from above
1 μL of T4 PNK buffer (www.neb.com)
1 μL of ATP (10 mM stock)
1 μL of T4 PNK (www.neb.com)
-
7
Allow reaction to proceed in a thermocycler with the following protocol:
30 min at 37 °C
10 min at 70 °C (to inactivate PNK)
Quick cool to 4 °C
Phosphorylated oligonucleotides can be stored at −20 °C until ready for use.
3.2.4 Ligation and transformation
We use a 2X stock of Takara T4 DNA Ligase (DNA ligation kit, Version 2.1) and homemade TOP10 competent cells.
-
8
In a 0.5 mL tube, combine the following:
1 μL BbsI digested vector
4 μL phosphorylated annealed oligonucleotides
5 μL 2X Takara T4 ligase
-
9
As a control, set up the same reaction as above but substitute 4 μL of molecular biology grade H2O for the oligonucleotides.
-
10
Allow ligation to proceed for 1 h at room temperature.
-
11
Hand-thaw a frozen aliquot of competent bacteria and keep on ice.
-
12
Add the entire 10 μL reaction mixture to the competent bacteria and incubate on ice for 20–30 min.
-
13
Heat shock the bacteria for 1 min at 42 °C and return to ice for at least 1 min.
-
14
Plate the bacteria on prewarmed ampicillin (or carbenicilin) agar plates and incubate at 37 °C for 16 h. A successful ligation should produce few (if any) colonies on a control (vector alone) plate and many-fold more colonies on an experimental (vector with insert) plate.
-
15
Select a colony from the experimental plate and prepare a 1–5 mL culture in lysogeny broth containing ampicillin. Shake at 37 °C for at least 16 h and perform a plasmid purification as normal.
-
16
To check for correct oligonucleotide insertion, sequence the plasmid using the U6 forward primer (5′-ACTATCATATGCTTACCGTAAC-3′).
-
17
PX459 plasmid DNA containing a correctly cloned gRNA can be stored at −20 °C until ready for use.
3.3 DESIGN OF A REPAIR TEMPLATE
HDR can be exploited to generate defined edits at the site of a DSB introduced by SpCas9. As a substrate for HDR, cells are provided with an exogenous repair template containing the desired alteration. Repair templates can be single-stranded (ssDNA) or double-stranded DNA (dsDNA) with homology arms flanking the cut site. ssDNA repair templates have higher efficiency of HDR than identical dsDNA templates (Lin, Staahl, Alla, & Doudna, 2014). Therefore, for small insertions or point mutations we use single-stranded oligonucleotides with approximately 80 nucleotides of homology on either side of the cut site (Figure 2(B)). The ssDNA repair template can be designed to be complementary to either the sense or antisense strand. Along with the desired modification, a repair template should also possess a mutation in the PAM site that will prevent re-cutting by SpCas9 after HDR. In addition, to simplify the downstream screening of cell line clones, we strongly recommend introducing a silent restriction site along with the desired mutation. In the example below, we outline the steps for designing the ssDNA oligonucleotide repair template for introducing a point mutation (Figure 2(B)).
-
1
Identify the specific site of cleavage by SpCas9 in the genomic target sequence. This will be 3-nts upstream of the PAM sequence (Figures 1 and 2(A)).
-
2
Select 80 nucleotides on either side of the cut site to act as the homology for the repair template. Make the following three modifications to the sequence:
3.3.1 Insertion of the point mutation
-
3
Identify the codon for the amino acid to be mutated and change the sequence to code for a new amino acid.
3.3.2 Mutation of the PAM site
-
4
Identify the PAM in the repair template and replace one or both of the G bases with a C or T to create a silent mutation. For SpCas9, the “NGG” PAM sequence can be mutated to anything other than “NAG” to prevent SpCas9 cleavage (Hsu et al., 2013; Jiang, Bikard, Cox, Zhang, & Marraffini, 2013). If it is not possible to make a silent point mutation in the PAM sequence then at least four silent point mutations should be introduced into the targeting sequence of the gRNA to prevent re-cutting. On the whole, SpCas9 is less tolerant of mismatches that lie close to the PAM (Hsu et al., 2013).
3.3.3 Insertion of a restriction enzyme cleavage site
To rapidly screen clonal cell lines (see below), it is desirable to introduce a silent restriction enzyme site into the repair template.
-
5
Use the online restriction enzyme site finder WatCut, (http://watcut.uwaterloo.ca/template.php) to identify a region of the repair template that will allow the creation of a restriction enzyme site that does not change the coding sequence. Introduce the mutation into the repair template. To form a reliable marker to track insertion of the point mutation, the restriction site should be positioned as close as possible to the mutation. In addition, we recommend choosing a restriction enzyme that is not blocked by CpG methylation and shows good activity in a range of restriction enzyme buffers.
-
6
Order the ssDNA repair oligonucleotide from IDT (http://www.idtdna.com/site). In our experience polyacrylamide gel electrophoresis purification is not necessary.
3.4 TRANSFECTION AND SCREENING
For genome editing experiments we recommend selecting a stably diploid cell line, as this simplifies the process of achieving homozygous gene targeting. While we have successfully targeted aneuploid cell lines using the CRISPR/SpCas9 system, determining the genotype of the resulting clones is more complex. Since puromycin is used to achieve rapid killing of cells that do not receive the PX459 expression vector, it is important that the chosen cell line is also puromycin sensitive. An alternative approach is to use the PX458 expression vector (available from Addgene, vector #48138) that co-expresses SpCas9, gRNA, and GFP, allowing fluorescent transfected cells to be directly sorted into individual wells of a 96-well plate.
There are a number of methods for delivering DNA to mammalian cells in culture. Here, we describe a method using Roche’s X-tremeGENE 9 transfection reagent. Depending on the cell line to be used, other DNA delivery methods (e.g., nucleofection) may be required.
3.4.1 Day 1
-
1
Seed cells for transfection at 2 × 105 cells/well in 2 mL of media in a six-well plate. Seed at least three wells per transfection, with one of these wells serving as a nontransfected control. Transfections are carried out in the presence of serum and, if desired, antibiotics.
3.4.2 Day 2
-
2
Change media on cells 30 min prior to transfection.
-
3
In a 1.5 mL tube prepare the following for each transfected well in the order written below:
100 μL serum-free media at room temperature
3 μL X-tremeGENE 9 transfection reagents
1 μg of total DNA (20:1 molar ratio of repair template:PX459 plasmid)
-
4
Flick tube gently 10 times to mix.
-
5
Incubate at room temperature for 15–20 min.
-
6
Add the transfection mixture drop-wise to cells.
-
7
Return cells to the incubator.
3.4.3 Day 4
The PX459 plasmid contains a puromycin resistance marker to select for transfected cells.
-
8
Change media on all cells (including controls) with fresh media containing puromycin (1–5 μg/mL is usually sufficient).
-
9
Return cells to the incubator.
Cells in the untransfected control wells should all die within 1–2 days in puromycin. When all control cells are dead proceed with limiting dilution. Cells should not remain in puromycin more than 2–3 days as the PX459 plasmid does not integrate into the genome and resistance to puromycin is lost over time.
3.4.4 Limiting dilution
After puromycin selection, single-cell clones should be isolated and screened to identify those that have undergone HDR. Either single-cell sorting or dilution cloning may be used to isolate single cells. Below we outline how to obtain single clones using dilution cloning.
Cells are diluted into 96-well plates to obtain wells containing a single cell. Since the clonogenic survival of cell lines varies greatly, we recommend seeding multiple 96-well plates with varying cell densities (1, 5, 10, and 50 cells/well). A 96-well plate that has growth in 10% of the wells will have a ~90% probability of a given well having a single colony.
Add 15 mL of puromycin-free media to a sterile reagent reservoir.
Add the desired number of cells to the media. For example, to achieve 10 cells/ well, 1000 cells would be added to the media.
Mix the cells in the media by pipetting up and down five times with a 10 mL pipette.
Using a multichannel pipette, add 150 μL of media to each well of the 96-well plate.
Repeat Steps 1–4 for each cell density.
Wrap the 96-well plates in plastic film and return to the incubator.
It will take 2–4 weeks for single cell to grow into colonies large enough to isolate sufficient quantities of genomic DNA.
3.4.5 Genomic DNA extraction
To extract genomic DNA from a small number of clones, we use the Sigma GenElute Mammalian Genomic DNA Miniprep Kit (G1N350). For extracting genomic DNA from a large number of clones, we use a protocol adapted to use with 96-well plates and outlined below.
Using a tissue culture microscope, identify wells containing a single colony. Transfer 96 individual clones into 24-well plates. Return cells to the incubator.
When the clones are confluent, trypsinize cells in 200 μL of 0.05% trypsin.
Remove 160 μL of the trypsinized cell suspension and place into a single well of a 96-well plate with U-bottom wells.
Add 1 mL of media to the cells remaining in the 24-well plates and return plates to the incubator.
Spin the 96-well plate at 2000 RPM in a swinging-bucket rotor for 10 min to pellet cells.
To remove supernatant, quickly invert the plate to remove media and remove excess liquid by blotting on paper towels.
Resuspend cells in each well with 150 μL phosphate buffered saline (PBS) and spin at 2000 RPM for 10 min. Remove PBS as in Step 6.
To lyse cells, add 50 μL of lysis buffer (10 mM Tris–HCl, pH 7.5, 10 mM of ethylene diamine tetraacetic acid, 0.5% of sodium dodecyl sulfate, 10 mM of NaCl, 1 mg/mL of proteinase K) and seal the plate with parafilm. Put plate into a humidified chamber at 60 °C overnight. A humidified chamber can be created by placing a few inches of water in a small plastic container with a sealable lid. The 96-well plate is placed in the sealed container on top of a test tube rack so that it rests above the water level.
The next day, remove the plate from the humidified chamber and cool to room temperature.
Add 100 μL of ice-cold EtOH/NaCl mix (75 mM of NaCl in ~100% EtOH; it forms a cloudy solution) to precipitate DNA and mix well.
Incubate at room temperature for 30 min.
Spin at 4000 RPM in a swinging-bucket rotor for 20 min to pellet precipitated DNA.
Decant liquid as in Step 6.
Rinse pellet with 150 μL cold 70% EtOH and spin for 10 min at 4000 RPM.
Decant liquid as in Step 6.
Repeat washing step (#14–15) and air-dry DNA for 10 min.
Add 50 μL TE, pH 8.0 (10 mM of Tris–HCl, pH 8.0, 1 mM EDTA) to genomic DNA pellet.
Cover plate with parafilm and incubate at 50 °C for 2 h.
Genomic DNA is ready for further screening or may be stored at 4 °C until ready for use.
3.4.6 Screening
HDR can be tracked by PCR amplification of genomic DNA followed by restriction enzyme digest. Forward and reverse primers are designed to bind to regions of the genome outside of the homology arms of the repair template to amplify a region of 250–1000-nt (Figure 3(A)). Primers are designed using the Primer3 program (biotools.umassmed.edu). We recommend screening at least three different forward and reverse primers in all possible combinations to identify a primer pair that yields a strong and specific PCR product from low quantities of DNA (Figure 3(B)). In most cases, the PCR protocol outlined below produces good results. However, PCR optimization may be required.
FIGURE 3. Screening of the Plk4AS Allele.
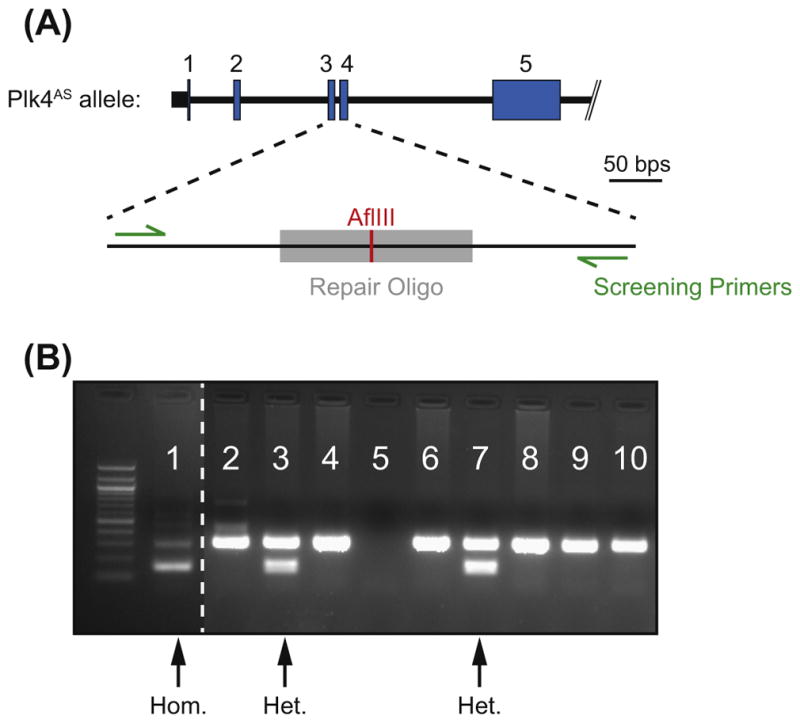
(A) Forward and reverse screening primers (green (light gray in print versions)) should be designed to bind outside of the sequence of the repair template (gray). PCR-amplified Plk4AS alleles will be digested with the restriction enzyme AflIII whereas Plk4WT alleles will not. (B) Genomic DNA extracted from individual clones is PCR-amplified and subject to AflIII digestion. Clone 1 shows near complete digestion of the PCR product and is homozygous for the Plk4AS allele. Clones 3 and 7 show partial digestion of the PCR products and are heterozygous for the Plk4AS allele. Remaining clones were either not cut by SpCas9 or repaired through non-homologous end joining (NHEJ). The genomic DNA extraction or PCR failed for clone 5.
3.4.7 PCR amplification
-
1
For each clone, add 2 μL of genomic DNA to a single well of a 96-well plate.
-
2
Prepare a master mix (100 reaction per plate) as follows:
a. Molecular biology grade H2O 1120 μL b. 5X GC buffer (www.neb.com) 400 μL c. Forward primer (10 μM stock) 100 μL d. Reverse primer (10 μM stock) 100 μL e. dNTPs (stock with each dNTP at 10 mM) 40 μL f. Phusion polymerase (www.neb.com/) 40 μL -
3
Add 18 μL of the master mix to each well, cover the plate with a piece of sealing film and perform a PCR reaction using the following parameters:
a. 98 °C 30 s b. 98 °C 10 s c. 57 °C 15 s d. 72 °C 2 kb/min e. Repeat steps b to d 34X f. 72 °C 5 min g. 12 °C hold
The PCR product is ready for restriction enzyme digest (below).
3.4.8 Restriction enzyme digest
To determine whether a clone underwent HDR, the PCR product is digested with the restriction enzyme that was introduced into the repair template. If the genome underwent HDR with the repair template, digestion products smaller than the length of the amplicon will be observed (Figure 3(B)). Prepare a digestion for each clone’s PCR product as below:
8.5 μL genomic PCR product
1 μL 10X restriction enzyme buffer (www.neb.com)
0.5 μL restriction enzyme
Note that for some restriction enzymes it is possible to perform the digestion directly in PCR buffer.
-
4
Incubate at 37 °C for 2 h.
-
5
Add 10 μL of a DNA loading dye to each restriction enzyme digest and remaining undigested PCR products and load the entire samples into wells of a 2% agarose DNA gel.
-
6
Run gel and image with a gel documentation system.
There are three possible results for each reaction (Figure 3(B)).
Only an undigested band: There are two possibilities here (Figure 3(B), Lanes 2, 4, 6, 8, 9, and 10): (1) Both alleles are wild type. In this case SpCas9 may have failed to cut, or the DSB was repaired by HDR using the sister chromatid, rather than the repair template. (2) One or both alleles are mutated. The DSB was repaired through (NHEJ) resulting in the creation of InDels that prevent further SpCas9 cutting.
Both undigested and digested bands: There are two possibilities here that are not mutually exclusive (Figure 3(B), Lanes 3 and 7). (1) The clone is heterozygous. In this case, the SpCas9/gRNA complex cleaved the genomic DNA and repaired one allele by HDR using the repair template. The other allele is either wild type or has been repaired by NHEJ. In our experience, many cells repair one allele by HDR using the repair template and the second allele undergoes NHEJ producing InDels. InDels frequently result in frameshift mutations that generate a null allele. (2) The cells are polyclonal. This may occur when two or more parental cells grow in a single well.
Only (or mostly) digested bands: This clone is homozygous for insertion of the point mutation (Figure 3(B), Lane 1).
3.4.9 Sequencing clones
To verify insertion of the desired mutation, the undigested PCR product can be cloned into a vector for sequencing. We recommend using the ZeroBlunt® TOPO® Cloning Kit from Invitrogen, which allows for easy cloning of blunt-end PCR products into the pCR-Blunt II-TOPO vector. We usually sequence ~10 clones to ensure coverage of both alleles.
Clones that are homozygous for the insertion of the point mutation can be taken forward for further analysis. However, heterozygous clones that contain a point mutation in one allele and a frameshift mutation in the second allele may also prove useful if the frameshift mutation creates a null allele.
4. FUNCTIONAL ANALYSIS
Depending on the point mutation introduced and the predicted phenotype, a variety of assays can be conducted. In the case of an AS kinase, treatment with bulky ATP-analogs (1NM-PP1, 3 MB-PP1) should inhibit kinase activity. In this case, cells can be treated with varying concentrations (0.1–20 μM) of a bulky ATP-analog and assayed for kinase activity.
CONCLUSION
The ease of use and high efficiency of the CRISPR/Cas9 system has propelled its use in genome engineering in a wide array of organisms and cultured cells (Hsu et al., 2014). However, there is competition between NHEJ and HDR for DSB repair (Heyer, Ehmsen, & Liu, 2010). While NHEJ is well suited for disrupting gene expression, introducing precise alterations in the genome requires efficiently engaging the HDR machinery. Increasing the frequency with which DSBs are repaired by HDR will greatly facilitate genome editing applications. An additional consideration for using the CRISPR/SpCas9 system is the potential of off-target cutting and mutagenesis. This concern is elevated in cell lines where there is no opportunity to breed out unintended mutations. Evaluating and improving the specificity of CRISPR/SpCas9 genome editing remains an important future goal.
References
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407(6802):395–401. doi: 10.1038/35030148. http://dx.doi.org/10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–1512. doi: 10.1126/science.1178811. http://dx.doi.org/10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–964. doi: 10.1126/science.1159689. http://dx.doi.org/10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244(4910):1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Molecular and Cellular Biology. 1995;15(4):1968–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–761. doi: 10.1534/genetics.110.120717. http://dx.doi.org/10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. http://dx.doi.org/10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. doi: 10.1038/nature09886. http://dx.doi.org/10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468(7320):67–71. doi: 10.1038/nature09523. http://dx.doi.org/10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. http://dx.doi.org/10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. http://dx.doi.org/10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. http://dx.doi.org/10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 2013;31(9):827–832. doi: 10.1038/nbt.2647. http://dx.doi.org/10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. 2013;31(3):233–239. doi: 10.1038/nbt.2508. http://dx.doi.org/10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. http://dx.doi.org/10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;4 doi: 10.7554/eLife.04766. http://dx.doi.org/10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, et al. Structural basis for selective inhibition of Src family kinases by PP1. Chemistry & Biology. 1999;6(9):671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. http://dx.doi.org/10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nature Reviews Genetics. 2010;11(3):181–190. doi: 10.1038/nrg2749. http://dx.doi.org/10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nature Biotechnology. 2007;25(7):778–785. doi: 10.1038/nbt1319. http://dx.doi.org/10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nature Biotechnology. 2011;29(2):143–148. doi: 10.1038/nbt.1755. http://dx.doi.org/10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(Pt 3):733–740. doi: 10.1099/mic.0.023960-0. http://dx.doi.org/10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi: 10.1126/science.1178817. http://dx.doi.org/10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130(3):451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. http://dx.doi.org/10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Molecular and Cellular Biology. 1994;14(12):8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin N, Sugarman E, Haber JE. Genetic and physical analysis of double-strand break repair and recombination in Saccharomyces cerevisiae. Genetics. 1989;122(3):519–534. doi: 10.1093/genetics/122.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SA, Erdmann S, Mojica FJ, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biology. 2013;10(5):891–899. doi: 10.4161/rna.23764. http://dx.doi.org/10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435(7042):646–651. doi: 10.1038/nature03556. http://dx.doi.org/10.1038/nature03556. [DOI] [PubMed] [Google Scholar]