

Abstract
Small molecule-induced protein degradation is an attractive strategy for the development of chemical probes. One method for inducing targeted protein degradation involves the use of PROTACs, heterobifunctional molecules that can recruit specific E3 ligases to a desired protein of interest. PROTACs have been successfully used to degrade numerous proteins in cells, but the peptidic E3 ligase ligands used in previous PROTACs have hindered their development into more mature chemical probes or therapeutics. We report the design of a novel class of PROTACs that incorporate small molecule VHL ligands to successfully degrade HaloTag7 fusion proteins. These HaloPROTACs will inspire the development of future PROTACs with more drug-like properties. Additionally, these HaloPROTACs are useful chemical genetic tools, due to their ability to chemically knockdown widely used HaloTag7 fusion proteins in a general fashion.
Introduction
The use of small molecules to induce targeted protein degradation is an emerging strategy for the development of novel therapeutics and biological probes.1-3 Current small molecule therapeutics, such as enzyme inhibitors and receptor antagonists target specific protein activities, while leaving other activities, (such as scaffolding functions or other enzymatic functions in multidomain proteins) intact; on the other hand, protein degraders have the power to abrogate all of the functions of a drug target at once, including scaffolding functions which are difficult to target with small molecule inhibitors. As biological probes, chemical knockdown through the use of a protein degrader gives a greater degree of temporal control than genetic knockdown strategies.
PROTACs are a class of heterobifunctional molecules that link a ligand for a protein of interest (POI) to an E3 ligase ligand. This binding event recruits the E3 ligase to the POI, inducing its ubiquitination and subsequent degradation by the proteasome.4, 5 PROTACs have been developed that successfully target a wide variety of proteins6-11; however their development as therapeutics and biological probes has been severely hindered by the use of large, peptidic E3 ligase ligands. Previous attempts to design a small molecule PROTAC using a small molecule ligand (based on nutlin)12 led to degradation of the AR,13 but was less effective than peptidic PROTACs.5 Recently, the Hashimoto lab has successfully used bestatin to recruit cIAP1 to degrade CRABPs in cells.14-17 However, bestatin is commonly used as an aminopeptidase inhibitor, which can lead to off-target effects.18 Furthermore, ligands for IAPs often induce degradation of the E3 ligase itself,19, 20, (an effect observed with some bestatin hybrids)16 that complicates their use in PROTACs. Furthermore, both peptidic and small molecule PROTACs suffer from low potency, commonly requiring concentrations in excess of 10 μM to achieve maximal knockdown.
In an attempt to resolve these issues, we sought to design small molecule ligands for VHL, an E3 ligase which has previously been targeted in numerous PROTACs with peptidic ligands.5, 7, 9, 11 We recently described the synthesis of potent ligands for VHL based upon a key hydroxyproline residue. Crystallographic evidence indicated that these ligands contained numerous sites that were solvent exposed, suggesting possible linker positions.21-23
With these ligands in hand, we sought to develop PROTACs that were capable of effectively degrading HaloTag7 fusion proteins (Figure 1). HaloTag is a modified bacterial dehalogenase that covalently reacts with hexyl chloride tags; HaloTag fusion proteins have seen wide use as a method to bioorthogonally label proteins in vivo.24 We have previously developed an alternative degradation technology that uses hydrophobic tags to mimic protein unfolding, leading to the degradation of HaloTag2 fusion proteins.25 However, these molecules were far less capable of degrading proteins fused to HaloTag7,26 a variant developed by Promega to increase the protein’s stability.27 Due to HaloTag7’s resistance towards alternative methods of induced degradation and the existence of a potent small molecule ligand capable of accommodating long linkers, we concluded that HaloTag7 fusion proteins would make an ideal model system to develop novel small molecule HaloPROTACs. Furthermore, due to the wide availability of HaloTag7 fusion proteins, an effective degrader of HaloTag7 could prove to be a powerful tool in chemical genetic studies.
Figure 1.

Schematic depiction of a bifunctional HaloPROTAC containing chloroalkane (which binds HaloTag7 fusion proteins) and a hydroxyproline derivative which binds VHL. These two motifs would serve to bring the HaloTag7 fusion protein into proximity with the E3 ligase, VHL, leading to the degradation of the HaloTag7 fusion protein by the proteasome.
Results and Discussion
Design of HaloPROTACs
Initially, we sought to develop chloroalkane-containing PROTACs (HaloPROTACs) building off of the acyl amine moiety (Degradation Inducing Moiety A), as this corresponds to the N-terminal linkage position of previously synthesized peptidic PROTACs targeting VHL (Scheme 1).5 We synthesized HaloPROTAC1 and HaloPROTAC2, containing different linker lengths. We then tested their ability to degrade GFP-HaloTag7, (stably expressed in HEK 293 cells) by flow cytometry, measuring changes in mean fluorescence intensity. This assay (previously developed to test hydrophobic tags)26 was chosen as our primary assay rather than immunoblotting as it offered more sensitive and reliable quantitation. Furthermore, the relatively high throughput nature of the assay allowed us to rapidly test multiple treatment conditions and obtain more complete dose response curves. While 24 hour treatment with HaloPROTAC1 led to less than 20% degradation, the longer HaloPROTAC2 led to nearly 70% degradation of GFP-Halotag7 at 2.5 μM (Figure 2A). Furthermore, we observed reduced degradation at higher concentrations, a phenomenon commonly observed with ternary complexes due to auto-inhibition of the ternary complex, in favor of the two binary complexes.28
Scheme 1.

Synthesis of HaloPROTACs containing Degradation Inducing Moiety A and Degradation Inducing Moiety B. Full synthetic details are found in the Supporting Information.
Figure 2.

The average fluorescence per cell compared to vehicle control was measured by flow cytometry after 24 hour treatment with the indicated compounds and concentrations. All compounds were tested in triplicate unless otherwise noted. A) HaloPROTACs containing Degradation Inducing Moiety A lead to nearly 70% degradation of GFP-HaloTag7, when sufficiently long linkers are used. B) HaloPROTAC3 (tested in quintuplicate), which contains Degradation Inducing Moiety B, leads to 90% degradation of GFP-HaloTag7 at 625 nM. GFP-HaloTag7 degradation was measured by flow cytometry after 24 hour treatment with HaloPROTAC and normalized to 24 hour treatment with DMSO. Error bars depict the Standard Error of the Mean (SEM).
We then synthesized HaloPROTAC3 and HaloPROTAC4 in order to test an alternative linkage position off of the phenyl ring (Degradation Inducing Moiety B). Gratifyingly, we observed that HaloPROTAC3 was able to induce 90 ± 1 % degradation of GFP-Halotag at 625 nM. We then calculated the half maximal degradation concentration, or DC50, to be 19 ± 1 nM, making HaloPROTAC3 one of the most potent PROTACs described to date. HaloPROTAC4 was modestly less effective, inducing over 70% degradation (Figure 2B). As the PROTACs containing the phenol linkage proved to be more effective at inducing degradation of GFP-HaloTag7, we chose to focus on them for the remainder of the study; however, it is likely that efficacy of each linkage position will be target dependent and that in some cases, PROTACs with the amide linkage may prove superior.
Effects of Linker Length and Affinity for VHL on HaloPROTAC Activity
Having identified HaloPROTAC3 as a highly potent and efficacious degrader of GFP-HaloTag7, we sought to determine the system’s tolerances of variations in linker lengths and affinity for VHL (Figure 3A). We found that HaloPROTAC5, HaloPROTAC6 and HaloPROTAC7 (which contained shorter linkers) had virtually no effect on levels of GFP-HaloTag7 (< 20% degradation).The shorter linker lengths likely lead to negative cooperativity in binding due to sterics, preventing the PROTACs from simultaneously binding to GFP-HaloTag7 and VHL. HaloPROTAC4 and HaloPROTAC8, which contained longer linkers had significant knockdown (although less effective than HaloPROTAC3). Furthermore, the HaloPROTACs with longer linkers had significant auto-inhibition at higher concentrations, whereas essentially no auto-inhibition was observed with HaloPROTAC3, which had an intermediary linker length. One possible explanation is that at the intermediary length, there could positive cooperativity for HaloPROTAC3 binding to GFP-HaloTag7 and VHL; such cooperativity in ternary complexes has been predicted to lead to a “width expansion” of the maximal effect.28 While the exact linker lengths of future PROTACs are expected to be highly target dependent, it seems likely that similar trends may be observed. A possible strategy for the development of future PROTACs would therefore be to initially synthesize and test compounds with slightly longer linkers, then to gradually reduce length until a drop-off in activity is observed.
Figure 3.

A) A study of linker length with Degradation Inducing Moiety B shows that three ethylene glycol units are optimal for the degradation of GFP-HaloTag7. B) Structures of HaloPROTACs that have weaker affinity for VHL. C) Reducing the affinity for VHL attenuates their ability to induce degradation of GFP-HaloTag7, although the effect is not necessarily linear. GFP-HaloTag7 degradation was measured by flow cytometry after 24 hour treatment with HaloPROTAC and normalized to 24 hour treatment with DMSO. All compounds were tested in triplicate except for HaloPROTAC3 (quintuplicate) and HaloPROTAC9 and HaloPROTAC10 (quadruplicate). Error bars depict the SEM.
We then sought to examine the effect of affinity for VHL on HaloPROTAC activity. Using the fluorescence polarization assay previously reported in the development of VHL ligands,21 we found HaloPROTAC3 to bind to VHL with an IC50 of 0.54 ± 0.06 μM (Supplemental Figure 1). Replacement of the valine-isoindolinone moiety with the previously described isoxazole moiety (HaloPROTAC9) led to a significant decrease in VHL binding (Figure 3B). This reduction in binding attenuated its activity in cells, leading to only 45% degradation of GFP-HaloTag7 (Figure 3C). A more modest reduction in VHL affinity was obtained with the alanine-isoindolinone moiety of HaloPROTAC10, which had similar maximal degradation to HaloPROTAC3, but slightly decreased cellular potency (DC50 = 36 ± 4 nM). Finally, ent-HaloPROTAC (the enantiomer of HaloPROTAC3, containing D-hydroxyproline and D-valine residues) was found to have no detectable binding to VHL and led to no significant degradation of GFP-HaloTag7 in cells. These data highlight the dependency of protein degradation on VHL binding, although it is likely that other factors such as cell permeability may affect efficacy.
Mechanistic Studies
We next sought to confirm that our HaloPROTACs worked through the expected mechanism; that is, their ability to recruit VHL to GFP-HaloTag7 led to their degradation by the proteasome and that they did not share the VHL-independent mechanism of our previous reported hydrophobic tags.25, 26 An ideal control compound was therefore the enantiomers of the specific HaloPROTACs as they would have identical physical properties (such as hydrophobicity, solubility and passive membrane permeability) but would lack the ability to bind to VHL. We synthesized five key ent-HaloPROTACs with both amide and phenol linkers of various lengths and found that none resulted in any significant degradation of GFP-HaloTag7, providing strong evidence that the HaloPROTACs work through a VHL dependent mechanism (Figure 4A). This is further supported by the finding that pretreatment with excess ent-HaloPROTAC3 could prevent HaloPROTAC3-mediated degradation, indicating that ent-HaloPROTAC3 binds to GFP-HaloTag7, but does not induce its degradation (Figure 4B).
Figure 4.
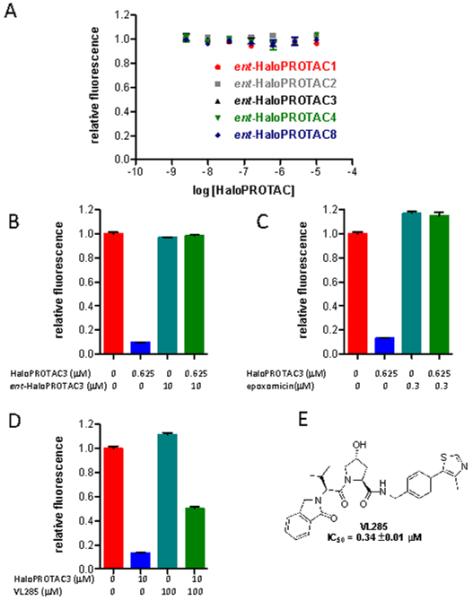
A) The enantiomers of HaloPROTACs (containing D-amino acid residues) which do not bind VHL do not induce degradation of GFP-HaloTag7, supporting the necessity of VHL binding for activity. B) Pre-treatment with excess ent-HaloPROTAC3 (1 hour) prevents degradation of GFP-HaloTag7 by HaloPROTAC3 after 24 hours. C) Pre-treatment with epoxomicin (4 hours) prevents degradation of GFP-HaloTag7 by HaloPROTAC3 after 20 hours. D)Treatment with VL285 attenuates the ability of HaloPROTAC3 to induce the degradation of GFP-HaloTag7. E) Structure of VL285. All error bars depict SEM. All compounds were tested in triplicate.
To confirm that the degradation of GFP-HaloTag7 was proteasome dependent we used the specific proteasome inhibitor epoxomicin.29, 30 Treatment with 300 nM epoxomicin completely prevented HaloPROTAC mediated degradation of GFP-HaloTag7 (Figure 4C). Finally, cotreatment with excess VL285, the core VHL ligand from which HaloPROTAC3 is derived, was able to significantly reduce HaloPROTAC3 mediated activity to 50% degradation, further implicating VHL in the observed degradation (Figure 4D, E).
Use of HaloPROTAC3 to Degrade GFP-HaloTag7
Having developed HaloPROTAC3 and established that it functions through a VHL and proteasome dependent mechanism, we sought to further characterize its ability to degrade GFP-HaloTag7. We compared HaloPROTAC3 to HyT36, the most efficacious hydrophobic tag for the degradation of HaloTag7 fusion proteins. We compared the two compounds and found HaloPROTAC3 to be dramatically more potent and efficacious (DC50 = 19 ± 1 nM, max degradation = 90 ± 1 %) than HyT36 (DC50 = 134 ± 7 nM, max degradation = 56 ± 1 %) (Figure 5A). We then studied the kinetics of HaloPROTAC3 mediated degradation and found that 50% of GFP-HaloTag7 was degraded between 4 and 8 hours (Figure 5B). Treatment of HaloPROTAC3 for 24 hours, followed by a 24 hour washout led to significant recovery of GFP-HaloTag7 (Figure 5C). To confirm the flow cytometry results, we examined the ability of HaloPROTAC3 to knockdown GFP-HaloTag7 with fluorescence microscopy. Treatment with 625 nM HaloPROTAC3 led to a striking decrease in fluorescence compared to DMSO alone or the control, ent-HaloPROTAC3 (Figure 6). No significant differences in cell density or morphology were observed between different treatment conditions under phase contrast (Supplemental Figure 2).
Figure 5.

A) Comparison of HaloPROTAC3 (quintuplicate) to Hyt36 (triplicate) shows that HaloPROTAC3 is significantly more potent and efficacious. B) HaloPROTAC3 leads to 50% degradation of GFP-HaloTag7 within 4 to 8 hours. C) Significant recovery from 24 hour treatment with HaloPROTAC3 is observed after a 24 hour washout. All data was repeated in triplicate. Error bars depict the SEM.
Figure 6.

Fluorescent microscopy shows drastic loss of fluorescence upon 24 hour treatment with HaloPROTAC3 but not the inactive ent-HaloPROTAC3. Comparison of fluorescence images to phase contrast view is shown in Supplemental Figure 2.
Future Uses of HaloPROTAC as a Probe in Chemical Genetics Studies
While GFP-HaloTag7 was chosen as a model system, due to the ease of analysis of GFP degradation, HaloPROTAC3 can lead to the degradation of other HaloTag7 fusion proteins, allowing for its possible use as a chemical probe. HaloPROTAC3 is well suited for this as it has low toxicity (Supplemental Figure 3) and does not affect HIF (an endogenous target of VHL) stability (Supplemental Figure 4). This system could therefore function as an alternative to the widely used Shield1 system, developed by Wandless and coworkers.31, 32 Shield1 binds to and stabilizes mutant FKBP12 (and mutant FKBP12 fusion proteins), which rapidly subjected to proteasome mediated degradation in the absence of Shield1.31, 32 This system has proved quite general and has been widely used and modified33-35 in numerous chemical genetics studies. The HaloPROTAC3 system would induce degradation of fusion proteins, rather than stabilizing them like Shield (although one report describes a system where Shield1 does lead to the destabilization of a modified FKBP12 protein),36 allowing for an orthogonal approach. Additionally, while use of the Shield system generally requires cloning steps to generate the fusion protein of interest, plasmids containing HaloTag7 fused with 20,000 human genes proteins are commercially available37 allowing for degradation of numerous proteins in a screen.
To test the generality of HaloTag7 protein degradation, we first confirmed by immunoblotting the flow cytometry data for the degradation of GFP-HaloTag7 by HaloPROTAC3. Once again, we observed significant degradation by 50 nM and virtually complete degradation by 500 nM. Next, we sought to broaden the scope for HaloPROTACS to other cytosolic proteins fused with HaloTag7. For this purpose, we chose ERK-1 and MEK-1, which are key kinases involved in the Mitogen-Activated Protein Kinase (MAPK) pathway. Dysregulation of the MAPK pathway is implicated in a wide variety of cancers, and several of its components are validated therapeutic targets in various forms of the disease.38, 39, Treatment with 500 nM HaloPROTAC3 was able to induce nearly complete knockdown of both HaloTag7-ERK1 and HaloTag7-MEK1, suggesting that HaloPROTAC3 induced degradation of HaloTag7 fusion proteins is general for HaloTag7 fused proteins located in the cytosol (Figure 7). These results suggest the possibility of using HaloPROTAC3 to study the chemical knockdown of HaloTag fusion proteins. However, such studies would require either the use of gain-of-function mutant, or would require the additional step of genetic knockdown of the endogenous fusion proteins.
Figure 7.
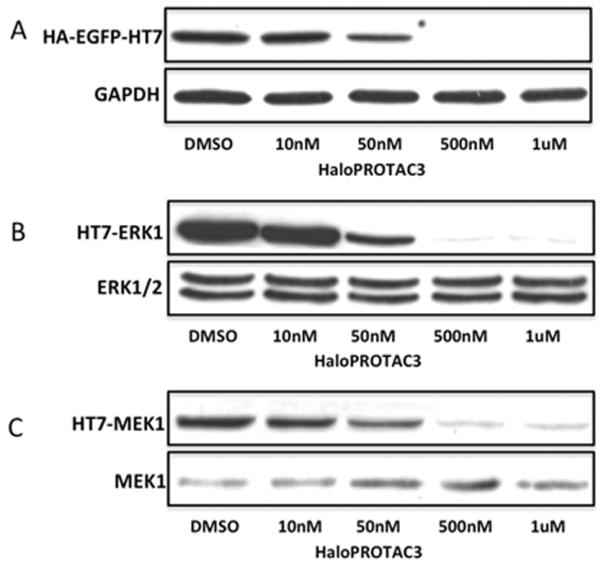
Immunoblotting confirms that nearly complete degradation of A) GFP-HaloTag7 is observed after 24 hour treatment with 500 nM HaloPROTAC3, with significant degradation at 50 nM HaloPROTAC3. HaloPROTAC3 can lead to degradation of other HaloTag7 fusion proteins such as B) HaloTag7-ERK1 and HaloTag7-MEK1. As expected, endogenous ERK and MEK are not degraded.
Conclusion
Using ligands for the E3 ligase VHL, we have successfully developed two new degradation-inducing moieties for use in small molecule PROTACs. Both Degradation Inducing Moiety A (in HaloPROTAC2) and Degradation Inducing Moiety B (in HaloPROTAC3) are capable of inducing the degradation of GFP-HaloTag7 in cell-based assays. Furthermore, we have shown that both act through a VHL dependent mechanism. The activity of these degradation-inducing moieties, (along with the accompanying studies on linker length) will prove indispensable in the development of future PROTACs against additional targets. As these new PROTACs lack long peptidic chains, they possess improved drug-like properties, helping to advance the field from a “parlor trick” into the realm of useful chemical probes and possibly therapeutics.
The most active compound, HaloPROTAC3 is capable of inducing 90% degradation of GFP-HaloTag7 proteins, with a DC50 of 19 nM, making this compound one of the most potent PROTACs described to date. We have shown that this degradation is not limited to GFP-HaloTag7, but appears to be general for cytosolic HaloTag7 fusion proteins. While HaloTag7 fusion proteins are not endogenous proteins, they are widely used in biological studies (although not all fusions with HaloTag retain the same activity as their endogenous counterparts) and are readily available reagents. Therefore, we expect HaloPROTAC3 to have wide use in chemical genetics studies.
Methods
A. Cell culture
All cells were cultured at 37°C and 5% CO2 in DMEM and supplemented with 10% FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. HEK 293 Flp-In cells stably expressing GFP-HaloTag7 were obtained as described in the literature.26 To generate HEK293T cells stably expressing HT7-ERK1 or HT7-MEK1, each of the HT7 fusion constructs was cloned into the retroviral pBabe-puro vector. The pHTN vector was used as HT7 template, and the Proquest human spleen cDNA library was used as a source of ERK-1 and MEK-1 cDNA. Viral particles were generated in GP2-293 cells, with a pVSV-G vesicular stomatitis virus retroviral vector and the corresponding pBabe-puro plasmid. After 48 hr, viral supernatants were harvested, passed through a 0.45 μm filter, and added directly to the growth media of target 293T cells that had been supplemented with polybrene (8 μg/mL). HaloPROTACs were dissolved in DMSO as 1,000× stock solutions and added to cells at final DMSO concentration 0.1%. Competition experiments (HaloPROTAC3 with ent-HaloPROTAC3, epoxomicin or VL285) had a final concentration of 0.2% DMSO. All HaloPROTAC treatments were for 24 hours, unless otherwise noted.
B. Flow Cytometry
Flow cytometry experiments were done in an analogous method to that used in the study of the degradation of HaloTag7-GFP using hydrophobic tags.26 HEK 293 FlpIn cells expressing GFP-HaloTag7 were treated with HaloPROTACs for 24 hr. The cells were then detached from the plate with trypsin (0.15 mL) and suspended in DMEM (0.85 mL). Intracellular EGFP fluorescence was then measured on the FL1 channel of a FACSCalibur (BD Biosciences). The resulting data was quantified in terms of mean fluorescence intensity using CellQuestTM (BD Biosciences). The fluorescence was then normalized by dividing by the average fluorescence of the vehicle (0.1% DMSO) alone control, which was set to 1. The normalized data were then plotted in Prism 5.0, allowing the DC50 to be determined. Error bars indicate standard error of the mean (SEM), with all data being repeated 3 to 5 times. For washout experiments, after treatment, media was removed, then replaced with fresh media (without any compounds) and incubated for 30 min. After 30 min, the media was again replaced to remove any residual compound remaining.
C. Immunoblotting
Compounds were dissolved in DMSO and added to cells at a final DMSO concentration of 0.1%. Cells were lysed in Radioimmunoprecipitation buffer (1%Tx-100, 0.1% SDS, 0.5% Sodium deoxycholate, 150mM NaCl, 25mM Tris pH 8) supplemented with a protease inhibitor cocktail (Roche). Lysates were normalized with a BCA assay and 20ug of each sample was loaded on a 10% Bis-Tris gel for SDS-PAGE. Proteins were transferred on to nitrocellulose membrane, which was blocked with 5% milk/TBST and probed with either anti-ERK1 (1:2000), MEK-1 (1:1000), or HA (1:5000) antibodies.
Supplementary Material
Acknowledgments
This project was supported by the DoD (W81XWH-12-1-0484) and the NIH (RO1AI084140 to CMC; F32GM10052101 to JLG; T32GM067543 to DLB).
Footnotes
Supporting Information. Chemical, biochemical and biological procedures, characterization of novel compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Author Contributions
All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Buckley, Crews Small Molecule Control of Intracellular Protein Levels Through Modulation of the Ubiquitin Proteasome System, Angew. Chem. Int. Ed. 2014;53:2312. doi: 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakamoto KM. Protacs for treatment of cancer, Pediatr. Res. 2010;67:505–508. doi: 10.1203/PDR.0b013e3181d35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banaszynski LA, Wandless TJ. Conditional control of protein function. Chem. Biol. 2006;13:11–21. doi: 10.1016/j.chembiol.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto K, Kim K-B, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneekloth JS, Jr, Fonseca FN, Koldobskiy M, Mandal AK, Deshaies RJ, Sakamoto K, Crews CM. Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 6.Sakamoto K, Kim K-B, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular & Cellular Proteomics. 2003;2:1350. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Lee H, Puppala D, Choi E-Y, Swanson H, Kim K-B. Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool. ChemBioChem. 2007;8:2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K-B, Crews CM, Deshaies RJ, Sakamoto KM. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyrus K, Wehenkel M, Choi E-Y, Swanson H, Kim K-B. Two-Headed PROTAC: An Effective New Tool for Targeted Protein Degradation. ChemBioChem. 2010;11:1531–1534. doi: 10.1002/cbic.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cyrus K, Wehenkel M, Choi E-Y, Han H-J, Lee H, Swanson H, Kim K-B. Impact of linker length on the activity of PROTACs. Mol. BioSyst. 2011;7:359–364. doi: 10.1039/c0mb00074d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 13.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh Y, Ishikawa M, Kitaguchi R, Okuhira K, Naito M, Hashimoto Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg. Med. Chem. Lett. 2012;22:4453–4457. doi: 10.1016/j.bmcl.2012.04.134. [DOI] [PubMed] [Google Scholar]
- 15.Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, Hashimoto Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorg. Med. Chem. 2011;19:3229–3241. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 16.Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic Acid-binding proteins. J. Am. Chem. Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 17.Okuhira K, Ohoka N, Sai K, Nishimaki-Mogami T, Itoh Y, Ishikawa M, Hashimoto Y, Naito M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Letters. 2011;585:1147–1152. doi: 10.1016/j.febslet.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J. Antibiot. 1976;29:97–99. doi: 10.7164/antibiotics.29.97. [DOI] [PubMed] [Google Scholar]
- 19.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 20.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJA, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew. Chem. Int. Ed. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Molle I, Thomann A, Buckley DL, So Ernest C., Lang S, Crews CM, Ciulli A. Dissecting Fragment-Based Lead Discovery at the von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol. 2012;19:1300–1312. doi: 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 25.Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tae HS, Sundberg TB, Neklesa TK, Noblin DJ, Gustafson JL, Roth AG, Raina K, Crews CM. Identification of Hydrophobic Tags for the Degradation of Stabilized Proteins. ChemBioChem. 2012;13:538–541. doi: 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohana RF, Encell LP, Zhao K, Simpson D, Slater MR, Urh M, Wood KV. HaloTag7: a genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expression and Purification. 2009;68:110–120. doi: 10.1016/j.pep.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sin N, Kim K-B, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Total synthesis of the potent proteasome inhibitor epoxomicin: a useful tool for understanding proteasome biology. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 31.Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nature Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau HD, Yaegashi J, Zaro BW, Pratt MR. Precise control of protein concentration in living cells. Angew. Chem. Int. Ed. 2010;49:8458–8461. doi: 10.1002/anie.201003073. [DOI] [PubMed] [Google Scholar]
- 34.Shoulders MD, Ryno LM, Cooley CB, Kelly JW, Wiseman RL. Broadly Applicable Methodology for the Rapid and Dosable Small Molecule-Mediated Regulation of Transcription Factors in Human Cells. J. Am. Chem. Soc. 2013 doi: 10.1021/ja402756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoulders MD, Ryno Lisa M., Genereux Joseph C., Moresco James J., Tu Patricia G., Wu C, Yates John R., Su Andrew I., Kelly Jeffery W., Wiseman RL. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Reports. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonger KM, Chen L-C, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. http://www.genecopoeia.com/tech/halo-tag/
- 38.Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103–119. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.