

Abstract
Gestational trophoblastic diseases (GTD) are a group of pregnancy-related disorders representing rare human tumours. They encompass premalignant disorders including complete (CHM), partial hydatidiform mole (PHM), exaggerated placental site (EPS), and placental-site nodule (PSN) as well as malignant disorders (also known as “gestational trophoblastic neoplasia [GTN]”) including invasive mole, choriocarcinoma (CC), placenta-site trophoblastic tumour (PSTT), and epitheloid trophoblastic tumours (ETT) (Fig. 1). Originally, GTD develop from abnormal proliferation of trophoblastic tissue and form botryoid arranged vesicles. Premalignant moles are usually treated by suction curettage while persistent and recurrent moles and malignant forms require systemic therapy with methotrexate or combination chemotherapy consisting of etoposide, actimomycin D, methotrexate, vincristine, and cyclophosphamide (EMA-CO). β-human chorion gonadotropin (β-hCG) plays a crucial role in diagnosis and monitoring therapeutic effects. Since the definitive diagnosis cannot be obtained by histology in most cases, persistent or recurrent disease is diagnosed by elevated or persistent serum levels of β-hCG. While curing rates are described to be as high as 98 %, GTD may initially present, recur, or end up as a metastasising systemic disease. This underlines the importance of a regular and consistent follow-up after treatment.
Key words: gestational trophoblastic disease, hydatidiform moles, molar pregnancy, gestational neoplasia, human chorion gonadotropin
Abstract
Zusammenfassung
Gestationsbedingte Trophoblasterkrankungen (GTE) gehören zu der Gruppe seltener menschlicher Tumoren, die im Rahmen einer Schwangerschaft entstehen. Diese Gruppe umfasst prämaligne Veränderungen wie Blasenmole und Partialmole, hyperplastische Implantationsstelle des Plazentabetts (exaggerated placental site, EPS) und Plazentabettknötchen (placental site nodule, PSN) sowie maligne Erkrankungen (auch gestationsbedingte Trophoblastneoplasien [GTN] genannt) wie invasive Molen, Chorionkarzinom, Plazentabetttumor (placental site trophoblastic tumor, PSTT) und epitheloider Trophoblasttumor (ETT) (Abb. 1). Die Entstehung von GTE ist durch die abnorme Proliferation von Trophoblastzellen gekennzeichnet, die traubenförmig angeordnete Bläschen bilden. Prämaligne Molen werden zumeist mit Saugkürettage therapiert. Die Behandlung persistierender und rezidivierender Molen sowie maligner Formen besteht aus einer systemischen Therapie mit Methotrexat oder einer Kombinationschemotherapie aus Etoposid, Actinomycin-D, Methotrexat, Vincristin, und Cyclophosphamid (EMA-CO). β-humanes Choriongonadotropin (β-hCG) spielt eine wichtige Rolle bei der Diagnose und Kontrolle der Therapie. Da eine histologische Untersuchung in den meisten Fällen keine endgültige Diagnose liefert, beruht die Diagnose von Persistenz und Rezidiv auf erhöhten (persistierenden) Serumkonzentrationen von β-hCG. Obwohl die in der Literatur angegebenen Heilungsraten hoch sind mit bis zu 98 %, können GTE nach dem ersten Auftreten rekurrieren oder in eine systemische Erkrankung mit Metastasierung übergehen. Dies unterstreicht die Bedeutung einer regelmäßigen und konsequenten Nachsorge.
Schlüsselwörter: gestationsbedingte Trophoblasterkrankung, Blasenmole, molare Schwangerschaft, gestationsbedingte Neoplasie, humanes Choriongonadotropin
Introduction
Gestational trophoblastic diseases (GTD) are a rare complication of pregnancy caused by defective differentiation of the trophoblast. Trophoblastic tissue does not underlie common regulatory mechanisms avoiding neoplastic growth. As a result, complete hydatidiform moles (CHM) and partial hydatidiform moles (PHM) or malignant disorders known as invasive mole, choriocarcinoma (CC), placenta-site tumour (PSTT) and epitheloid trophoblastic tumour (ETT) can develop due to insufficient control mechanisms 1. Symptoms differ and may range from uterine bleeding to metabolic disease such as β-hCG triggered hyperthyreosis. Additionally, lutein-cysts of the ovaries can occur as a consequence of increased β-hCG resulting in ovarian hyperstimulation. Colour-coded Duplex sonography can be used as a diagnostic tool since these tumours are typically hypervascularised (Fig. 2). This review covers the epidemiology, pathology and clinical management of GTD.
Fig. 2.
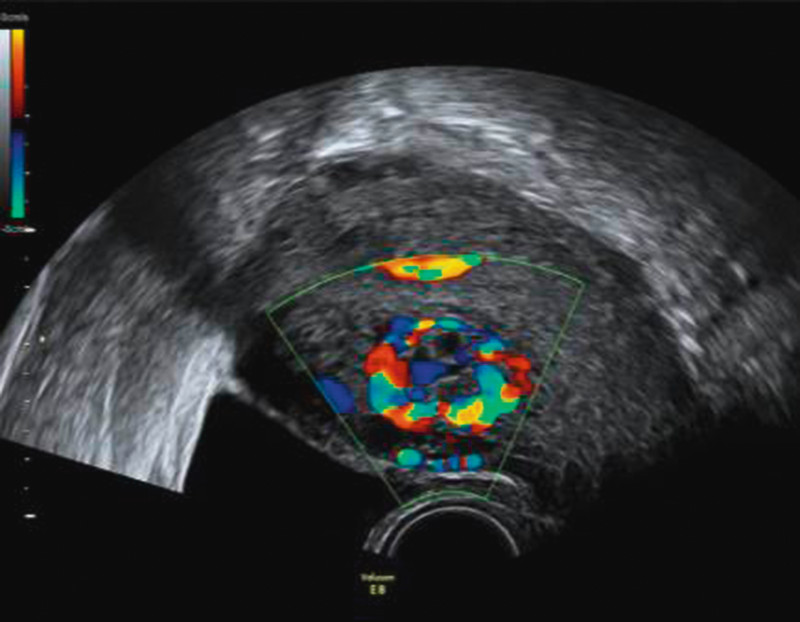
Duplex ultrasonography of a GTN following a CHM.
Fig. 1.
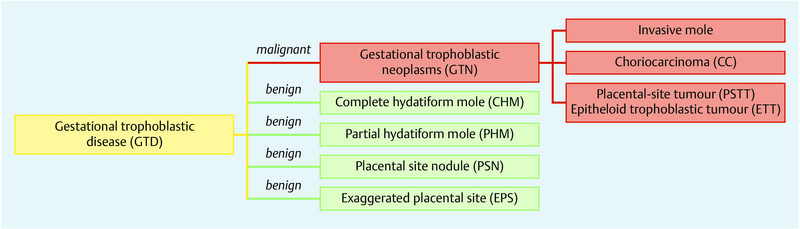
Classification of gestational trophoblastic disease.
Epidemiology
The incidence and epidemiology of GTD differs worldwide. While the incidence is approximately 1–3 in 1000 pregnancies for CHM and 3 in 1000 pregnancies for PHM in North America and Europe, both conditions appear to be diagnosed more often in Asia and Latin America. This could be due to differences in hospital systems and population data, availability of pathological expertise or genetic effects 1, 2. The risk for molar pregnancy is increased by 1–2 % after one, and by 15–20 % after two prior molar pregnancies, respectively. Promiscuity has no effect on the incidence 1. The occurrence of a molar pregnancy strongly correlates with maternal age especially for women younger than 16 years and even more pronounced for those above 45 years 3. The incidence of CC and PSTT/ETT is estimated to be 1 in 50 000 pregnancies but may even be higher as it can arise from any pregnancy 4, 5. Furthermore the influence of hormonal factors such as a late menarche, the usage of oral contraceptives and light menstrual flow have been linked to the increased risk of GTN 1.
Genetics and Pathology
GTD represents the result of an aberrant human pregnancy with an abnormal karyotype due to an incorrect fertilisation and, finally, abnormal proliferation of placental villi 1. In most cases the fertilisation of an empty ovum with one haploid sperm leads to a CHM containing a duplicated karyotype of paternal origin 6, 7, 8 (Fig. 3 a). Alternatively, fewer cases of a dispermic fertilisation of an empty ovum have been described 9 (Fig. 3 b). The loss of maternal chromosomes can occur either before or right after conception 1. Though nuclear DNA is entirely paternal in CHM, mitochondrial DNA is still of maternal origin 10. In addition, recurrent CHM is linked to an autosomal recessive disease with biparental karyotype (BiCHM), termed as familial recurrent hydatidiform mole (FRHM) 11 (Fig. 3 c). Thereby, in women with recurrent pregnancy loss mutations in NLRP7 gene (OMIM 609 661) and less frequently in KHDC3 L gene (OMIM 611 687) could be detected 12. It is assumed that NLRP7 plays an important role in the regulation of maternal imprinting in oocytes. Since these women are unable to conceive naturally, the only way to become pregnant is by oocyte donation 12, 13, 14, 15. In contrast to CHM, PHM is caused by the fertilisation of an ovum by two sperms, leading to a triploid karyotype in most cases 16, 17, 18 (Fig. 3 d).
Fig. 3 a.
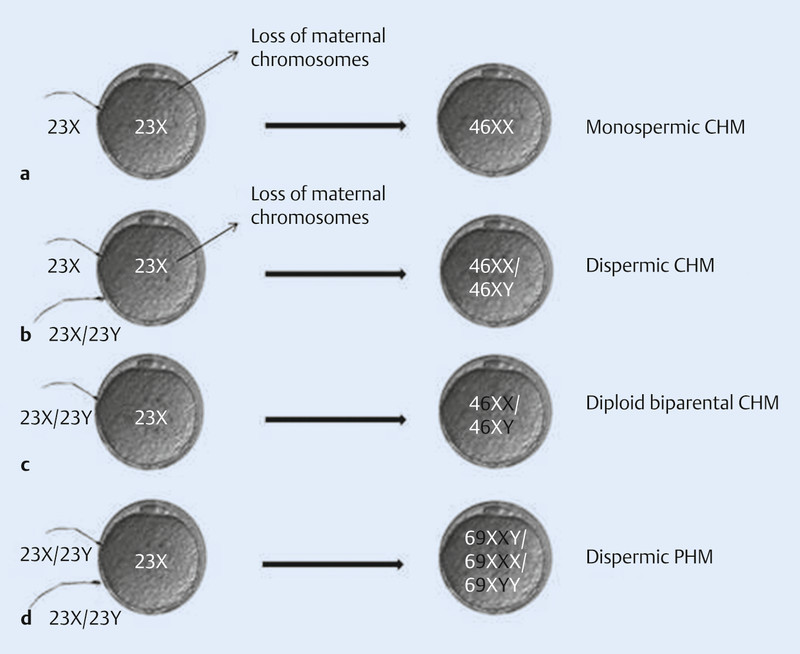
to d Pathogenesis of CHM, PHM and rare familial recurrent hydatidiform mole; CHM = complete hydatidiform mole, PHM = partial hydatidiform mole. Paternal (white) and maternal (black) derived genes are shown.
All HM and CC arise from villous trophoblast, whereas PSTT are formed by interstitial trophoblast 1. Although various morphological characteristics are known to distinguish between CHM and PHM, they are very similar in early stages of pregnancy requiring a subtle histopathological review 19. CHM (Fig. 4) shows a pathognomonic architecture, described as botryoid structure induced by abnormal trophoblast hyperplasia, stromal hypercellularity, stromal karyorrhectic debris, and collapsed villous blood vessels. Contrarily, early PHM (Fig. 5) present as patchy villous hydrops with scattered abnormally shaped irregular villi, trophoblastic pseudoinclusions, and patchy trophoblast hyperplasia 20, 21, 22.
Fig. 4.
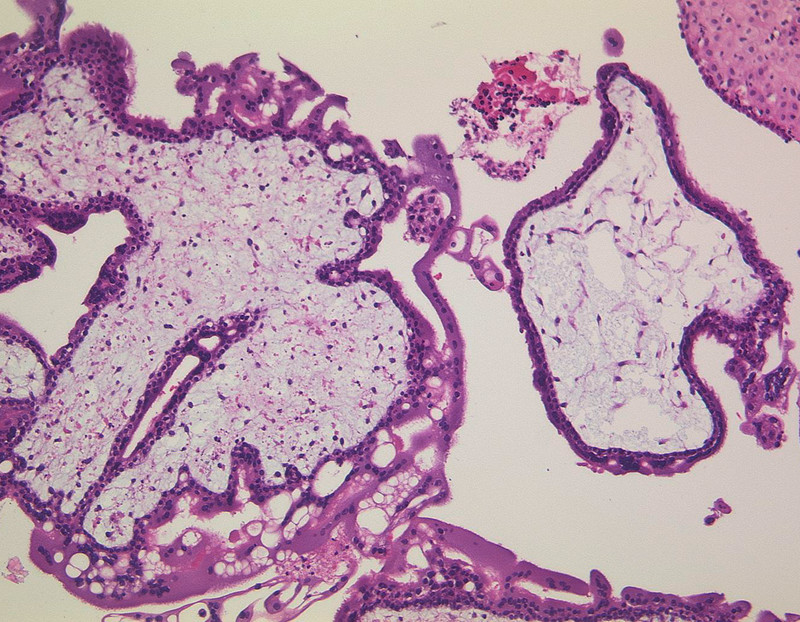
Complete hydatidiform mole, haematoxin-eosin staining, original magnification × 100 (with kind permission of Prof. Gabbert, Department of Pathology, Heinrich-Heine-University Düsseldorf).
Fig. 5.
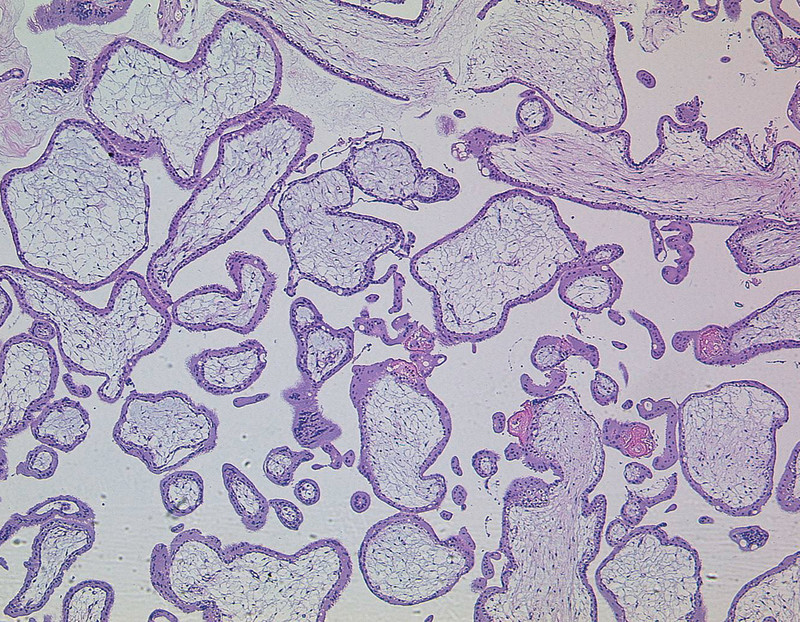
Partial hydatidiform mole, haematoxin-eosin staining, original magnification × 50 (with kind permission of Prof. Gabbert, Department of Pathology, Heinrich-Heine-University Düsseldorf).
Occasionally, other aberrant pregnancies like trisomy, monosomy, maternally derived triploidy and translocations can mimic similar histological features and may cause confusion. Immunostaining with P57kip2 is an established method to distinguish between CHM and PHM as well as non-molar pregnancies. Since P57kip2 is expressed exclusively by maternal chromosomes, immunostaining is positive in PHM and non-molar pregnancies and negative in CHM 3. Alternatively, in situ hybridisation, DNA ploidy analysis by flow cytometry or molecular genotyping can be performed to differ between PHM or CHM and non-molar hydropic abortions 23, 24, 25. Nevertheless, there is no predictive marker for GTN. Keeping this in mind, a routine monitoring of β-hCG after a GTD is crucial to detect persistence and potential malignant transformation 19.
An invasive mole describes the case of a CHM infiltrating the myometrium rather than a PHM infiltrating the myometrium. In contrast to a CC, an invasive mole shows chorionic villi, CC does not 27.
CC are epithelial tumours derived from trophoblastic elements. It is of particular importance that CC can occur after any kind of pregnancy. However, it is mainly diagnosed subsequent to a HM. Haematogenous spread, especially to the lungs and to the vagina, is based on the aggressive and invasive growth of this highly malignant tumour 26. Histologically, CC shows a biphasic picture with central necrosis. CC (Fig. 6) show parts of cytotrophoblast-like cells and multinucleated, pleomorphic syncytiotrophoblast-like parts missing formed chorionic villi 27.
Fig. 6.
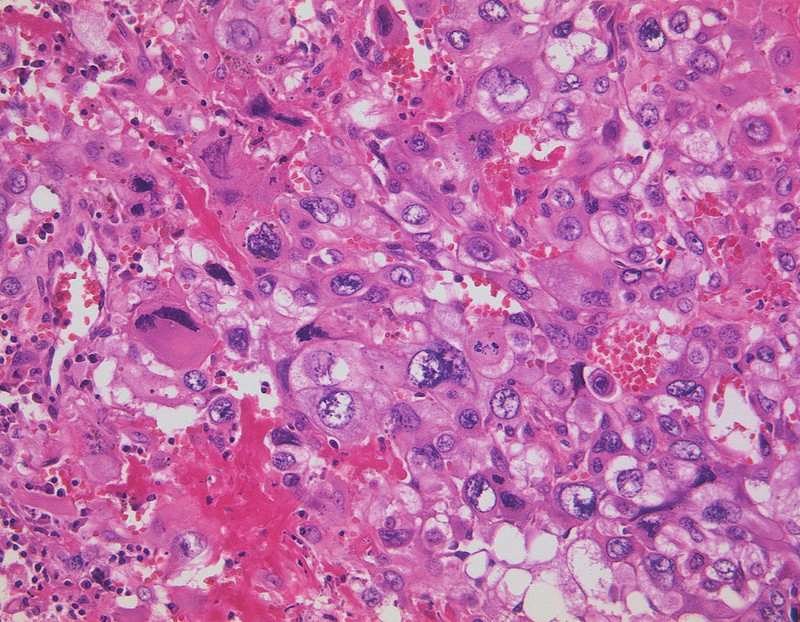
Choriocarcinoma, haematoxin-eosin staining, original magnification × 200, (with kind permission of Prof. Gabbert, Department of Pathology, Heinrich-Heine-University Düsseldorf).
Intraplacental CC following an uncomplicated term pregnancy is less common, however, metastastic disease can often be traced back to this circumstance. Taking a biopsy of metastatic CC after a previous pregnancy should be carefully considered to avoid life-threatening haemorrhage due to a high vascularity. Apart from that, a precise differentiation between a gestational and other non-gestational tumours as for example gastric or lung cancer can only be obtained through a pathological review 28.
PSTT differ in their clinical behaviour as they show less necrosis and haemorrhage with a lower β-hCG level. In the histological examination monomorphic infiltrating nests and parts of interstitial trophoblasts can be found. Additionally, PSTT shows positive immunostaining for human placental lactogen (hpl) and other extravillous trophoblast markers 29. ETT is a variant of PSTT whose clinical features are similar to those of PSTT even though their immunohistochemical features differ 30.
Diagnosis
GTD causes a wide spectrum of different symptoms. The most frequent clinical symptom is unexpected vaginal bleeding. Nowadays, imaging including ultrasonography is able to diagnose GTD even before the onset of symptoms such as anaemia, hyperemesis, pre-eclampsia, uterine enlargement, hyperthyreoidism and respiratory distress 31, 32. In addition to an enlarged uterus, the classic indicators for GTD are the ultrasonographic display of CHM (“snowstorm” pattern) and theca lutein ovarian cysts, as well as abnormally increased β-hCG serum levels not correlating with gestational age.
It is important to keep in mind that only 40–60 % of CHM and especially PHM are detected by ultrasonography. Meanwhile, 10 % of suspected CHM and PHM based on ultrasound turn out to be non-molar hydropic abortions on histological review 33. Therefore, a histological review of the material of any non-viable pregnancy is mandatory 21, 32. β-hCG surveillance plays an important role in the clinical management of women with GTD. For example, in women diagnosed with GTD, repeated measurements of serum β-hCG levels should be performed for at least 3 to 4 weeks until they drop below detection threshold 34. This enables an early detection of progression to GTN by persistent or increasing β-hCG levels 1. However, due to the high similarity of the alpha unit of β-hCG to the pituitary glycoprotein hormones, assays for specific detection of the beta subunit should ideally be used 1.
Since persistent CHM/PHM, CC, PSTT, and ETT can occur even years after any pregnancy, the diagnosis is challenging under these circumstances. Besides vaginal bleeding and dysmenorrhea, further unspecific symptoms as for example nausea or haemoptysis may only be diagnosed after tumour spread to different organs. PSTT and ETT can be differentiated from CC as they grow slower, produce lower β-hCG levels, and have a higher incidence of lymphatic spread 19.
Treatment of Hydatidiform Moles
The gold standard in treating HM is suction dilation and curettage performed under ultrasonographic vision to avoid uterus perforation 35. The role of prostaglandins in priming of the cervix remains contradictory. In some cases an increased risk of trophoblastic embolisation, especially to the lung, has been described as a potential effect of prostaglandins causing uterine contractions. All rhesus D-negative patients should obtain prophylactic anti-D rhesus immunglobulin due to the expression of the rhesus D factor on trophoblasts 1. In cases of life-threatening haemorrhage, hysterectomy is recommended 36. A recent study describes twin pregnancies consisting of a normal fetus and a co-existing HM in one of 20 000 to 100 000 pregnancies. Although these pregnancies are of high risk with an increased risk for spontaneous abortion, a case series of 77 pregnancies suggests that remarkable 40 % of these cases result in the birth of a healthy baby 37.
Follow-up of Hydatidiform Moles
Patients with a molar pregnancy and plateauing or rising β-hCG levels should be referred to specialized centres. Even if the risk of developing a GTN following PHM is not higher than 0.5–2 %, a CHM results in 15–20 % in a malignant disease 4, 38, requiring constant β-hCG measurements. Besides a variety of different protocols for follow-up, the recommendation of the International Federation of Gynecology and Obstetrics (FIGO) has been generally accepted. According to FIGO, women with PHM should be followed up by weekly β-hCG controls to record a normal level of β-hCG in two consecutive measurements, followed by monthly check-ups for the next 3 to 6 months. CHM requires monthly check-ups for an entire year. Table 1 summarises the follow-up of CHM as well as PHM.
Table 1 Follow-up of hydatidiform moles modified from Ka Yu Tse et al. (Obstetrics, Gynaecology and Reproductive Medicine, 2015 39).
| CHM | PHM | |
|---|---|---|
| Duration of serum/urine β-hCG controls | 1 year | 3–6 months |
| Frequency of β-hCG measurement | weekly until 3 × negative, then monthly | weekly until 2–3 × negative, then monthly |
| Prophylactic chemotherapy | no | no |
| Risk of subsequent GTD | 1–2 % following one previous mole15–18 % following two previous moles | |
| Chance for GTN | 15–20 % | 0.5–2 % |
Additionally, patients should be instructed to use contraception for a period of least one year to enable conclusive follow-up processes. To avoid perforation of the uterus intrauterine devices (IUDs) should not be used for contraception. The usage of hormones during pending elevated β-hCG levels can be recommended 28. Patients having a histology of a molar pregnancy should be reminded of checking β-hCG levels 6 and 10 weeks after each subsequent pregnancy to detect a possible relapse of previous molar diseases 28.
Treatment Indications
The diagnosis of GTN and the subsequent indication for systemic chemotherapy is made by three or more equivalent elevated β-hCG levels over a period of at least 3 weeks or a rise in β-hCG of 10 % or greater. Histological verification of CC and metastases (especially brain, liver or lung) of excluded other origins as well as radiologically abnormal findings larger than 2 cm are further reasons for chemotherapeutic treatment. Additionally women with heavy vaginal bleeding, gastrointestinal haemorrhage requiring transfusions or β-hCG levels above 20 000 IU/l after 4 weeks following HM evacuation should receive chemotherapy 1, 40.
Staging
The diagnosis of HM or GTN is based on histological examination and surveillance of β-hCG. Moreover, patients should receive a pelvic duplex ultrasonography to size the extent and to visualize the vascularity of the disease 41, 42. GTN most frequently spreads to the lung. Therefore a chest CT thorax should be performed to detect pulmonary metastases. In case of a chest CT thorax showing suspicious findings, patients need a complete staging including whole-body CT scan and MRI of the pelvic and the brain (Fig. 7) 19. These staging investigations include a contrast enhanced CT of the chest and the abdomen, a MRI of the brain and a doppler ultrasound of the pelvis. The individual prognosis of a patient with GTN can be calculated by using the FIGO prognostic score (Table 2) in combination with an anatomical staging system (Table 3). The anatomical staging system can also be applied in patients with PSTT and ETT. The scoring system predicts the possibility of developing a single-drug chemotherapy resistance. A score less than 6 indicates a low risk and a score higher than 7 suggests a high risk for a primary resistance to monochemotherapy. A prognostic scoring system as well as an anatomical staging are helpful to compare data between different medical institutions 1.
Fig. 7.
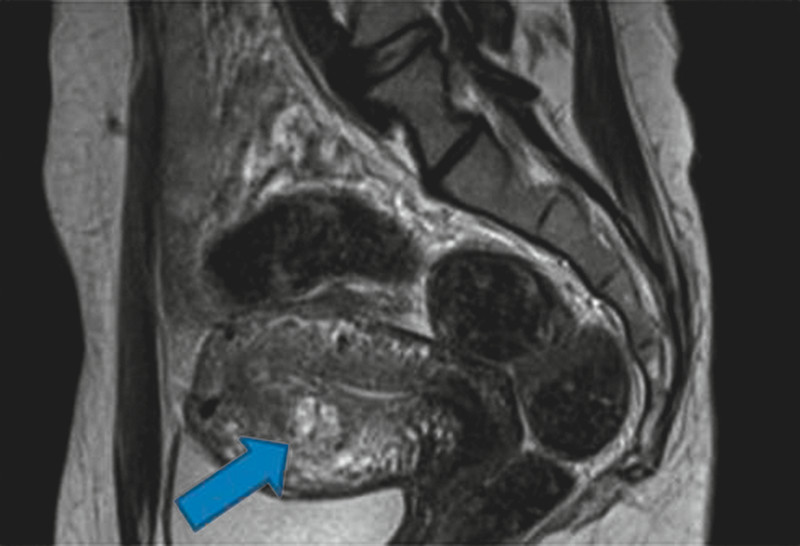
Magnetic resonance imaging of an intramural GTN (with kind permission of Prof. Antoch, Department of Radiology, Heinrich-Heine-University Düsseldorf).
Table 2 FIGO risk score for gestational trophoblastic neoplasia (reprinted from 41, Copyright 2014 with permission from Current Oncology Reproduction).
| 0 | 1 | 2 | 4 | |
|---|---|---|---|---|
| Calculation of patientʼs risk score by adding single factors:0–6 points → low-risk group≥ 7 points → high-risk group | ||||
| Age (years) | < 40 | ≥ 40 | ||
| Antecedent pregnancy | mole | abortion | term | |
| Interval from antecedent pregnancy to chemotherapy (months) | < 4 | 4–6 | 7–12 | > 12 |
| hCG (IU/l) | < 103 | 103–104 | 104–105 | > 105 |
| Number of metastases | 0 | 1–4 | 5–8 | > 8 |
| Site of metastases | lung | spleen/kidney | gastrointestinal tract | brain/liver |
| Largest tumour mass (cm) | 3–5 | > 5 | ||
| Previous chemotherapay | monotherapy | combined therapy | ||
Table 3 FIGO anatomical staging used for PSTT/ETT (reprinted from 41, Copyright 2014 with permission from Current Oncology Reproduction).
| Stage | Description |
|---|---|
| I | Disease confined to the uterus |
| II | Disease extending into the pelvis |
| III | Disease spread to lungs with or without known genital involvement |
| IV | All other metastatic sites (liver, kidney, spleen, brain) |
Treatment of Low-Risk Disease
Patients having less than 7 points based on the FIGO scoring system belong to the low-risk group. In this group a nearly 100 % cure rate is achieved by single-agent systemic chemotherapy. Therefore, a second dilatation and suction curettage may be considered. It has been demonstrated that a disease located in the uterine cavity rather than in the myometrium with β-hCG levels below 5000 IU/l could benefit from a second intervention 1.
Various single agent chemotherapy regimens are available to treat low-risk GTN. Of all approved agents, methotrexate (MTX) is the most commonly used with a good therapeutic index 41. There are no safety data available to establish universal dose regimens. However, the following protocol is the most commonly used (Table 4): 50 mg MTX i.m. on days 1, 3, 5, and 7, followed by 15 mg folinic acid (FA) orally 30 hours after administration of MTX. This so-called “folinic acid rescue” is crucial to prevent negative side effects of MTX such as mouth ulcers, sore eyes, and pleuritis, caused by its role as an inhibitor of the folate metabolism 43, 44. An increased risk of bleeding after MTX injection has been described so that patients should receive their first dose of MTX in an inpatient setting. Subsequent injections can be given on an outpatient basis. Another agent used as single agent chemotherapy for low-risk disease is Actinomycin D (ActD), an antitumour antibiotic. ActD is approved for patients resistant to MTX treatment with β-hCG levels below 300 IU/l. Patients lacking remission after treatment with MTX and β-hCG levels greater than 300 should be treated according to the protocols for high-risk disease 45. Once the first cycle of chemotherapy has been administered resulting in normal β-hCG levels, 2–3 consolidation cycles should be administered to prevent relapse. However, only 2 consolidation cycles may eventually raise the risk of relapse to 50 % 46. Thus, patients with GTN should receive 3 consolidation cycles.
Table 4 Treatment for low-risk disease (< 6 point, reprinted from ref. 41, Copyright 2014 with permission from Current Oncology Reproduction).
| Agent | Regimen | |
|---|---|---|
| Methotrexate | 50 mg i. m. (day 1, 3, 5, 7) | biweekly repeated |
| Folinic acid | 15 mg p. o. (30 h after each MTX-injection) | biweekly repeated |
Treatment of High-Risk and Drug-Resistant Disease
Patients with a FIGO score greater than 6 points have a high risk to develop resistance to single-agent systemic chemotherapy. Polychemotherapy regimen with etoposide, MTX and ActD alternating with cyclophosphamide plus vincristine (EMA-CO) as first line therapy for patients with high-risk disease is well accepted across the world (Table 5). Despite the high effectiveness of EMA-CO, up to 30 to 40 % of the patients develop progress or relapse. These patients should receive additional multi-agent, platinum-based chemotherapy (Table 6). Among different risk factors, patients presenting with metastases outside the lung or vagina and patients having not received standard chemotherapy regimens, have an even higher risk for progressive or recurrent disease. As recommended for low-risk disease, 2–3 consolidation cycles should be applied to ensure persistent therapeutic success 47.
Table 5 Treatment for high-risk disease (reprinted from 41, Copyright 2014 with permission from Current Oncology Reproduction).
| Agent | Regimen | |
|---|---|---|
| Day 1 | etoposide | 100 mg/m2 intravenously over 30 min |
| actinomycin D | 0.5 mg intravenous bolus | |
| methotrexate | 300 mg/m2 intravenously over 12 h | |
| Day 2 | etoposide | 100 mg/m2 intravenously over 30 min |
| actinomycin D | 0.5 mg intravenous bolus | |
| folinic acid | 15 mg intravenously or orally, every 12 h for 4 doses, starting 24 h after the start MTX infusion | |
| Day 8 | vincristine | 1 mg/m2 intravenous bolus |
| cyclophosphamide | 600 mg/m2 over 30 min |
Table 6 Treatment for relapsed GTN (reprinted from ref. 41, Copyright 2014 with permission from Current Oncology Reproduction).
| Agent | Regimen | |
|---|---|---|
| Day 1 | dexamethasone | 20 mg orally 12 and 6 h before paclitaxel |
| cimetidine | 30 mg in 100 ml NaCl intravenously over 30 min | |
| chlorphenamine | 10 mg intravenous bolus | |
| paclitaxel | 135 mg/m2 in 250 ml NaCl intravenously over 3 h | |
| mannitol | 10 % in 500 ml intravenously over 3 h | |
| cisplatin | 60 mg/m2 in 1 l NaCl intravenously over 3 h | |
| post-hydration | 1 l NaCl + 20 mmol KCL + 1 g MgSO4 intravenously oder 2 h | |
| Day 15 | dexamethasone | 20 mg orally 12 and 6 h before paclitaxel |
| cimetidine | 30 mg in 100 ml NaCl intravenously over 30 min | |
| chloramphenamine | 10 mg intravenous bolus | |
| paclitaxel | 135 mg/m2 in 250 ml NaCl intravenously over 3 h | |
| etoposide | 150 mg/m2 in 1 l NaCl intravenously over 1 h |
PSTT/ETT
As mentioned before, PSTT/ETT can vary in their clinical presentation as well as in their growth behaviors. Above, primary treatment has to be distinguished from invasive molar disease or CC. In contrast to CC, primary hysterectomy with removal of suspicious pelvic and abdominal lymph nodes including a bilateral adnexectomy in postmenopausal women is the treatment of choice for PSTT/ETT. Patients with risk factors such as a high mitotic rate or metastatic disease will need adjuvant chemotherapy either with EP/EMA or TE/TP. Relapses beyond 4 years after last pregnancy have a particular high mortality risk, so that patients may profit from high-dose chemotherapy combined with stem cell support 1, 48.
Follow-up of GTN
Subsequent to chemotherapy, a follow-up including post-treatment images has to be initiated to guarantee a continuous monitoring of clinical processes. Duplex ultrasonography plays an important role in the follow-up of low-risk disease and careful surveillance with serum β-hCG is crucial. Most relapses occur within the first year after completion of chemotherapy. A generally approved schedule of β-hCG surveillance indicates monitoring on a weekly basis for 6 weeks after chemotherapy followed by biweekly measurements until 6 months after chemotherapy. Afterwards, a biannual measurement of β-hCG for a period of 5 years is sufficient 19. Although women getting pregnant within the first year after a GTN have a favorable outcome, a subsequent pregnancy is recommended after one year at the earliest. Meanwhile, safe oral contraception should be prescribed. Fortunately, women with GTN, having received chemotherapeutic treatment are still fertile in up to 80 % without having an increased rate of congenital anomalies 49.
Conclusions
In conclusion, GTD are a rare group of trophoblast-derived diseases with an overall favorable survival rate. Overall survival rates of 95 to 100 % can be retained despite raising drug-resistant diseases 50. Nevertheless, some patients may develop a malignant disease resulting in death. Therefore, patients should be treated in specialized centers experienced in the management of women with GTD. In contrast to other European Countries, as for example the United Kingdom, a centralized care of women with GTD is not offered in Germany. Establishing specialized centers for treatment of GTD could also simplify research of the disease, especially for genetic causes such as the biCHM. Furthermore, novel and supplementary therapeutic approaches are needed to cure even advanced or heavily pretreated women.
Footnotes
Conflict of Interest None.
References
- 1.Seckl M J, Sebire N J, Berkowitz R S. Gestational trophoblastic disease. Lancet. 2010;376:717–729. doi: 10.1016/S0140-6736(10)60280-2. [DOI] [PubMed] [Google Scholar]
- 2.Palmer J R. Advances in the epidemiology of gestational trophoblastic disease. J Reprod Med. 1994;39:155–162. [PubMed] [Google Scholar]
- 3.Ngan S, Seckl M J. Gestational trophoblastic neoplasia management: an update. Curr Opin Oncol. 2007;19:486–491. doi: 10.1097/CCO.0b013e3282dc94e5. [DOI] [PubMed] [Google Scholar]
- 4.Seckl M J, Fisher R A, Salerno G. et al. Choriocarcinoma and partial hydatidiform moles. Lancet. 2000;356:36–39. doi: 10.1016/S0140-6736(00)02432-6. [DOI] [PubMed] [Google Scholar]
- 5.Fisher R A, Lawler S D, Povey S. et al. Genetically homozygous choriocarcinoma following pregnancy with hydatidiform mole. Br J Cancer. 1988;58:788–792. doi: 10.1038/bjc.1988.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kajii T, Ohama K. Androgenetic origin of hydatidiform mole. Nature. 1977;268:633–634. doi: 10.1038/268633a0. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita K, Ishikawa M, Shimizu T. et al. HLA antigens in husband-wife pairs with trophoblastic tumour. Gynecol Oncol. 1981;12:68–74. doi: 10.1016/0090-8258(81)90096-2. [DOI] [PubMed] [Google Scholar]
- 8.Fisher R A, Newlands E S. Gestational trophoblastic disease. Molecular and genetic studies. J Reprod Med. 1998;43:87–97. [PubMed] [Google Scholar]
- 9.Pattillo R A, Sasaki S, Katayama K P. et al. Genesis of 46,XY hydatidiform mole. Am J Obstet Gynecol. 1981;141:104–105. [PubMed] [Google Scholar]
- 10.Azuma C, Saji F, Tokugawa Y. et al. Application of gene amplification by polymerase chain reaction to genetic analysis of molar mitochondrial DNA: the detection of anuclear empty ovum as the cause of complete mole. Gynecol Oncol. 1991;40:29–33. doi: 10.1016/0090-8258(91)90080-o. [DOI] [PubMed] [Google Scholar]
- 11.Fisher R A, Hodges M D, Newlands E S. Familial recurrent hydatidiform mole: a review. J Reprod Med. 2004;49:595–601. [PubMed] [Google Scholar]
- 12.Murdoch S, Djuric U, Mazhar B. et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38:300–302. doi: 10.1038/ng1740. [DOI] [PubMed] [Google Scholar]
- 13.Judson H, Hayward B E, Sheridan E. et al. A global disorder of imprinting in the human female germ line. Nature. 2002;416:539–542. doi: 10.1038/416539a. [DOI] [PubMed] [Google Scholar]
- 14.Fisher R A, Hodges M D, Rees H C. et al. The maternally transcribed gene p 57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Hum Mol Genet. 2002;11:3267–3272. doi: 10.1093/hmg/11.26.3267. [DOI] [PubMed] [Google Scholar]
- 15.Kinoshita T, Wang Y, Hasegawa M. et al. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1beta secretion. J Biol Chem. 2005;280:21720–21725. doi: 10.1074/jbc.M410057200. [DOI] [PubMed] [Google Scholar]
- 16.Szulman A E, Surti U. The syndromes of hydatidiform mole. I. Cytogenetic and morphologic correlations. Am J Obstet Gynecol. 1978;131:665–671. doi: 10.1016/0002-9378(78)90829-3. [DOI] [PubMed] [Google Scholar]
- 17.Lawler S D, Fisher R A, Dent J. A prospective genetic study of complete and partial hydatidiform moles. Am J Obstet Gynecol. 1991;164:1270–1277. doi: 10.1016/0002-9378(91)90698-q. [DOI] [PubMed] [Google Scholar]
- 18.Lage J M, Mark S D, Roberts D J. et al. A flow cytometric study of 137 fresh hydropic placentas: correlation between types of hydatidiform moles and nuclear DNA ploidy. Obstet Gynecol. 1992;79:403–410. doi: 10.1097/00006250-199203000-00016. [DOI] [PubMed] [Google Scholar]
- 19.Seckl M J, Sebire N J, Fisher R A. et al. Gestational trophoblastic disease: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 06:vi39–vi50. doi: 10.1093/annonc/mdt345. [DOI] [PubMed] [Google Scholar]
- 20.Sebire N J, Makrydimas G, Agnantis N J. et al. Updated diagnostic criteria for partial and complete hydatidiform moles in early pregnancy. Anticancer Res. 2003;23(2C):1723–1728. [PubMed] [Google Scholar]
- 21.Sebire N J, Fisher R A, Rees H C. Histopathological diagnosis of partial and complete hydatidiform mole in the first trimester of pregnancy. Pediatr Dev Pathol. 2003;6:69–77. doi: 10.1007/s10024-002-0079-9. [DOI] [PubMed] [Google Scholar]
- 22.Sebire N J, Seckl M J. Gestational trophoblastic disease: current management of hydatidiform mole. BMJ. 2008;337:a1193. doi: 10.1136/bmj.a1193. [DOI] [PubMed] [Google Scholar]
- 23.Fisher R A, Newlands E S. Rapid diagnosis and classification of hydatidiform moles with polymerase chain reaction. Am J Obstet Gynecol. 1993;168:563–569. doi: 10.1016/0002-9378(93)90494-4. [DOI] [PubMed] [Google Scholar]
- 24.Makrydimas G, Sebire N J, Thornton S E. et al. Complete hydatidiform mole and normal live birth: a novel case of confined placental mosaicism: case report. Hum Reprod. 2002;17:2459–2463. doi: 10.1093/humrep/17.9.2459. [DOI] [PubMed] [Google Scholar]
- 25.Osada H. A complete hydatidiform mole coexisting with a normal fetus was confirmed by Variable Number Tandem Repeat (VNTR) polymorphism analysis using polymerase chain reaction. Gynecol Oncol. 1995;56:90–93. doi: 10.1006/gyno.1995.1015. [DOI] [PubMed] [Google Scholar]
- 26.Ozalp S S, Oge T. Gestational trophoblastic diseases in Turkey. J Reprod Med. 2013;58:67–71. [PubMed] [Google Scholar]
- 27.Arima T, Imamura T, Amada S. et al. Genetic origin of malignant trophoblastic neoplasms. Cancer Genet Cytogenet. 1994;73:95–102. doi: 10.1016/0165-4608(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 28.Fisher R A, Savage P M, MacDermott C. et al. The impact of molecular genetic diagnosis on the management of women with hCG-producing malignancies. Gynecol Oncol. 2007;107:413–419. doi: 10.1016/j.ygyno.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 29.Baergen R N, Rutgers J L, Young R H. et al. Placental site trophoblastic tumour: A study of 55 cases and review of the literature emphasizing factors of prognostic significance. Gynecol Oncol. 2006;100:511–520. doi: 10.1016/j.ygyno.2005.08.058. [DOI] [PubMed] [Google Scholar]
- 30.Shih I M, Kurman R J. Epithelioid trophoblastic tumour: a neoplasm distinct from choriocarcinoma and placental site trophoblastic tumour simulating carcinoma. Am J Surg Pathol. 1998;22:1393–1403. doi: 10.1097/00000478-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 31.Hou J L, Wan X R, Xiang Y. et al. Changes of clinical features in hydatidiform mole: analysis of 113 cases. J Reprod Med. 2008;53:629–633. [PubMed] [Google Scholar]
- 32.Fowler D J, Lindsay I, Seckl M J. et al. Histomorphometric features of hydatidiform moles in early pregnancy: relationship to detectability by ultrasound examination. Ultrasound Obstet Gynecol. 2007;29:76–80. doi: 10.1002/uog.3880. [DOI] [PubMed] [Google Scholar]
- 33.Fowler D J, Lindsay I, Seckl M J. et al. Routine pre-evacuation ultrasound diagnosis of hydatidiform mole: experience of more than 1000 cases from a regional referral center. Ultrasound Obstet Gynecol. 2006;27:56–60. doi: 10.1002/uog.2592. [DOI] [PubMed] [Google Scholar]
- 34.Seckl M J, Gillmore R, Foskett M. et al. Routine terminations of pregnancy–should we screen for gestational trophoblastic neoplasia? Lancet. 2004;364:705–707. doi: 10.1016/S0140-6736(04)16898-0. [DOI] [PubMed] [Google Scholar]
- 35.Berkowitz R S, Goldstein D P. Chorionic tumours. N Engl J Med. 1996;335:1740–1748. doi: 10.1056/NEJM199612053352306. [DOI] [PubMed] [Google Scholar]
- 36.Xia Z F, Song H Z, Tang M Y. Risk of malignancy and prognosis using a provisional scoring system in hydatidiform mole. Chin Med J (Engl) 1980;93:605–612. [PubMed] [Google Scholar]
- 37.Sebire N J, Foskett M, Paradinas F J. et al. Outcome of twin pregnancies with complete hydatidiform mole and healthy co-twin. Lancet. 2002;359:2165–2166. doi: 10.1016/S0140-6736(02)09085-2. [DOI] [PubMed] [Google Scholar]
- 38.Hancock B W, Nazir K, Everard J E. Persistent gestational trophoblastic neoplasia after partial hydatidiform mole incidence and outcome. J Reprod Med. 2006;51:764–766. [PubMed] [Google Scholar]
- 39.Tse K Y, Chan K KL, Tam K F. et al. Current management of gestational trophoblastic disease. Obstet Gynaecol Reprod Med. 2015;25:12–21. [Google Scholar]
- 40.Kohorn E I. Negotiating a staging and risk factor scoring system for gestational trophoblastic neoplasia. A progress report. J Reprod Med. 2002;47:445–450. [PubMed] [Google Scholar]
- 41.Froeling F E, Seckl M J. Gestational trophoblastic tumours. Curr Oncol Rep. 2014;16:408. doi: 10.1007/s11912-014-0408-y. [DOI] [PubMed] [Google Scholar]
- 42.Agarwal R, Strickland S, McNeish I A. et al. Doppler ultrasonography of the uterine artery and the response to chemotherapy in patients with gestational trophoblastic tumours. Clin Cancer Res. 2002;8:1142–1147. [PubMed] [Google Scholar]
- 43.McNeish I A, Strickland S, Holden L. et al. Low-risk persistent gestational trophoblastic disease: outcome after initial treatment with low-dose methotrexate and folinic acid from 1992 to 2000. J Clin Oncol. 2002;20:1838–1844. doi: 10.1200/JCO.2002.07.166. [DOI] [PubMed] [Google Scholar]
- 44.Garrett A P, Garner E O, Goldstein D P. et al. Methotrexate infusion and folinic acid as primary therapy for nonmetastatic and low-risk metastatic gestational trophoblastic tumours. 15 years of experience. J Reprod Med. 2002;47:355–362. [PubMed] [Google Scholar]
- 45.Sita-Lumsden A, Short D, Lindsay I. et al. Treatment outcomes for 618 women with gestational trophoblastic tumours following a molar pregnancy at the Charing Cross Hospital, 2000-2009. Br J Cancer. 2012;107:1810–1814. doi: 10.1038/bjc.2012.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lybol C, Sweep F C, Harvey R. et al. Relapse rates after two versus three consolidation courses of methotrexate in the treatment of low-risk gestational trophoblastic neoplasia. Gynecol Oncol. 2012;125:576–579. doi: 10.1016/j.ygyno.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Lurain J R. Treatment of gestational trophoblastic tumours. Curr Treat Options Oncol. 2002;3:113–124. doi: 10.1007/s11864-002-0057-5. [DOI] [PubMed] [Google Scholar]
- 48.Schmid P, Nagai Y, Agarwal R. et al. Prognostic markers and long-term outcome of placental-site trophoblastic tumours: a retrospective observational study. Lancet. 2009;374:48–55. doi: 10.1016/S0140-6736(09)60618-8. [DOI] [PubMed] [Google Scholar]
- 49.Woolas R P, Bower M, Newlands E S. et al. Influence of chemotherapy for gestational trophoblastic disease on subsequent pregnancy outcome. Br J Obstet Gynaecol. 1998;105:1032–1035. doi: 10.1111/j.1471-0528.1998.tb10271.x. [DOI] [PubMed] [Google Scholar]
- 50.Kohorn E I. Worldwide survey of the results of treating gestational trophoblastic disease. J Reprod Med. 2014;59:145–153. [PubMed] [Google Scholar]