

Abstract
Sparing of the extraocular muscles in muscular dystrophy is controversial. To address the potential role of utrophin in this sparing, mdx:utrophin+/− and mdx:utrophin−/− mice were examined for changes in myofiber size, central nucleation, and Pax7-positive and MyoD-positive cell density at intervals over their life span. Known to be spared in the mdx mouse, and contrary to previous reports, the extraocular muscles from both the mdx:utrophin+/− and mdx:utrophin−/− mice were also morphologically spared. In the mdx:utrophin+/− mice, which have a normal life span compared to the mdx:utrophin−/− mice, the myofibers were larger at 3 and 12 months than the wild type age-matched eye muscles. While there was a significant increase in central nucleation in the extraocular muscles from all mdx:utrophin+/− mice, the levels were still very low compared to age-matched limb skeletal muscles. Pax7- and MyoD-positive myogenic precursor cell populations were retained and similar to age-matched wild type controls. These results support the hypothesis that utrophin is not involved in extraocular muscle sparing in these genotypes. In addition, it appears these muscles retain the myogenic precursors that would allow them to maintain their regenerative capacity and normal morphology over a lifetime even in these more severe models of muscular dystrophy.
Keywords: extraocular muscle, muscular dystrophy, satellite cell, MyoD, Pax7, utrophin
1. INTRODUCTION
In Duchenne muscular dystrophy (DMD) all tissues are devoid of dystrophin, with skeletal muscles the focus of the primary pathology. Despite the ubiquitous absence of dystrophin, not all skeletal muscles are equally affected. Extraocular muscles (EOM), along with a small number of other craniofacial muscles are preferentially spared from muscle pathology. Other muscles that are spared include the intrinsic laryngeal muscles [1] and various sphincteric muscles [2]. Not only are the EOM structurally spared from DMD pathology [3], the eye movements are also normal [4].
The EOM are a set of six muscles in each orbit that control eye position and eye movements, with an unusual pattern of sparing and/or preferential involvement in a large number of skeletal muscle disorders [5]. The EOM are spared not only in Duchenne muscular dystrophy, but also in other forms of muscular dystrophy, including sarcoglycan deficiency disorders [6], some forms of myotonic dystrophy [7], and limb-girdle muscular dystrophy amongst others [8]. It is currently unclear what the exact mechanism of this preferential sparing is, although many mechanisms have been proposed. Altered handling of intracellular calcium [9], increased antioxidant levels [10], altered nNOS levels [11], and up-regulation of the transmembrane protein α7β1 integrin [12] were examined as possible mechanisms to account for EOM sparing in DMD, but none proved to be responsible. Given that no single pathway had proven mechanistic, it was proposed that intrinsic differences in the EOM account for their preferential sparing [12].
Another potential mechanism proposed to explain EOM sparing in DMD is up-regulation of its autosomal homolog utrophin. Utrophin is located at the neuromuscular and myotendinous junctions in normal adult skeletal muscle [13]. It is also expressed in regenerating myofibers in dystrophic muscles [13-14]. As is true for dystrophin, utrophin contains both F-actin and β-dystroglycan binding domains [15, 16]. In addition, increased expression of utrophin in the skeletal muscles of mdx mice was shown to result in a functional improvement in muscle function [17-19]. To further test this hypothesis a mouse lacking dystrophin and utrophin (mdx:utrophin−/−) was developed [20, 21]. It was shown to have a much more severe phenotype than the mdx mouse and an earlier onset of pathology. However, the mdx:utrophin−/− mice die prematurely between the ages of 6 and 20 weeks, making it difficult to obtain or maintain colonies [22]. A series of recent studies examined the mdx:utrophin+/− mouse, and showed that their histopathology and muscle function were intermediate in the continuum from mdx to mdx:utrophin−/− mouse [23-26]. It was suggested that the time course of the pathology in the mdx:utrophin+/− mice more closely mimics that of DMD patients, and therefore was proposed as a better model for assessing longer term therapies for effectiveness.
The potential relationship of EOM sparing and utrophin expression has been controversial. Utrophin was shown to be increased in mdx EOM in one study [27], and unchanged relative to normal wild type (WT) mouse EOM in another [9]. In addition, a previous study examined the EOM pathology of the mdx:utrophin−/− mice and found that the EOM developed significant pathology in the absence of both dystrophin and utrophin [28]. In order to resolve this controversy, the EOM disease profile in the mdx:utrophin+/− and mdx:utrophin−/− mouse models of DMD, which express altered levels of utrophin, was determined, and these were compared to WT control EOM and, where possible, mdx EOM. Utrophin levels were examined immunohistochemically in all genotypes. The extent of pathology was assayed by traditional markers of dystrophinopathies, including myofiber cross-sectional area, centrally located myonuclei, and fibrosis, which are indicative of the degeneration and regeneration events common in DMD. The density of Pax7-positive satellite cells and MyoD-positive myogenic precursor cells were also assayed in these four genotypes in order to better understand the regenerative potential in these DMD disease models. In adult skeletal muscle, Pax7 is a marker for satellite cells; [29 - 31], while MyoD plays an essential role in myogenic cell specification and differentiation [30-32]. If utrophin is involved in the sparing of the EOM in DMD, significant evidence of disease pathology should be present in both mdx:utrophin+/− and mdx:utrophin−/− EOM, and should be more severe in the mdx:utrophin−/− EOM.
2. METHODS
2.1 Animals
All experiments were performed in accordance with NIH guidelines for use of animals in research and were approved by the Institutional Animal Care and Usage Committee at the University of Minnesota. C57BL/10 (WT) mice were purchased from Harlan Laboratories (Madison, WI). Dystrophic mice (mdx, mdx:utrophin+/−, and mdx:utrophin−/−), originating from Washington University (ECR 42) were maintained as a colony at the University of Minnesota through mdx:utrophin+/− breeding pairs. Animals were housed by Research Animal Resources at the University of Minnesota, were raised in 12-hour light/dark cycles, and were allowed to feed and drink ad libitum. All mice were sacrificed by CO2 asphyxiation.
2.2 Immunochemistry Methods
Immediately following sacrifice, orbital exenteration was performed to remove the globe with the EOM attached in situ. In addition, limb muscles were removed and handled similarly to the EOM samples. Globe and muscles were embedded in tragacanth gum and frozen in 2-methylbutane chilled to a slurry on liquid nitrogen. Sections were prepared at 12μm using a cryostat and stored at −80°C until stained. All histochemistry was performed using our standard laboratory methods. One set of slides was stained with hematoxylin and eosin. Immunohistochemical identification of specific proteins was performed by incubating the microslides in 10% normal serum in phosphate buffered saline (PBS) containing 0.1% Triton X-100, followed by incubation for one hour at room temperature with an antibody to one of the following: Pax7 (1:3000; Aviva Systems Biology, San Diego, CA), MyoD (1:400; Santa Cruz, Santa Cruz, CA), collagen I (1:1000; abcam, Cambridge, MA), or collagen IV (1:1000; abcam). This was followed by a PBS rinse and sequential incubation in reagents from the ABC VectaElite kit (Vector Labs, Burlingame, CA) labeled with peroxidase, and developed with diaminobenzidine with heavy metals and hydrogen peroxide.
Sections were also immunostained for the expression of utrophin (1:200, Santa Cruz, Santa Cruz, CA). After an overnight incubation in primary antibody to utrophin, the slides were rinsed in PBS, and incubated in goat anti-rabbit AF488 secondary antibody (1:2000; Jackson Immunoresearch, West Grove, PA) for one hour. After a rinse in PBS, they were coverslipped with Vectashield mounting medium.
2.3 Morphometric Analysis Methods
Morphometric analysis was performed using Bioquant Life Science software (Bioquant, Nashville, TN). A minimum of 3 slides were counted for each muscle examined from each animal, with a minimum of 200 myofibers counted in both the orbital and global layers per slide. The data from each slide were averaged, with an N of 6 for each genotype and each experimental time point. Mean cross-sectional area and presence of central nucleation were determined. For central nucleation, data are presented as percent of centrally nucleated myofibers per total fiber number. Pax7 and MyoD data are presented as percent of positive cells per total fiber number. Collagen I and IV densities were determined by a semi-automated feature of the morphometry program Bioquant (Bioquant, Nashville, TN) that determines the total collagen-positive area as a percent of total muscle area. Three fields were analyzed and averaged per mouse, with an N of 4-6 mice per experimental time point and genotype.
2.4 Statistical Analysis
Data are expressed as mean ± SEM. All data were analyzed for statistical significance using analysis of variance (ANOVA) followed by a Tukey's or Dunn's multiple comparison test. Statistics were performed using Prism software (GraphPad Software Inc., San Diego, CA). Data were considered statistically significantly different if p<0.05.
3. RESULTS
3.1 Verification of Mouse Genotype and Changes in Utrophin Expression
Genotyping confirmed that the WT and mdx mice had two wild type utrophin alleles as evidenced by the presence of a single 650bp PCR product, the mdx:utrophin+/− (het) mice had both a wild type utrophin allele and a mutant utrophin allele as evidenced by the presence of a 650bp and a 450-bp PCR product, and the mdx:utrophin−/− (dko) mice had two mutant utrophin alleles as evidenced by the presence of a single 450bp PCR product (Supplemental Figure 1A). Western blot analysis showed that the mdx mouse muscles had significantly increased levels of utrophin compared to WT controls for both the triceps and EOM. While the levels of utrophin in the mdx:utrophin+/− mouse muscles were more heterogeneous compared to the WT muscles, there was no significant difference in the levels of utrophin protein in the mdx:utrophin+/− mouse muscles compared to the WT muscles. Utrophin levels in the mdx:utrophin+/− triceps muscles were significantly reduced compared to that seen in the mdx triceps muscles, but only trended toward lower levels in the EOM. The utrophin protein was absent in the muscles of the mdx:utrophin−/− mice (Supplemental Figure 1B, C).
The differential levels of utrophin immunostaining on EOM sections from WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice confirmed the western blot data (Figure 1). In the WT mice, expression of utrophin was mainly at presumed neuromuscular junctions [13], and therefore somewhat sparsely and regionally located (Figure 1A). In the EOM sections from mdx mice, utrophin expression was seen on many myofibers in cross-section (Figure 1B). In the mdx:utrophin+/− EOM, while there was more utrophin expression around fibers than in the EOM from WT mice, it appeared to be decreased compared to the expression pattern seen in the EOM from the mdx mice (Figure 1C). As expected, the EOM from the dko mice were negative for utrophin expression (Figure 1D). This qualitative assessment confirmed the quantitative assessment using western blots (Supplemental Figure 1). As expected from previous studies [9, 27], utrophin expression patterns were irregularly located within the muscle sections, but overall the levels were highest in the EOM from the mdx mouse.
Figure 1.
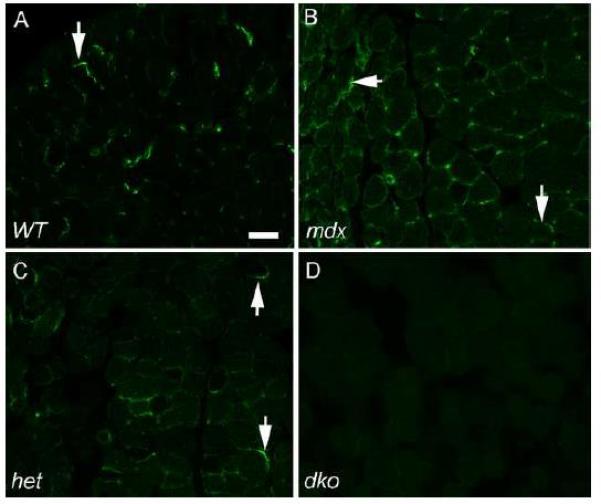
Utrophin Expression in Extraocular Muscle Cross-sections in WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− Mice. A) Expression in EOM from WT mouse. Arrow indicates positive immunostaining. As expected from the literature, utrophin was found in the areas of the EOM containing neuromuscular junctions and expressed in lower levels in areas more distant from the end plate zone (not shown). B) Expression in EOM from mdx mouse. Increased numbers of myofibers expressed utrophin compared to the WT muscle, and its distribution was wider in terms of both numbers of fibers and in terms of expression proximal or distal to the main endplate zone (not shown). Arrows indicate positive immunostaining. C) Expression in EOM from mdx:utrophin+/− (het) mouse. Arrow indicates positive immunostaining. Note that there are far fewer fibers showing utrophin expression than in the mdx EOM cross-sections, but more than was seen in the WT EOM sections. D) There was no expression of utrophin in the mdx:utrophin−/− (dko) mouse EOM. Bar is 30 μm.
3.2 EOM Morphology in mdx:utrophin+/− and mdx:utrophin−/− Mice
One of the hallmarks of dystrophinopathy is repeated cycles of degeneration and regeneration of myofibers. This on-going process can be identified by alterations in mean myofiber cross-sectional areas and the presence of centrally located nuclei within individual myofibers. The morphology of the EOM from the mdx:utrophin+/− and mdx:utrophin−/− mice was examined and compared to that of WT control mice (Figure 2). The EOM from both the mdx:utrophin+/− (Figure 2B) and mdx:utrophin−/− (Figure 2C) displayed normal morphology, and their appearance was similar to the EOM from WT control mice (Figure 2A), with little evidence of pathological changes such as central nucleation, fiber necrosis, fibrosis, and inflammatory cell infiltrate (Figure 2A-C). While the EOM were spared from pathology in both these genotypes, the nearby levator palpebrae superioris (Figure 2D) showed significant central nucleation of myofibers at levels similar to that seen in limb skeletal muscles from these mice (Figure 2E-G). Normal limb muscles have a cobblestone appearance with homogeneity in individual myofiber cross-sectional areas (Figure 2E). In the limb muscles from the mdx:utrophin+/− mice, the majority of myofibers were centrally nucleated, and there was a marked heterogeneity of myofiber size (Figure 2F). In the limb muscles from the mdx:utrophin−/− mouse, evidence of accelerated degeneration/regeneration was seen, as evidenced by the calcified fibers, and increased myofiber degeneration/regeneration as evidenced by the presence of many centrally nucleated basophilic fibers (Figure 2G). Thus, in comparison to the severe pathology in the limb muscles in the mouse models of muscular dystrophy, the EOM were structurally spared.
Figure 2.
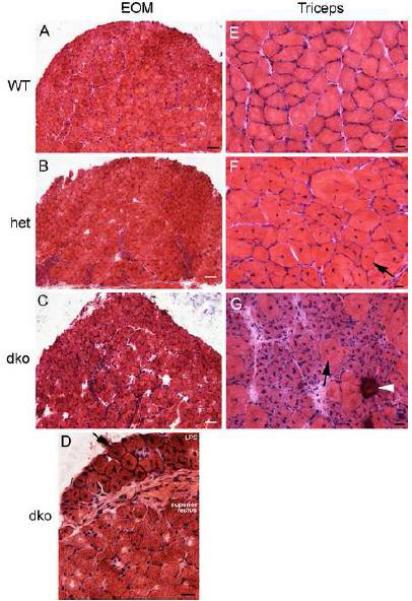
Sparing of the Extraocular Muscles (EOM) in mdx:utrophin+/− (het) and mdx:utrophin−/− (dko) Mice but not the Levator Palpebrae Superioris Muscle or Triceps Skeletal Muscles. Photomicrographs of muscle specimens stained with hematoxylin and eosin obtained from mice at 3 months of age, except the mdx:utrophin−/− muscles which were obtained from 2 month old mice. Extraocular muscles from A. a wild type (WT) control EOM, B. an mdx:utrophin+/− (het) EOM, and C. an mdx:utrophin−/− (dko) EOM demonstrate that there was little sign of muscle pathology in the mouse models of muscular dystrophy. Bar for A-C is 100 μm. D. Photomicrograph of the levator palpebrae superioris (LPS) and the superior rectus muscle of an mdx:utrophin−/− (dko) mouse. Note that the LPS was significantly affected, with central nucleation (arrow) in almost every myofiber present, while the superior rectus was normal in appearance. E. Photomicrographs of normal triceps muscle from a wild type (WT) mouse, F. an mdx:utrophin+/− (het) triceps muscle, and G. an mdx:utrophin−/− (dko) triceps muscle demonstrate dystrophic pathology. There were many centrally nucleated myofibers in the mdx:utrophin+/− limb muscle specimen. The pathology was even more in the triceps muscle from the dko mouse. This included basophilic myofibers suggestive of new regeneration and accelerated protein synthesis, central nucleation (arrow), and a calcified fiber (arrowhead). For D-G, bar is 20μm.
3.3 EOM Morphometry in WT, mdx, mdx:utrophin+/− and mdx:utrophin−/− Mice at Intervals Over their Life Span
In order to assess if there was increased heterogeneity of myofiber size in the EOM from the mouse models of DMD, mean myofiber cross-sectional areas were assessed in the orbital and global layers of the EOM at intervals over the lifetime of WT and mdx:utrophin+/− mice, at 2 months for the mdx:utrophin−/− mice, and at 3, 6, and 12 months for mdx mice. In the leg muscles of the mdx:utrophin+/− mice over time [26], there was a decrease in myofiber cross-sectional area with aging [26]. There were no overt changes in the myofibers in the EOM in either the mdx:utrophin+/− or mdx:utrophin−/− mice. The mean cross-sectional area of the mdx:utrophin+/−mouse orbital layer myofibers was significantly larger than the age-matched WT controls only at 12 months (Figure 3A), while areas were significantly larger in the global layer myofibers at 3 and 12 months in the mdx:utrophin+/− mice (Figure 3B). Only at the 12 month time point were the mdx global layer fibers significantly larger than the WT control. This phenomenon of increasing myofiber size in EOM was seen in previous studies after a number of diverse perturbations including chemical denervation by botulinum toxin [33]. These results mirror the resistance of the EOM to developing a dystrophic phenotype relative to fiber size.
Figure 3.
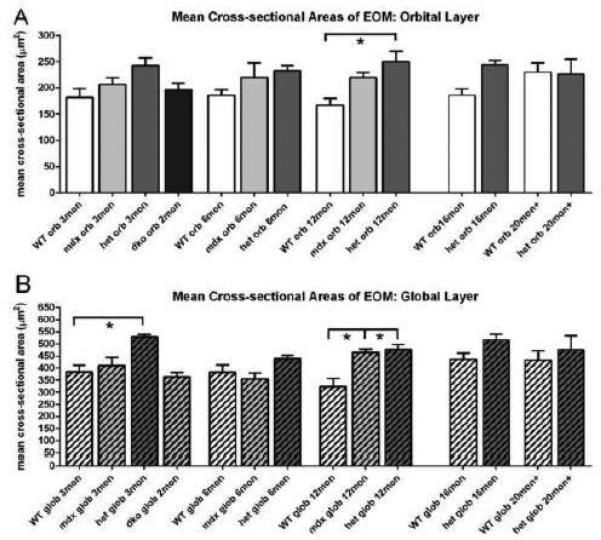
Cross-sectional Areas in Extraocular Muscles from WT, mdx, mdx:utrophin+/− (het), and mdx:utrophin−/− (dko) Mice. A. Quantification of mean myofiber cross-sectional area in the orbital layer of WT, mdx, mdx:utrophin+/− (het), and mdx:utrophin−/− (dko) at indicated intervals showed that there were few differences between any of the genotypes at any of the ages examined. The only significant difference was between the WT orbital layer myofibers and the mdx/utrophin+/− myofibers at 12 months. B. Quantification of mean myofiber cross-sectional area in the global layer of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice at indicated intervals showed that there were few differences between any of the genotypes at any of the ages examined. The only significant differences were between the WT global layer myofibers and the mdx/utrophin+/− myofibers at 3 months and the WT global layer myofibers and both the mdx and mdx/utrophin+/− myofibers at 12 months. Data are expressed as mean ± SEM. * indicates significant difference from WT control.
In the orbital layer of the EOM of all four mouse genotypes examined in this study, no significant differences were seen in the percentages of myofibers with central nucleation in the orbital layer (Figure 4A). While there was apparent heterogeneity in density of central nucleation in the mdx:utrophin−/−, it did not reach significance. However, in the global layer the mean density of centrally nucleated myofibers was significantly greater in all the mdx:utrophin+/− mice compared to their age-matched WT controls at 3, 6, 12, 16, and 20+ months, with an average of 4-fold greater numbers of centrally nucleated fibers (Figure 4B). It should be noted that the percentage of centrally nucleated myofibers in the mdx:utrophin+/− EOM never exceeded 3%, while, in contrast, centrally nucleated myofibers were found in 57% of all myofibers in the triceps muscles from the same mdx:utrophin+/− mice (Figure 4C) [26]. Thus, while significantly different from age-matched WT control EOM myofibers, this represents an extremely low incidence of central nucleation. Again, this supports the ability of the EOM to retain normalcy in the mdx:utrophin+/− and mdx:utrophin−/− mice compared to limb muscle despite the reduction or lack of utrophin [26].
Figure 4.
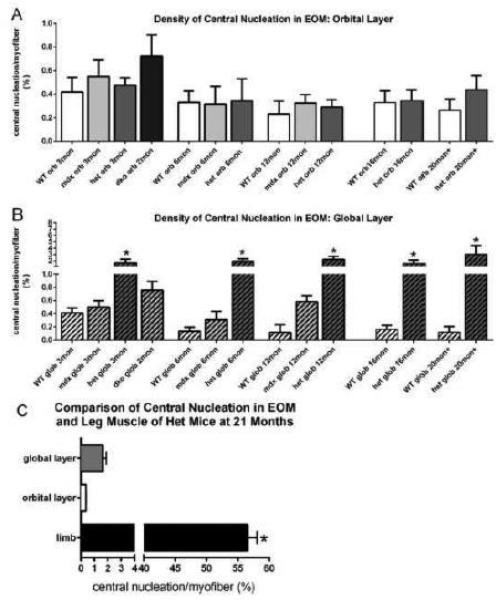
Central Nucleation in Extraocular Muscles from WT, mdx, mdx:utrophin+/−(het), and mdx:utrophin−/− (dko) Mice. A. Quantification of central nucleation density in the orbital layer of the EOM in WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− at indicated intervals showed that in the orbital layer there were no significant differences between any of the genotypes at any of the ages examined. B. Quantification of the density of central nucleation in the global layer of the EOM in WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice at indicated intervals showed that for each set of age-matched muscles, there were significantly more myofibers with centrally located nuclei in the mdx:utrophin+/− (het) than in WT control global layer myofibers at that time point. * indicates significant difference from WT control. C. Comparison of the density of central nucleation in the EOM orbital and global layers compared to triceps muscles from the mdx:utrophin+/− mouse. Note that the density of centrally located nuclei in EOM was approximately 30-fold lower than what was seen in the triceps muscles of age-matched mdx:utrophin+/− mice. Data are expressed as mean ± SEM. * indicates significant difference from EOM.
3.4 Fibrosis in WT, mdx, mdx:utrophin+/− and mdx:utrophin−/− Mice at Intervals Over their Life Span
In limb skeletal muscles, fibrosis is one of the long term sequelae of DMD. The EOM from WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice were examined for expression levels of collagen I and IV (Figure 5,6). These two collagen isoforms were chosen from a larger superfamily of collagen molecules [34], as they represent two of the major forms found in skeletal muscle. Collagen I is in the family of fibril-forming collagens, and is thought to provide rigidity and tensile strength to muscle. Collagen IV is a basement membrane collagen and is contained in the basement membrane surrounding individual myofibers. We have previously demonstrated their localization patterns in the extraocular muscles [35]. When the density of collagen I was quantified, there was no significant difference between the muscles from mdx compared to WT mice at any of the ages examined. Interestingly, the EOM from both the mdx:utrophin+/− and mdx:utrophin−/− mice had greater collagen I density at 3 months, at 20.64 ± 2.9% and 16.5 ± 1.5% respectively compared to 0.2 ± 0.8% for WT (Figure 5). This can be seen microscopically in the greater density around the myofibers in the dystrophic mice EOM (Figure 5B, C) compared to WT (Figure 5A). At 6 and 12 months, this density had decreased in the mdx:utrophin+/− EOM to control levels, but at 16 and 20 months collagen I density again was significantly increased compared to their age-matched WT controls, at 17.73 ± 1.3% and 19.0 ± 2.7% compared to 10.27 ± 0.2% and 12.2 ± 0.5% in WT muscles. This suggested a moderate fibrotic change in the EOM of these dystrophic mice. This was in contrast to the very extensive fibrosis that developed in the limb muscles of the mdx:utrophin+/− mice where collagen levels reached up to 50% of the total tissue area [26]. Interestingly, there were no significant differences in the density of collagen IV between any of mouse genotypes at any of the ages examined (Figure 6). This demonstrates differential control over collagen protein expression in these genotypes, and the lack of fibrosis is another manifestation of the differences between limb muscles and EOM in muscular dystrophy.
Figure 5.
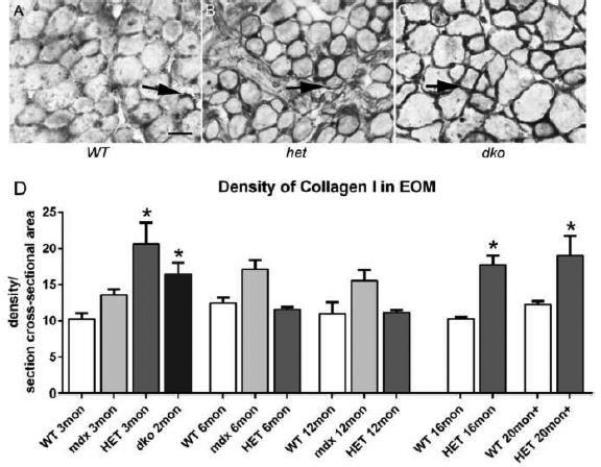
Density of Collagen I in WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− Mice. A. Photomicrographs of collagen I immunostaining in the EOM of WT (A), mdx:utrophin+/− (het) (B), and mdx:utrophin−/− (dko) mice (C). Arrows indicate collagen I which was significantly increased in these two genotypes compared to WT control. Bar is 50μm. D. Quantification of collagen I density in the EOM of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− at indicated intervals shows that collagen I was upregulated in the EOM of both mdx:utrophin+/−, and mdx:utrophin−/− EOM. Data are expressed as mean ± SEM. * indicates significant difference from WT control.
Figure 6.
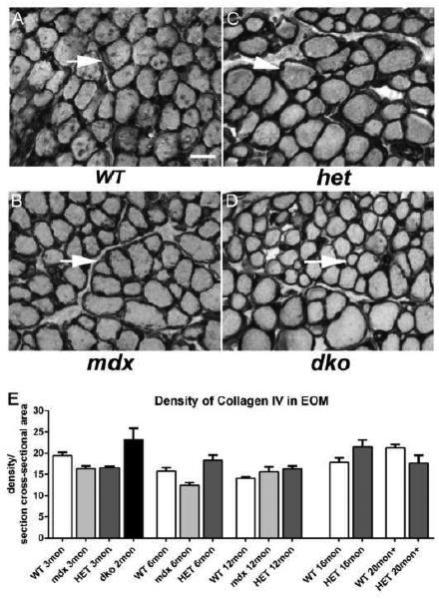
Density of Collagen IV in WT, mdx, mdx:utrophin+/− (het), and mdx:utrophin−/− (dko) Mice. Photomicrographs of collagen IV immunostaining in the EOM of WT (A), mdx (B), mdx:utrophin+/− (het) (C), and mdx:utrophin−/− (dko) mice (D). Arrows indicate collagen IV around individual myofibers. The collagen IV levels in the disease models did not appear to differ significantly from levels in WT control mice. Bar is 50μm. E. Quantification of collagen IV density in the EOM of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− at indicated intervals show that there were no significant changes in the level of collagen IV in the four genotypes examined. Data are expressed as mean ± SEM.
3.5 Pax7 Cell Density in mdx, mdx:utrophin+/−, and mdx:utrophin−/− EOM at Intervals Over their Life Span
In order to assess if the retention of normal morphology might be related to numbers of Pax7 satellite cells in the EOM in mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice, levels of Pax7-positive cell density in these genotypes were compared to age-matched WT controls. Pax7 identifies satellite cells within skeletal muscle and is easily seen with immunochemical techniques (Figure 7A) [29]. When compared to age-matched WT controls, no significant differences were seen in the Pax7-positive cell density in the orbital or global layers of any of the genotypes at any of the ages examined (Figure 7B, C). In the mdx:utrophin+/− mice, Pax7-cell density in the orbital layer at 16 months was significantly greater than at 3 or 6 months of age and in the EOM of mice at 20 months there was a significantly greater density of Pax7-positive cells than at 3, 6 or 12 months of age. In addition in WT mice, the density of orbital layer Pax7-positive cells was significantly greater at 20 months than at 3 months (Figure 7B). The absence of a significant difference in Pax7-positive cell density between the genotypes examined suggested that the population was not significantly depleted. Future studies should focus on sparing of the EOM in the mdx or mdx:utrophin+/− mouse in mouse depleted of Pax7-positive cells in order to adequately address the role this specific population plays in EOM sparing. It should be noted that 18Gy irradiation of the triceps and EOM of mdx:utrophin+/−mice produced a permanent depletion of Pax7-positive cells in the irradiated triceps, but only a temporary decrease and then a rebound to normal density of Pax7-positive cells in the irradiated EOM [36].
Figure 7.
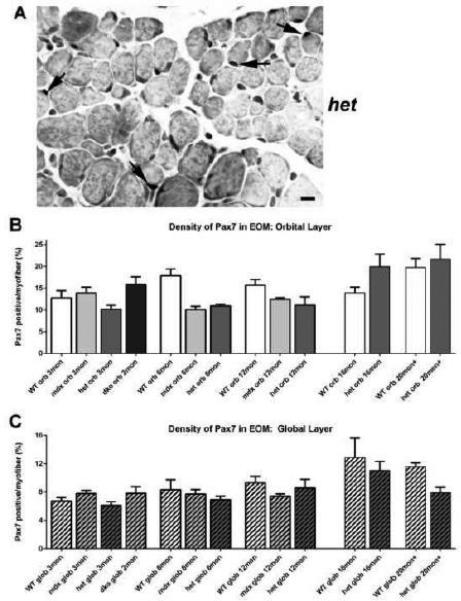
Pax7-Positive Satellite Cell Density in EOM from WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− Mice. A. EOM from an mdx:utrophin+/− (het) mouse immunostained for the expression of Pax7, with several positive nuclei indicated by the black arrows. Bar is 20μm. B. Quantification of Pax7-positive cell density in the orbital layer of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− (dko) at indicated intervals showing no significant differences in Pax7-positive nuclear density at any of the ages or genotypes examined. C. Quantification of Pax7-positive satellite cell density in the global layer of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− at indicated intervals. There were no significant differences when compared to age-matched WT controls. Data are expressed as mean ± SEM.
3.6 MyoD Cell Density in WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− EOM at Intervals Over their Life Span
The MyoD-positive cell population represents myogenic precursor cell commitment to the myogenic lineage [32, 37]. Our previous studies demonstrated the presence of this population in normal, adult EOM [38-40], and even in aging EOM [41]. The density of these activated precursor cells was assessed in the WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− EOM over time (Figure 8A-D). Compared to age-matched WT control EOM, no alteration in the density of the MyoD-positive population was seen in the orbital layer in any of the genotypes at any of the ages examined (Figure 8A-C). The global layer generally has a lower density of MyoD-positive cells in WT EOM compared to the density in the orbital layer [38-41]. When compared to age-matched WT controls, MyoD-positive cell density in the global layer was significantly increased in the mdx and mdx:utrophin−/− mice only at 3 months of age, to 13.5 ± 0.8% and 12.6 ± 2.9% respectively compared to 7.3 ± 0.3% for WT, after which all genotypes showed similar MyoD-positive cell density at all additional ages examined (Figure 8A-C). It is unclear why there was only a significantly increased density of MyoD-positive cells in the mdx and mdx:utrophin−/− mice early in disease progression and only in the global layer. It may be that a steady state is achieved between on-going continuous remodeling and activation of myogenic precursor cells. Future studies will need to address this question.
Figure 8.
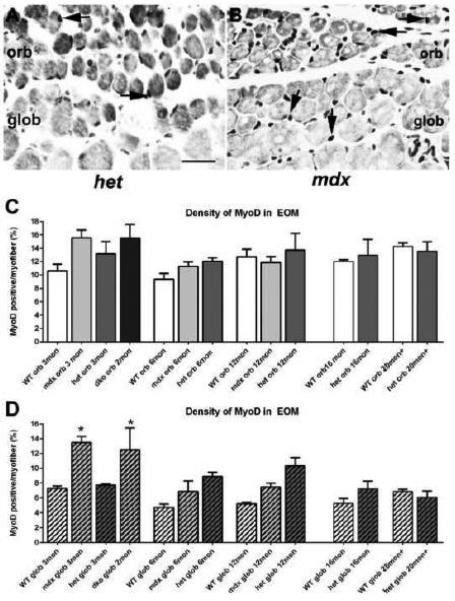
: MyoD-Positive Cell Density in the EOM of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− Mice. A. EOM from an mdx:utrophin+/− (het) mouse immunostained for the expression of MyoD, with two positive nuclei in the orbital layer indicated by the black arrows. B. EOM from an mdx mouse immunostained for the expression of MyoD, with two positive nuclei in the orbital layer and two positive nuclei in the global layer indicated by the black arrows. Bar is 20μm. C. Quantification of MyoD-positive cell density in the orbital layer of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− (dko) at indicated intervals showed that there was no significant difference in the density of MyoD-positive cells in any of the genotypes at any of the ages examined. D. Quantification of MyoD-positive cell density in the global layer of WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− at indicated intervals showed a significant increase in density of MyoD-positive nuclei in the mdx andmdx:utrophin−/− EOM compared to WT control global layer. No other significant differences were seen. Data are expressed as mean ± SEM. * indicates significant difference from age-matched WT control.
4. DISCUSSION
The EOM are morphologically spared both in the mdx:utrophin+/− mouse, which lacks dystrophin and is haploinsufficient for utrophin, as well as in the mdx:utrophin−/− mouse, which lacks both dystrophin and utrophin. They maintain their normal cross-sectional areas, and actually become slightly larger over the life span of these mice, have minimal increases in centrally located myonuclei, and show minimal changes in collagen density compared to age-matched WT control EOM. In addition, they also maintain a high level of Pax7-positive satellite cells and MyoD-positive cells.
The sparing of the mdx:utrophin+/− and mdx:utrophin−/− EOM in the present study is at odds with a previous report that the EOM are not spared in the mdx:utrophin−/− mouse [28]. The general sense in the muscle community is that they are not spared, and the present analysis, which encompasses the life span through 20 months of age in the mdx:utrophin+/− mouse as well as examination of mdx:utrophin−/− mice firmly refutes this earlier study. While this study used the mouse strain developed at Washington University (Grady strain) [20] as opposed to the strain produced at the University of Oxford (Deconinck strain) [21] used in the previous report, the literature suggests that the strain used in the present study may in fact express a more severe pathology. Not only does the Grady strain of mdx:utrophin−/− mice die earlier (14 weeks as opposed to 20 weeks) [20, 21], but they have been shown to exhibit more severe cardiac pathology [42]. While these differences in phenotype may be attributable to different genetic backgrounds of the mice, they may also arise from the different gene knockout strategies used. The Grady strain is null for all isoforms of utrophin, while the Deconinck strain only has the largest utrophin isoform inactivated.
The rationale for the potential role of utrophin in sparing skeletal muscle from pathology is relatively strong. Utrophin up-regulation has been described in the regenerating myofibers of the mdx mouse [13, 14]. Additionally, as noted previously, when utrophin expression was up-regulated in the limb and body muscles of the mdx mouse, many of the pathological features decreased [17-19]. However, when the literature on utrophin changes in the EOM of the mdx mouse is examined, there is good support for why utrophin up-regulation is unlikely to be mechanistic for EOM sparing in both the mdx mouse and in DMD. The observed up-regulation of utrophin [27, 28] was not universally seen when examined by different laboratories [9]. Subsequently, in one study, and contrary to our results, western blots showed that utrophin was not increased in the mdx EOM [12]. Not all the myofibers in EOM had up-regulated utrophin levels [12], yet all of the myofibers were spared when examined histologically. In our hands, the proteins level of utrophin in the EOM of mdx mice was extremely variable, and the explanation for this variability is unclear. One possible explanation is that in normal EOM from WT mice, the normal levels of dystrophin is itself extremely variable, with some fibers completely lacking dystrophin [43]. Irrespective of the differential expression levels of utrophin, the EOM were still morphologically spared in the complete absence of utrophin in the mdx:utrophin−/− mouse. This suggests that while it can be protective when increased in limb muscles from muscular dystrophy mouse models [17-19], it does not appear to be responsible for the sparing of the EOM. It should be noted that the EOM differ quite significantly from limb skeletal muscles in terms of fiber types, patterns of innervation, and up-regulation of various signaling factors, any of which might contribute to the sparing of EOM in a variety of degenerative conditions [44-47]. Certainly in the mdx:utrophin−/− mice, where utrophin is absent, the lack of pathology supports the view that up-regulation of utrophin levels is not responsible for EOM sparing in DMD.
Fibrosis is well known to increase as the duration of disease progresses in children with DMD and related animal models [48]. In addition, it was shown that the expression patterns and amount of collagen within the muscle tissue was different in affected and spared skeletal muscles in mdx mice [49]. Thus it is not surprising that very little increase in fibrosis was seen in the EOM from the three dystrophic genotypes in our study.
This, then, raises the basic question: by what mechanism is the EOM from the mdx, mdx:utrophin+/− and mdx:utrophin−/− mice, as well as the EOM in human patients with DMD, Becker's, limb-girdle, sarcoglycan-deficient, and actin-related dystrophinopathies spared? The EOM have a number of properties that support the hypothesis that intrinsic differences in the EOM are responsible for their sparing in muscular dystrophies in general. While limb and body skeletal muscles are dependent on Pax3 expression early in development, in its absence the EOM develop normally [50]. Further experimental evidence suggests that muscle stem cells appear to play a critical role in the sparing of EOM in dystrophinopathies and related muscle diseases. While the limb muscles of the mdx:utrophin+/− and mdx:utrophin−/− mice showed progressively decreased myofiber cross-sectional area [26], the EOM not only maintained their fiber size as they aged, but the myofibers actually increased in cross-sectional area. There is precedence for this type of “growth” response in EOM when denervated or injured. One example is the chemodenervation from the paralytic agent, botulinum toxin A. While limb skeletal muscle atrophied during the effective period after botulinum toxin A treatment, the EOM actually either maintained or increased their myofiber cross-sectional areas [33, 51]. Concomitant with increased EOM myofiber size after various types of injury, stretch, or chemodenervation, significant increases were seen in myogenic precursor cell proliferation and density of Pax7- and MyoD-positive cells after these perturbations [33, 52, 53]. A similar response was seen after high dose gamma irradiation of EOM and limb skeletal muscle; while the leg muscles atrophied the EOM mean myofiber cross-sectional areas actually increased [36].
We hypothesize that one potential mechanism for retaining the remarkable resiliency for myofiber size maintenance in the EOM might be an inherent population of myogenic precursor cell. As shown in the present study, the EOM from the mdx:utrophin+/− mice retained the density of their Pax7- and MyoD-positive cells, even in the aging EOM. It is interesting to note that at 20 months, the density of Pax7-positive cells was significantly increased compared to 3 and 6 months. This sustained level of Pax7 and MyoD cell density even during aging in the EOM is not surprising; continuous myofiber remodeling coupled with a high density of Pax7- and MyoD-positive cells was previously demonstrated in uninjured, adult EOM of multiple species compared to limb skeletal muscles [38-40]. This was seen even at significantly advanced ages [41]. Several hypotheses can be put forth that might explain the enhanced survivability in terms of numbers and/or the enhanced regenerative potential of myogenic precursor cells in the EOM in contrast to the decline seen in both numbers and myogenic potential in aging limb skeletal muscles and in muscular dystrophy [54. 55]. First, we have shown that the density of specific identified populations of myogenic precursor cells in the EOM can be up to 10 fold greater than limb muscles from age-matched animals [56, 57]. In addition, identified populations of EOM derived myogenic precursor cells showed an enhanced rate of proliferation [57] and fusion [56]. These studies were recently repeated and again showed significantly increased rates of proliferation in EOM myogenic precursor cells, confirming our earlier results [58]. This hypothesis is also supported by a seminal study by the Kardon laboratory showing the continuous addition of satellite cells to EOM over many months [59]. In the present study, the Pax7 and MyoD positive precursor cell pools were retained in the EOM from all three dystrophic genotypes, even in the aging dystrophic mice. This is in contrast to the Pax7 and MyoD populations in aging WT and mdx:utrophin+/− limb muscles, where their density was first static, and ultimately decreased in the aging and dystrophic limb skeletal muscles [26]. The potential role of myogenic precursor cells in the sparing of EOM is supported by previous work showing that EOM and laryngeal muscles are both spared in DMD and other dystrophinopathies [1], and both these groups of muscles retain the unusual capacity to undergo continuous and extensive remodeling throughout normal life [37-40, 60,61].
Using flow cytometric analysis to identify specific subpopulations of myogenic precursor cells from EOM from WT, mdx, mdx:utrophin+/−, and mdx:utrophin−/− mice, the EOM myogenic precursor cells were shown to be more proliferative and more resistant to oxidative, irradiation, and toxic stress [53, 56-57, 62]. The present results are consistent with this hypothesis, that the EOM are morphologically and functionally maintained due to maintenance of highly proliferative myogenic precursor cells. In a related study, high dose gamma irradiation temporarily reduced the density of both Pax7- and MyoD-positive myogenic precursor cells in mdx:utrophin+/− EOM, resulting in a dystrophic phenotype. Interestingly, the irradiated EOM completely recovered both normal morphology and normal numbers of Pax7 and other myogenic precursor cells by two months post-irradiation [53].
A number of genes and transcription factors have been implicated in normal EOM development [63-65]. The distinct differences in the early genetic control over the formation of somite-derived muscle and EOM support the hypothesis that there could be distinct myogenic precursor cells in each muscle. While limb and body musculature depends on Pax3 expression for differentiation, in its absence the EOM develop completely normally [49]. Conversely, Pitx2 expression is required for the normal development of EOM [65] as well as maintenance of normal properties of mature EOM [66, 67]. Pitx2-positive mononuclear cells derived from the EOM appear to have increased rates of proliferation compared to the same cells derived from limb muscle [55]. On-going studies are specifically focused on the role of Pitx2 in EOM sparing in muscular dystrophies.
5. CONCLUSIONS
In summary, the EOM of mdx:utrophin+/− mice, which are haploinsufficient for utrophin, and the mdx:utrophin−/− mice, which do not express utrophin, are spared morphologically over their life span. Contrary to what has been shown in limb skeletal muscles from these same genotypes [26], there does not appear to be a loss of Pax7- and MyoD-positive myogenic precursor cells in the EOM from these mice models of DMD.
Supplementary Material
Highlights.
Eye muscles are spared in muscular dystrophy models in the absence of dystrophin and utrophin.
Pax7- and MyoD-positive cells were retained and similar to age-matched wild type controls.
Thus, utrophin is not involved in extraocular muscle sparing in these genotypes.
ACKNOWLEDGEMENTS
Supported by NIH EY55137 (LKM) and EY11375 from the National Eye Institute, T32EY007133 (AAM), the 3M Science & Technology Fellowship (AAM), NIH P30-AR0507220, the University of Minnesota Foundation, the Minnesota Lions and Lionesses, and an unrestricted grant to the Department of Ophthalmology and Visual Neurosciences from Research to Prevent Blindness, Inc.
Abbreviations
- EOM
extraocular muscle
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors: No conflict of interest
REFERENCES
- 1.Marques MJ, Ferretti R, Vomero VU, Minatel E, Neto HS. Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve. 2007;35:349–353. doi: 10.1002/mus.20697. [DOI] [PubMed] [Google Scholar]
- 2.Vereecken RL, Verduyn H. The electrical activity of the paraurethral and perineal muscles in normal and pathological conditions. Br. J. Urol. 1970;2:457–463. doi: 10.1111/j.1464-410x.1970.tb04483.x. [DOI] [PubMed] [Google Scholar]
- 3.Karpati G, Carpenter S. Small caliber skeletal muscle fibers do not suffer deleterious consequences of dystrophic gene expression. Am. J. Med. Genet. 1986;25:653–658. doi: 10.1002/ajmg.1320250407. [DOI] [PubMed] [Google Scholar]
- 4.Kaminski HJ, al-Hakim M, Leigh RJ, Katirji MB, Ruff RL. Extraocular muscles are spared in advanced Duchenne dystrophy. Ann. Neurol. 1992;32:586–588. doi: 10.1002/ana.410320418. [DOI] [PubMed] [Google Scholar]
- 5.McLoon LK. In: The Extraocular Muscles. In: Adler's Physiology of the Eye. Kaufman P, Alm A, Levin LA, Nilsson S, Ver Hoeve J, Wu SM, editors. Mosby Press; Missouri: 2011. pp. 182–207. [Google Scholar]
- 6.Porter JD, Merriam AP, Hack AA, Andrade FH, McNally EM. Extraocular muscle is spared despite the absence of an intact sarcoglycan complex in gamma- or delta-sarcoglycan-deficient mice. Neuromuscul. Disord. 2001;11:197–207. doi: 10.1016/s0960-8966(00)00171-1. [DOI] [PubMed] [Google Scholar]
- 7.Di Costanzo A, Toriello A, Mottola A, Di Iorio G, Bonavita V, Tedeschi G. Relative sparing of extraocular muscles in myotonic dystrophy: an electrooculographic study. Acta Neurol. Scand. 1997;95:158–163. doi: 10.1111/j.1600-0404.1997.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 8.Andrade FH, Porter JD, Kaminski HJ. Eye muscle sparing by the muscular dystrophies: lessons to be learned? Microsc. Res. Tech. 2000;48:192–203. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<192::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 9.Khurana TS, Prendergast RA, Alameddine HS, et al. Absence of extraocular muscle pathology in Duchenne's muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J. Exp. Med. 1995;182:467–475. doi: 10.1084/jem.182.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragusa RJ, Chow CK, Porter JD. Oxidative stress as a potential pathogenic mechanism in an animal model of Duchenne muscular dystrophy. Neuromuscul. Disord. 1997;7:379–386. doi: 10.1016/s0960-8966(97)00096-5. [DOI] [PubMed] [Google Scholar]
- 11.Wehling M, Stull JT, McCabe TJ, Tidball JG. Sparing of mdx extraocular muscles from dystrophic pathology is not attributable to normalized concentration or distribution of neuronal nitric oxide synthase. Neuromuscul. Disord. 1998;8:22–29. doi: 10.1016/s0960-8966(97)00136-3. [DOI] [PubMed] [Google Scholar]
- 12.Porter JD, Merriam AP, Khanna S, et al. Constitutive properties, not molecular adaptations, mediate extraocular muscle sparing in dystrophic mdx mice. FASEB J. 2003;17:893–895. doi: 10.1096/fj.02-0810fje. [DOI] [PubMed] [Google Scholar]
- 13.Khurana TS, Watkins SC, Chafey P, et al. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- 14.Helliwell TR, Man NT, Morris GE, Davies KE. The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscul. Disord. 1992;2:177–184. doi: 10.1016/0960-8966(92)90004-p. [DOI] [PubMed] [Google Scholar]
- 15.Winder SJ, Hemmings L, Maciver SK, et al. Utrophin actin binding domain: analysis of actin binding and cellular targeting. J. Cell Sci. 1995;108:63–71. doi: 10.1242/jcs.108.1.63. [DOI] [PubMed] [Google Scholar]
- 16.Chung W, Campanelli JT. WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol. Cell Biol. Res. Commun. 1999;2:162–171. doi: 10.1006/mcbr.1999.0168. [DOI] [PubMed] [Google Scholar]
- 17.Tinsley JM, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 18.Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 19.Deconinck N, Tinsley J, De Backer F, et al. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat. Med. 1997;3:16–21. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- 20.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 21.Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 22.Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of an utrophin transgene rescues utrophin-dystrophin deficient mice. Nat. Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Rafael-Fortney JA, Huang P, et al. Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice. J. Neurol. Sci. 2008;264:106–111. doi: 10.1016/j.jns.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang P, Cheng G, Lu H, Aronica M, Ransohoff RM, Zhou L. Impaired respiratory function in mdx and mdx:utrn(+/−) mice. Muscle Nerve. 2011;43:263–267. doi: 10.1002/mus.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Putten M, Kumar D, Hulsker M, et al. Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. Neuromuscul. Disord. 2012;22:406–417. doi: 10.1016/j.nmd.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 26.McDonald AA, Hebert SL, Kunz MD, Ralles SJ, McLoon LK. Disease course in mdx:utrophin+/− mice: Comparison of three mouse models of muscular dystrophy. Physiol. Rep. 2015;3(4):e12391. doi: 10.14814/phy2.12391. doi: 10.14814/phy2.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 28.Porter JD, Rafael JA, Ragusa RJ, Brueckner JK, Trickett JI, Davies KE. The sparing of extraocular muscle in dystrophinopathy is lost in mice lacking utrophin and dystrophin. J. Cell Sci. 1998;111:1801–1811. doi: 10.1242/jcs.111.13.1801. [DOI] [PubMed] [Google Scholar]
- 29.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 30.Zammit PS, Relaiz F, Nagata Y, et al. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006;119:1824–1832. doi: 10.1242/jcs.02908. [DOI] [PubMed] [Google Scholar]
- 31.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cornelison DD, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 1997;191:270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- 33.Ugalde I, Christiansen SP, McLoon LK. Botulinum toxin treatment of extraocular muscles in rabbit results in increased myofiber remodeling. Invest. Ophthalmol. Vis. Sci. 2005;46:4114–4120. doi: 10.1167/iovs.05-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricard-Blum S, Ruggierio F. The collagen superfamily: from extracellular matrix to the cell membrane. Pathol. Biol. 2005;53:430–442. doi: 10.1016/j.patbio.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 35.Stager D, Jr., McLoon LK, Felius J. Postulating a role for connective tissue elements in inferior oblique muscle overaction. Trans. Am. Ophthalmol. Soc. 2013;111:119–132. [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald AA, Kunz MD, McLoon LK. Dystrophic changes in extraocular muscles after gamma irradiation in mdx:utrophin+/− mice. PLoS One. 2014;9(10):386424. doi: 10.1371/journal.pone.0086424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper RN, Tajbakhsh S, Mouly V, Cossu G, Buckingham M, Butler-Browne GS. In vivo satellite cell activation via MyF5 and MyoD in regenerating mouse skeletal muscle. J. Cell Sci. 1999;112:2895–2901. doi: 10.1242/jcs.112.17.2895. [DOI] [PubMed] [Google Scholar]
- 38.McLoon LK, Wirtschafter JD. Continuous myonuclear addition to single extraocular myofibers in uninjured adult rabbits. Muscle Nerve. 2002;25:348–358. doi: 10.1002/mus.10056. [DOI] [PubMed] [Google Scholar]
- 39.McLoon LK, Wirtschafter JD. Activated satellite cells are present in uninjured extraocular muscles of mature mice. Trans. Am. Ophthalmol. Soc. 2002;100:119–123. [PMC free article] [PubMed] [Google Scholar]
- 40.McLoon LK, Rowe J, Wirtschafter J, McCormick KM. Continuous myofiber remodeling in uninjured extraocular myofibers: myonuclear turnover and evidence for apoptosis. Muscle Nerve. 2004;29:707–715. doi: 10.1002/mus.20012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLoon LK, Wirtschafter J. Activated satellite cells in extraocular muscles of normal adult monkeys and humans. Invest. Ophthalmol. Vis. Sci. 2003;44:1927–1932. doi: 10.1167/iovs.02-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hainsey TA, Senapati S, Kuhn DE, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul. Disord. 2003;13:294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- 43.Pedrosa Domellöf F, Thornell LE. Cytoskeletal proteins in human extraocular muscles. Invest. Ophthalmol. Vis. Sci. 2005;46:4678. doi: 10.1167/iovs.62.2.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLoon LK, Park HN, Kim JH, Pedrosa-Domellöf F, Thompson LV. A continuum of myofibers in adult rabbit extraocular muscle: force, shortening velocity, and patterns of myosin heavy chain localization. J. Appl. Physiol. 2011;111:1178–1189. doi: 10.1152/japplphysiol.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu JX, Brännström T, Anderson PM, Pedrosa-Domellöf F. Distinct changes in synaptic protein composition at neuromuscular junctions of extraocular muscles versus limb muscles of ALS donors. PLoS One. 2013;8(2):e57473. doi: 10.1371/journal.pone.0057473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janbaz AH, Lindstrom M, Liu JX, Pedrosa Domellöf F. Intermediate filaments in human extraocular muscles. Invest. Ophthalmol. Vis. Sci. 2014;55:5151–5159. doi: 10.1167/iovs.14-14316. [DOI] [PubMed] [Google Scholar]
- 47.McLoon LK, Harandi VM, Brännström T, Andersen PM, Liu JX. Wnt and extraocular muscle sparing in amyotrophic lateral sclerosis. Invest. Ophthalmol. Vis. Sci. 2014;55:5482–5496. doi: 10.1167/iovs.14-14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldspink G, Fernandes K, Williams PE, Wells DJ. Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice Neuromuscul. Disord. 1994;4:183–191. doi: 10.1016/0960-8966(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 49.Porter JD, Merriam AP, Leahy P, Gong B, Khanna S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum. Mol. Genet. 2003;12:1813–1821. doi: 10.1093/hmg/ddg197. [DOI] [PubMed] [Google Scholar]
- 50.Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell. 1997;89:127–138. doi: 10.1016/s0092-8674(00)80189-0. [DOI] [PubMed] [Google Scholar]
- 51.Spencer RF, McNeer KW. Botulinum toxin paralysis of adult extraocular muscle. Structural alterations in orbital, singly innervated muscle fibers. Arch. Ophthalmol. 1987;105:1703–1711. doi: 10.1001/archopht.1987.01060120101035. [DOI] [PubMed] [Google Scholar]
- 52.Christiansen SP, McLoon LK. The effect of resection on satellite cell activity in rabbit extraocular muscle. Invest. Ophthalmol. Vis. Sci. 2006;47:605–613. doi: 10.1167/iovs.05-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christiansen SP, Antunes-Foschini RS, McLoon LK. Effects of recession versus tenotomy surgery without recession in adult rabbit extraocular muscle. Invest. Ophthalmol. Vis. Sci. 2010;51:5646–5656. doi: 10.1167/iovs.10-5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Decary S, Hamida CB, Mouly V, Barbet JP, Hentati F, Butler-Browne GS. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul. Disord. 2000;10:113–120. doi: 10.1016/s0960-8966(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 55.Renault V, Piron-Hamelin G, Forestier C, DiDonna S, Decary S, Hentati F, Saillant G, Butler-Browne GS, Mouly V. Skeletal muscle regeneration and the mitotic clock. Exp. Gerontol. 2000;35:711–719. doi: 10.1016/s0531-5565(00)00151-0. [DOI] [PubMed] [Google Scholar]
- 56.Kallestad KM, Hebert SL, McDonald AA, Daniel ML, Cu SR, McLoon LK. Sparing of extraocular muscle in aging and muscular dystrophies: A myogenic precursor cell hypothesis. Exp. Cell Res. 2011;317:873–885. doi: 10.1016/j.yexcr.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hebert SL, Daniel ML, McLoon LK. The role of Pitx2 in maintaining the phenotype of myogenic precursor cells in the extraocular muscles. PLoS One. 2013;8:e58405. doi: 10.1371/journal.pone.0058405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stuelsatz P, Shearer A, Li Y, et al. Extraocular muscle satellite cells are high performance myo-engines ret77aining efficient regenerative capacity in dystrophin deficiency. Dev. Biol. 2015;397:31–44. doi: 10.1016/j.ydbio.2014.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keefe AC, Lawson JA, Flygare SD, Fox ZD, Colasanto MP, Mathew SJ, Yandell M, Kardon G. Muscle stem cells contribute to myofibers in sedentary adult mice. Nat. Commun. 2015;14(6):7087. doi: 10.1038/ncomms8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goding GS, Al-Sharif K, McLoon LK. Myonuclear addition to uninjured laryngeal myofibers in adult rabbits. Ann. Otol. Rhinol. Laryngol. 2005;114:552–557. doi: 10.1177/000348940511400711. [DOI] [PubMed] [Google Scholar]
- 61.Shinners MJ, Goding GS, McLoon LK. Effect of recurrent laryngeal nerve section on the laryngeal muscles of adult rabbits. Otolaryngol. Head Neck Surg. 2006;134:413–418. doi: 10.1016/j.otohns.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 62.Kallestad KM, McLoon LK. Defining the heterogeneity of skeletal muscle-derived side and main population cells isolated immediately ex vivo. J Cellul. Physiol. 2010;222:676–684. doi: 10.1002/jcp.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown CB, Engleka KA, Wenning J, Lu M, Epstein JA. Identification of a hypaxial somite enhancer element regulating Pax3 expression in migrating myoblasts and characterization of hypaxial muscle Cre transgenic mice. Genes. 2005;41:202–209. doi: 10.1002/gene.20116. [DOI] [PubMed] [Google Scholar]
- 64.Tremblay P, Dietrich S, Mericskay M, Schubert FR, Li Z, Paulin D. A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Dev. Biol. 1998;203:49–61. doi: 10.1006/dbio.1998.9041. [DOI] [PubMed] [Google Scholar]
- 65.Diehl AG, Zareparsi S, Qian M, Khanna R, Angeles R, Gage PJ. Extraocular muscle morphogenesis and gene expression are regulated by Pitx2 gene dose. Invest. Ophthalmol. Vis. Sci. 2006;47:1785–1793. doi: 10.1167/iovs.05-1424. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Y, Cheng G, Dieter L, et al. An altered phenotype in a conditional knockout of Pitx2 in extraocular muscle. Invest. Ophthalmol. Vis. Sci. 2009;50:4531–4541. doi: 10.1167/iovs.08-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou Y, Liu D, Kaminski HJ. Pitx2 regulates myosin heavy chain isoform expression and multi-innervation in extraocular muscle. J. Physiol. 2011;589:4601–4614. doi: 10.1113/jphysiol.2011.207076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.