

Abstract
We have previously reported that treatment of rats subjected to permanent bilateral common carotid artery occlusion (pBCCAO), a model of chronic cerebral hypoperfusion (CCH), with S-nitrosoglutathione (GSNO), an endogenous nitric oxide carrier, improved cognitive functions and decreased amyloid-β accumulation in the brains. Since CCH has been implicated in tau hyperphosphorylation induced neurodegeneration, we investigated the role of GSNO in regulation of tau hyperphosphorylation in rat pBCCAO model. The rats subjected to pBCCAO had a significant increase in tau hyperphosphorylation with increased neuronal loss in hippocampal/cortical areas. GSNO treatment attenuated not only the tau hyperphosphorylation, but also the neurodegeneration in pBCCAO rat brains. The pBCCAO rat brains also showed increased activities of GSK-3β and Cdk5 (major tau kinases) and GSNO treatment significantly attenuated their activities. GSNO attenuated the increased calpain activities and calpain-mediated cleavage of p35 leading to production of p25 and aberrant Cdk5 activation. In in vitro studies using purified calpain protein, GSNO treatment inhibited calpain activities while 3-morpholinosydnonimine (a donor of peroxynitrite) treatment increased its activities, suggesting the opposing role of GSNO vs. peroxynitrite in regulation of calpain activities. In pBCCAO rat brains, GSNO treatment attenuated the expression of inducible nitric oxide synthase (iNOS) expression and also reduced the brain levels of nitro-tyrosine formation, thereby indicating the protective role of GSNO in iNOS/nitrosative-stress mediated calpain/tau pathologies under CCH conditions. Taken together with our previous report, these data support the therapeutic potential of GSNO, a biological NO carrier, as a neuro- and cognitive-protective agent under conditions of CCH.
Keywords: calpain, Cdk5, chronic cerebral hypoperfusion, S-nitrosoglutathione, p25, tau
Graphical Abstract
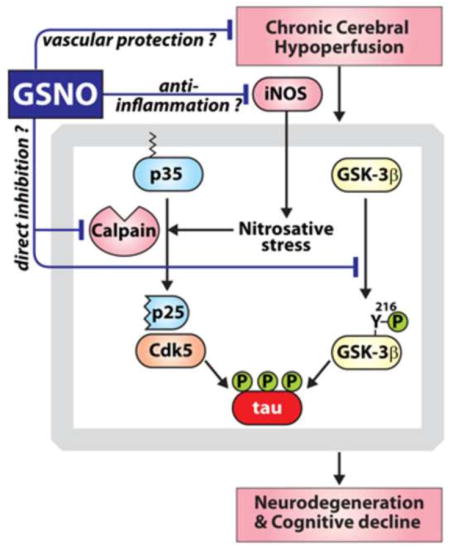
1. Introduction
Chronic cerebral hypoperfusion (CCH) is a pathological state that contributes to the establishment of various neurodegenerative diseases (Hartman et al., 2005; Masada et al., 1997; Pazos et al., 1999). Elder individuals are reported to have reduced cerebral blood flow (Bentourkia et al., 2000; Heo et al., 2010; Leenders et al., 1990), and any additional conditions causing dysfunction in cerebral blood flow, such as cerebral vascular disorders and hemodynamic and blood changes, can evoke CCH (Zhao and Gong, 2015). CCH has been implicated to cause neurodegeneration and cognitive impairment through multiple mechanisms, including induction of oxidative stress, amyloid-β (Aβ) accumulation and aggravation, tau hyperphosphorylation, synaptic dysfunction, neuronal loss, white matter lesion, and neuroinflammation (Zhao and Gong, 2015). For example, permanent bilateral common carotid occlusion (pBCCAO) of rats, the most frequently used animal model of CCH, causes central cholinergic dysfunction and increased oxidative damage, spatial learning and memory impairments, neuronal dysfunction, synaptic changes, inflammation, and Aβ aggregation (Wang et al., 2010; Won et al., 2013; Xi et al., 2014; Zhao and Gong, 2015). In a mouse model of Alzheimer’s disease (AD), pBCCAO was reported to increase neuronal Aβ levels and neuronal tau hyperphosphorylation at an epitope that is associated with AD-type paired helical filament (PHF) and neurofibrillary tangle (NFT) formation (Koike et al., 2010). Moreover, in rats, ischemia leads to the accumulation of hyperphosphorylated tau in neurons and the formation of filaments that resemble those present in human neurodegenerative tauopathies in AD (Gordon-Krajcer et al., 2007). These studies document that decreased or dysregulated cerebral blood flow plays a critical role in development and progression of Aβ and tau pathologies in AD.
Endogenous nitric oxide (NO) synthesized from NO synthase (NOS), plays a key role in numerous physiological and pathological processes including endothelium relaxation, neurotransmission; inhibition of platelet aggregation, and anti-tumor and anti-microbial processes (see review (Martinez-Ruiz et al., 2011)). NO is known to exert its biological effects via activation of soluble guanylyl cyclase (sGC) leading to the production of cGMP and subsequent activation of cGMP-activated protein kinase G (PKG) (Hammond and Balligand, 2012). NO also exerts its effects through cGMP-independent and redox-dependent mechanisms. For example, formation of peroxynitrite by reaction between NO and superoxide anion mediates various cellular signaling mechanisms via nitration/oxidation-dependent or independent mechanisms (Klotz et al., 2002; Liaudet et al., 2009; McAndrew et al., 1997). In addition, formation of low molecular mass S-nitrosothiols (e.g. S-nitrosocysteine or S-nitrosoglutathione/GSNO) by reaction between NO and reduced thiol-containing compounds (e.g. cysteine and glutathione/GSH) also regulates various signal transduction pathways via S-transnitrosylation of specific proteins (Baker et al., 2000; Gould et al., 2013; Hess et al., 1993; Zaman et al., 2006). GSNO is the most abundant endogenous intracellular S-nitrosothiol and has been suggested as a potential NO storage site or transport species in cells (Butler and Rhodes, 1997; Mayer et al., 1998). In addition, GSNO has been also implicated in anti-inflammation (Kim et al., 2014; Prasad et al., 2007; Won et al., 2013), anti-oxidation (Chiueh, 1999; Qian et al., 2012), and cerebrovascular and BBB protections (Aggarwal et al., 2014; Khan et al., 2012), while peroxynitrite has been implicated in various neuropathological processes, such as dysfunctions of blood brain barrier (BBB) and cerebral blood flow (Ding et al., 2014; Khan et al., 2012), Aβ and NFT pathologies (Kummer et al., 2011; Reyes et al., 2011) and inflammation (Hooper et al., 2000; Kutzing and Firestein, 2008).
Based on the beneficial efficacy of GSNO on neuro- and vascular-protection and anti-inflammation (Aggarwal et al., 2014; Khan et al., 2012; Kim et al., 2014; Prasad et al., 2007; Won et al., 2013), we recently evaluated the effect of GSNO treatment on cognitive-function in rats subjected to pBCCAO. The pBCCAO model is now recognized as a model for human CCH featuring cerebral amyloid angiopathy (CAA), mild cognitive impairment (MCI), vascular dementia (VaD), as well as late-onset AD (de la Torre et al., 2003; de la Torre and Aliev, 2005; Zlokovic, 2011). We reported that the rats subjected to pBCCAO had decreased spatial learning and memory performance with increased accumulation of Aβ peptide and expression of vascular inflammatory markers (ICAM-1 and VCAM-1 expression) (Won et al., 2013). GSNO treatment of pBCCAO rats for 2 months (50μg/kg/day) successfully improved the learning and memory performance and reduced the Aβ levels and ICAM-1/VCAM-1 expression in the brains of those rats (Won et al., 2013). In addition, we also observed in cultured microglia and endothelial cells that GSNO treatment also attenuated the cellular inflammatory responses, such as activations of NFκB and STAT1/3 and expressions of inducible form of NOS (iNOS), ICAM-1, and VCAM-1 under the inflammatory conditions and increased Aβ uptake by microglial and endothelial cells (Won et al., 2013). Moreover, GSNO treatment also inhibited β-secretase activity by S-nitrosylation of BACE1, thus reduced secretion of Aβ in cultured neurons (Kwak et al., 2011; Won et al., 2013), suggesting the potential role of GSNO mediated mechanisms in anti-inflammation and anti-amyloidogenesis.
Although GSNO treatment effectively improved the cognitive decline in pBCCAO rats with attenuating the brain Aβ accumulation and inflammation, the mechanisms underlying GSNO-mediated neuro and cognitive protection are largely unknown at present. There is growing evidence that chronic cerebral hypoperfusion (i.e. oligaemia) induces axonal damage via hyperphosphorylation of microtubule-associated protein tau (Fujita et al.; Koike et al., 2010; Zlokovic, 2011). Under several neurodegenerative disease conditions, excessive phosphorylation of tau in the proline-rich region (residues 172–251) and the C-terminal tail region (residues 368–441) results in self-assembly of tau to form straight filaments (PHF), which are involved in the pathogenesis of AD, frontotemporal dementia, and other tauopathies (Alonso et al., 2001; Hanger et al., 2009). In this study, we report that pBCCAO induced neurodegeneration in hippocampal and cortical areas of rat brains is associated with increasing tau hyperphosphorylation and that GSNO treatment not only attenuates neuronal degeneration, but also inhibits pathological tau hyperphosphorylation. We also report that GSNO inhibited GSK-3β and Cdk5 activities induced in pBCCAO brains which have been implicated in pathological tau hyperphosphorylation (Cruz et al., 2003; Imahori and Uchida, 1997; Lee and Tsai, 2003).
2. Results
2-1. Effect of GSNO treatment on neurodegeneration induced in rat brains following pBCCAO
Based on the reported beneficial efficacies of GSNO in cognitive protection and anti-amylodogenesis in rat pBCCAO model and in vitro cell culture model (Won et al., 2013), we next evaluated the therapeutic potential of GSNO on neuroprotection in rat model of pBCCAO by Nissl staining. In Nissl-stained brain sections, the intact neurons were defined as nonbasophilic neurons with both the pale nuclei and the discrete nucleoi (green arrow head in Fig. 1A) and the injured/degenerating neurons were defined as neurons with abnormal morphologies of massive shrunken and dark Nissl staining (red arrow head in Fig. 1A) as described previously (Konigsmark, 1970; Ooigawa et al., 2006). We observed that the rats subjected to pBCCAO had a decreased number of intact neurons and an increased number of injured/degenerating neurons in cortical and hippocampal (CA1 and dentate gyrus/DG) areas whereas GSNO (50μg/kg/day for 2 months) treatment significantly increased the number of intact neurons and decreased the injured/degenerating neurons in rats subjected pBCCAO. In support the observed neuroprotective efficacy of GSNO, we also observed that GSNO treatment significantly inhibited the losses in neuronal bodies and neurofilament as observed in immunofluorescent microscopy by staining of cortical sections for NeuN and 200 kDa neurofilament-H (Fig. 1B and C).
Fig. 1. Neuroprotective effect of GSNO treatment in rats under chronic cerebral hypoperfusion.
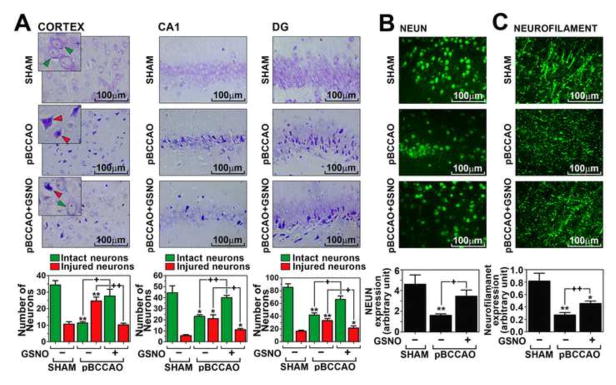
The distribution and number of intact neurons (nonbasophilic neurons with both the pale nuclei and the discrete nucleoi; green arrow head) and injured/degenerating neurons (neurons with abnormal morphologies of massive shrunken and dark Nissl staining; red arrow head) in cortical and hippocampal (dentate gyrus/DG and CA1) areas were analyzed by Nissl staining of paraffin-embedded sections of brains from rats subjected to pBCCAO (or sham operation) treated with GSNO (A). The loss of neurons and axons were also accessed by immunofluorescent staining for NeuN (B) and neurofilament-H (200 kDa) (C) in sections of cortical area and the intensities of immunofluorescence from different sections were analyzed by Image-Pro Plus and presented as bar graph. The vertical columns are the means of individual data and T-bars are the standard error mean. * p<0.01 and ** p<0.005 as compared to the sham group. + p<0.01 and ++p<0.005 as compared to untreated pBCCAO group.
2-2. Effect of GSNO treatment on pathological tau phosphorylation in rat brains following pBCCAO
Based on the observed GSNO-mediated neuroprotection (Fig. 1), we next examined the effect of GSNO treatment on pathological tau phosphorylation in rat model of pBCCAO. Excessive phosphorylation of tau in the proline-rich region (residues 172–251) and the C-terminal tail region (residues 368–441) is implicated in aberrant tau aggregate formation, also known as PHFs, under AD conditions (Hanger et al., 2009). We observed that the brains of pBCCAO rats also had increased tau phosphorylation in the proline-rich region (Ser202/Thr205) as well as C-terminal tail region (Ser396/Ser404) as compared to sham operated control rats as shown by Western analysis (Fig. 2A) and immunofluorescence staining of brain sections (Fig. 2B). These tau hyperphosphorylations in the brains of pBCCAO rats were markedly reduced in pBCCAO animals treated with GSNO. The inhibitory effect of GSNO on tau hyperphosphorylation was also observed in purified synaptosomes (Fig. 2C). In an ex vivo assay, semi-intact purified synaptosomes were treated with Aβ25–35 (40μM) in the presence or absence of GSNO (100μM). We observed that GSNO treatment attenuated Aβ25–35-induced tau hyperphosphorylation in purified synaptosomes. Overall, these data provide evidence for potential anti-tauopathy activity of GSNO in in vivo CCH model as well as ex vivo synaptosomal model of AD.
Fig. 2. Effect of GSNO treatment on pathological tau phosphorylation under chronic cerebral hypoperfusion.

A. The effect of GSNO on the phosphorylation of Ser396, Ser404, and Ser202/Thr205 which leading to paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) was analyzed by Western analysis using brain lysates extracted from sham and pBCCAO rats with/without GSNO treatment. Each band represents pooled brain tissues from experimental animals. B. The effect of GSNO on Ser396 phosphorylation was also analyzed by immunofluorescent staining and the intensities of immunofluorescence from different sections were analyzed by Image-Pro Plus and presented as bar graph. The vertical columns are the means of individual data and T-bars are the standard error mean. *** p<0.0001 as compared to the sham operated group. +++ p<0.0001 as compared to untreated pBCCAO group. C. The synaptosomes extracted from normal rat brains were pretreated with GSNO (100 μM) for 4 hrs and then treated with A β25–35 (40 μM) for 12 hrs and neuronal levels of phosphorylated tau (Ser396, Ser404, and Ser202/Thr205), total tau, and β-actin (loading control) were analyzed by Western blot analysis.
2-3. Regulation of GSK-3β activity by GSNO in rat brains following pBCCAO
Dysregulation of GSK-3β has been implicated in hyperphosphorylation of tau and PHF formation under AD conditions (Llorens-Martin et al., 2014). Activity of GSK-3β is regulated by phosphorylation of a conserved tyrosine residue on the activation loop of the kinase domain (Tyr216 in GSK-3β) that is required for kinase activity of GSK-3β (Cole et al., 2004) whereas phosphorylation of serine residue at an N-terminal pseudo-substrate domain (Ser9) prevents interaction of GSK-3β with substrates and thus inhibits its activity (Frame et al., 2001). In this study, we observed that the rats subjected to pBCCAO had increased enzyme activity (Fig. 3A) and phosphorylation at residueTyr216 (Fig. 3B) of GSK-3β in their brains. The pBCCAO rats treated with GSNO had reduced hyperphosphorylation of tau and phosphorylation at residueTyr216 of GSK-3β (Figs. 3A and B). However, phosphorylation of GSK-3β at residue of Ser9 was not affected in brains of either pBCCAO rats or pBCCAO rats treated with GSNO (Fig. 3B). These observations indicate that GSNO-mediated mechanisms regulate GSK-3β activity by attenuation of its Tyr216 phosphorylation.
Fig. 3. Effect of GSNO treatment on GSK-3β activation under chronic cerebral hypoperfusion.

A. GSK-3β activities were analyzed in sham, pBCCAO rats, and pBCCAO rats treated with GSNO by in vitro kinase assay as described under experimental procedure. B. In addition, the effect of GSNO treatment on the phosphorylation status of Tyr279 (GSK3α)/Tyr216 (GSK-3β) or Ser9 of GSK-3β was analyzed by Western blot using specific antibody. The vertical columns are the means of individual data and T-bars are the standard error mean. ***p<0.0001; and as compared to sham group. ++p<0.005 as compared to untreated pBCCAO group.
2-4. Regulation of calpain-mediated cleavage of p35 to p25 in brains of pBCCAO rat model
Calpain mediated cleavage of p35 leading to generation truncated p25 protein and followed Cdk5 activation have been implicated in pathological events leading to tau hyperphosphorylation and neurodegeneration (Cruz et al., 2003). Therefore, we next assessed whether GSNO inhibits calpain-mediated p35 proteolytic process to p25. Fig. 4A shows that the rats subjected to pBCCAO had elevated calpain activities and calpain-mediated proteolysis of p35 to p25 in the brains whereas GSNO treatment of pBCCAO rats attenuated the calpain activities as well as p35 proteolysis (Fig. 4A and B). Accordingly, GSNO treatment also attenuated the increased Cdk5 activities in the brains of pBCCAO rats (Fig. 4C). These observations indicate that GSNO-mediated mechanisms regulate the Cdk5 activity possibly via regulation of calpain activity.
Fig. 4. Effect of GSNO treatment on calpain activation, p35 cleavage to p25, and Cdk5 activation under chronic cerebral hypoperfusion.
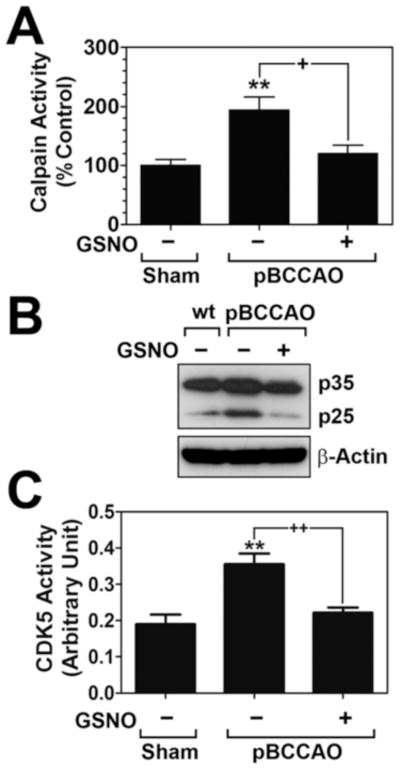
A. Calpain activities were analyzed in sham, pBCCAO rats, and pBCCAO rats treated with GSNO by in vitro protease assay as described under experimental procedure. B. In addition, the effect of GSNO treatment on the calpain-mediated proteolysis of p35, generating truncated p25 protein, was analyzed by Western blot. C. Cdk5 activities were analyzed in sham, pBCCAO rats, and pBCCAO rats treated with GSNO by in vitro kinase assay as described under experimental procedure. The vertical columns are the means of individual data and T-bars are the standard error mean. **p<0.005; and as compared to sham group. +p<0.01 as compared to untreated pBCCAO group.
Inducible form of NO synthase (iNOS) has been implicated in peroxynitrite-mediated brain injuries under various neurological disease conditions including pBCCAO (Kawano et al., 2007; Pannu and Singh, 2006). In brains of pBCCAO rats, we observed both the increased iNOS expression (Fig. 5A) and peroxynitrite formation (Fig. 5B) by Western analysis for iNOS and ELISA for nitro-tyrosine (N-Tyr), respectively. GSNO treatment attenuated the expression of iNOS as well as N-Tyr formation (Figs. 5A and B). Therefore, these data indicate the role of GSNO in inhibition of iNOS expression as well as peroxynitrite-induced nitrosative stress under CCH conditions. We made interesting observation that activity of purified calpain-1 is increased by 3-morpholinosyndnomine (SIN-1), a peroxynitrite donor, while calpain activity is inhibited by GSNO (Fig. 5C). These in vitro calpain activity assay using purified calpain protein indicate opposing role of two NO metabolites (GSNO vs. peroxynitrite) in regulation of calpain activity.
Fig. 5. Effect of GSNO treatment on peroxynitrite-mediated regulation of calpain activation.

A. Expressions of iNOS protein were analyzed in sham, pBCCAO rats, and pBCCAO rats treated with GSNO by Western blot using specific antibody. B. Productions of peroxynitrite in the brains were analyzed in sham, pBCCAO rats, and pBCCAO rats treated with GSNO by ELISA assay as described under experimental procedure. The vertical columns are the means of individual data and T-bars are the standard error mean. *p<0.01; and as compared to sham group. +p<0.01 as compared to untreated pBCCAO group. C. The effect of increasing concentrations of GSNO or SIN-1 (a donor of peroxynitrite) on enzyme activity of calpain was analyzed by in vitro protease assay using purified calpain-1. In each reaction mixture, GSNO or SIN-1 was treated for 60 min before assay. The data points on the line represent the mean values for each individual data. ***p<0.0001 as compared to the control.
3. Discussion
Aging is reported to reduce cerebral blood flow (Bentourkia et al., 2000; Heo et al., 2010; Leenders et al., 1990), so any additional conditions causing dysfunctions in cerebral perfusion could damage or kill vulnerable neurons (de la Torre, 2000; de la Torre, 2006), and thus cause cognitive impairment, VaD, and AD (Bookheimer et al., 2000; Iadecola, 2004; Knopman and Roberts, 2010). Therefore, protection of cerebrovasculature and improvement of cerebral blood flow have been a potential target for VaD as well as AD. In this study, we report that systemic GSNO treatment attenuates calpain/p25/Cdk5 and GSK3β mediated pathological tau hyperphosphorylation under disease conditions of chronic cerebral hypoperfusion leading to neuroprotection.
GSNO is the most abundant intracellular NO carrier molecule, but its cellular transport is not well understood at present. GSNO, itself, has limited cell membrane permeability but exogenous GSNO is reported to exert its cellular activities by formation of cell permeable S-nitrosocysteine via transnitrosylation and/or γ-glutamyl transpeptidase-mediated cleavage of GSNO to cell permeable S-nitroso-cysteinyl glycine dipeptide (Hogg et al., 1997; Lewis et al., 2005; Lipton et al., 2001). Exogenous GSNO is known to induce vaso-relaxation via activating potassium channels in aorta (Ceron et al., 2001). Moreover, it has a significant anti-platelet activity (de Belder et al., 1994) and was used therapeutically as a platelet-selective anti-thrombotic agent (Langford et al., 1994). In addition, the reported roles of GSNO in activation of cell survival signaling cascades mediated by dynamin-2, phosphatidylinositol 3-kinase (PI3K), extracellular signal-regulated kinase (ERK), and Bcl-2, and in inhibitions of proapoptotic caspase activation (Ju et al., 2005; Kang-Decker et al., 2007; Lowenstein, 2007; Zeigler et al., 2003) and pro-inflammatory cell signaling cascades (Kim et al., 2014; Prasad et al., 2007; Won et al., 2013) further suggest that exogenous GSNO targets various anti-inflammatory and cell survial signaling pathways and is protective in various neurological disease conditions, such as as stroke, traumatic brain injury, and spinal cord injury (Khan et al., 2005; Khan et al., 2011; Khan et al., 2015; Shunmugavel et al., 2012). In this study, we observed that systemic GSNO treatment attenuated the tau hyperphosphorylation and neurodegeneration in rat model of pBCCAO. Similar to studies with other CNS disease animal models (Khan et al., 2005; Khan et al., 2011; Khan et al., 2015; Shunmugavel et al., 2012). These observations indicate that GSNO-induced neuroprotection could be a consequence of its activities on cerebrovascular functions as well as direct contribution of GSNO-mediated signaling pathways for neurodegeneration. In support, we observed that GSNO treatment inhibited Aβ-induced tau hyperphosphorylation in purified synaptosomes (Fig. 2C). In addition, GSNO treatment inhibited calpain activity in in vitro assay system using purified calpain (Fig. 5C) and calpain-mediated p35 proteonlysis to p25 and Cdk5-mediated tau hyperphosphorylation in primary cultured rat neurons treated with Aβ (Annamalai et al., 2015). These studies indicate a direct role of neuronal GSNO in regulation of tau hyperphosphorylation-induced neuropathology in AD brain. However, relative contribution of above mechanisms is not known in systemic GSNO treatment-mediated regulation of neuronal tau hyperphosphorylation in pBCCAO rat model.
Increase in pathological tau hyperphosphorylation leading to neurotoxic PHF and NFT formation has been suggested as one of the mechanisms underlying neurodegeneration and cognitive decline under CNS hypoxic conditions (Gordon-Krajcer et al., 2007; Koike et al., 2010). We here report that GSNO treatment attenuated pathological tau hyperphosphorylation (Ser202/Thr205 and Ser396/Ser404) affording inhibition of neuronal degeneration. In addition, the observed inhibition of tau hyperphosphorylation correlated well with the decreased activities of GSK-3β and Cdk5 in the brains of pBCCAO rats treated with GSNO, suggesting that a role for GSNO-based mechanism in GSK-3β and Cdk5-mediated tau hyperphosphorylation under experimental CCH conditions. GSK-3β is regulated by its phosphorylation at an N-terminal Ser residue (Ser9) which acts as a pseudo-substrate and thus inhibits its kinase activity (Frame et al., 2001), calpain-mediated removal of N-terminal pseudo-substrate domain of GSK-3β generating truncated form of GSK-3β (30/40 kDa) (Goni-Oliver et al., 2007), and phosphorylation of a conserved Tyr216 residue on the activation loop of the kinase domain which increases kinase activity (Cole et al., 2004). In this study, we observed that both increased GSK-3β activity and phosphorylation of GSK-3β Tyr216 in pBCCAO rat brains were attenuated by GSNO treatment. On the other hand, GSNO had no effect on phosphorylation status of Ser9 and Ser9 phosphorylation has no effect on the activities of GSK-3β. In addition, there was no obvious alteration in molecular weight of GSK-3β, suggesting a role for GSNO mediated mechanism in regulation of GSK-3β activation via Tyr216 phosphorylation on active loop of GSK-3β kinase domain, but not via the truncation or phosphorylation (Ser9) of N-terminal pseudo-substrate domain. Along with GSK-3β, aberrant activation of Cdk5 has been also implicated in tau-pathology in AD (Cruz et al., 2003; Lee and Tsai, 2003). The kinase activity of Cdk5 is regulated by its binding with membrane associated p35 (Cruz et al., 2003). Upon the binding of Cdk5, p35 activates Cdk5 and undergoes degradation via ubiquitin-mediated proteolysis (Patrick et al., 1998). However, alternatively, p35 is also processed by calpain to p25 which is resistant to proteolysis under the pathological conditions and the resulted p25/Cdk5 complex dissociates from the membrane and interacts with different targets, such as tau (Cruz et al., 2003). Studies have reported that calpain-mediated p25 accumulation in brains of patients with AD induces prolonged activation and mislocalization of Cdk5 and thus hyperphosphorylation of tau (Lee et al., 2000). In this study, we observed that GSNO attenuated the calpain activities in pBCCAO rat brains (Fig. 4A), therefore, decreased p25 formation (Fig. 4B) and thus Cdk5 activation (Fig. 4C).
NO is now recognized as one of the key cell signaling mediators regulating physiological processes (Martinez-Ruiz et al., 2011). However, NO is also known to cause nitroso-oxidative damage by formation of peroxynitrite from reaction between NO and superoxide anion under inflammatory and oxidative disease conditions (Brown, 2007). In various neurological disease conditions including AD, the increased neuroinflammation and induction of iNOS gene are implicated in peroxynitrite and oxidative stress mediated neural tissue damages (Brown, 2007; Fernandez-Vizarra et al., 2004; Luth et al., 2002; Sayre et al., 2008). Brains of rats with pBCCAO express increased iNOS and nitrotyrosine and that GSNO treatment significantly attenuated both the iNOS expression and nitrotyrosine levels (Fig. 5A and B), suggesting a potential role of GSNO-mediated mechanisms in anti-inflammation and anti-nitrosative stress under CCH conditions. We previously reported that micromolar concentration of GSNO inhibits iNOS expression via S-nitrosylation-mediated inhibition of proinflammatory cell signaling, such as NFκB and Stat1 in cell culture studies (Prasad et al., 2007; Won et al., 2013). However, whether the systemic GSNO treatment of pBCCAO rats attenuates iNOS expression and nitrotyrosine levels by direct inhibition of NFκB and STAT1 activities or by indirect pathways, such as improvement of cerebrovascular function, is not known at present. Interestingly, we observed that treatment of purified calpain with SIN-1, a peroxynitrite donor, increased the calpain activity in in vitro assay using purified calpain, while GSNO treatment decreased the calpain activity (Fig. 5C), indicating direct opposing roles of peroxynitrite and GSNO in regulation of calpain. Since GSNO treatment decreased the brain nitrotyrosine levels (Fig. 5B), this suggests the possible role of GSNO treatment in attenuation of nitrosative stress induced calpain activation and thus tau hyperphosphorylation-mediated neurodegeneration under CCH conditions. Previous studies had also reported that calpain activity can be regulated by NO donors via S-nitrosylation of cysteine residue in their catalytic domains (Michetti et al., 1995; Samengo et al., 2012). However, whether exogenous GSNO treatment inhibits neuronal calpain activity directly through S-nitrosylation of calpain or indirectly via improving cerebrovascular function is not known at present.
Studies have reported that peroxynitrite and GSNO, different redox-related metabolites of NO, play opposing roles in various patholophysiological processes in the CNS. Peroxynitrite has been reported to play critical roles in induction of cerebrovascular morbidity and blood-brain barrier disruption (Faraci, 2006), oxidative/nitrosative damage of the CNS (Smith et al., 1997), Aβ aggregation (Kummer et al., 2011), and tau modification (Reyes et al., 2011; Zhang et al., 2006). However, GSNO has been reported to protect cerebrovascular function under brain ischemia conditions (Khan et al., 2005) and inhibit Aβ synthesis, pathological tau modification, inflammation, and cognitive deficits (Annamalai et al., 2015; Kwak et al., 2011; Won et al., 2013), suggesting GSNO as a neuroprotective effector of NO while peroxynitrite as pathological effector of NO. These findings suggest that understanding of beneficial vs. deleterious effectors of NO (e.g. GSNO vs. peroxynitrite), produced from NOS isoforms (e.g. nNOS, iNOS, and eNOS), would be critical for understanding NO mediated disease mechanisms as well as development of NO-based therapeutic approaches for AD and other neurological disease conditions.
In summary, these data report that GSNO inhibits tau hyperphosphorylation while decreasing iNOS expression and nitrosative stress and thus provides neuroprotection under chronic cerebral hypoperfusion. Although the detailed pharmacological action and mechanisms underlying GSNO-mediated neuroprotection are not known at present, however, the data described in this paper along with our previous reports (Annamalai et al., 2015; Won et al., 2013) support the therapeutic potential of GSNO-mediated mechanisms in neuro- and cognitive-protection under conditions of chronic cerebral hypoperfusion.
4. Experimental Procedures
4-1. Rat model of chronic cerebral hypoperfusion and GSNO treatment
As an animal model for human chronic cerebral hypoperfusion featuring mild cognitive impairment (de la Torre and Aliev, 2005; Ignarro, 1990), rats (250~270g body weight) received permanent occlusion of bilateral common carotid arteries (BCCAO). It is well established that the BCCAO of rats causes reduction of hippocampal blood flow (22~30%) and thus declines learning memory functions (de la Torre et al., 1995; de la Torre and Aliev, 2005; Ignarro, 1990; Tsuchiya et al., 1992). For permanent BCCAO or sham operation, the rats (n=6 per gourp) were anesthetized with Ketamine and Xylazine. Following a ventral midline incision, the common carotid arteries were bilaterally separated from the carotid sheath and vagus nerve and rigidly occluded with 3–0 silk sutures. The rats assigned to sham surgery were submitted to the same procedure without occlusion of the arteries. For post-procedural pain relief, the rats were administered buprenorphine (0.05 mg/kg, subcutaneous) twice daily for 3 days. Following surgery, the rats were fed ad libitum for 4 months to induce chronic hypoperfusion mediated changes and then treated daily with GSNO (50μg/kg, i.p.) or saline for 2 months. GSNO (World Precision Instruments Inc., Sarasota, FL) was dissolved in dimethylsulfoxide (DMSO) and kept in −80°C. Before the experiment, the concentration of GSNO was determined photometrically using a molar extinction coefficient of 900 M−1cm−1 at 336 nm as described previously (Stamler et al., 1992) and further diluted with phosphate buffered saline. Following the GSNO treatment, the rats were sacrified and each hemisphere was processed separately for immunohistological or biochemical studies. For subsequent studies, all samples were processed and investigated in a blinded manner.
4-2. Western Immunoblot analysis
Brain lysates were prepared with RIPA lysis buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl pH 8) with protease and phosphatase inhibitors. Protein concentration in lysates was determined by Lowry method using assay kit purchased from Bio-Rad (Richmond, CA). The equal amount of protein (50–100 μg) of each brain lysate was loaded per lane on polyacrylamide gels. To obtain representative Western blots, equal amounts of lysates (n=6) were pooled and loaded on the gel. Western immunoblot analysis was performed using antibodies against phospho-tau (p-tau) S396 (Abcam, Cambridge, MA), p-tau S404 (Abcam), p-tau S202/T205 (Pierce, Rockford, IL), pan-tau (Cell Signaling Technology), β-actin (Abcam), p35/p25 (Cell Signaling Technology, Danvers, MA), phospho-GSK-3β (p-GSK-3β) Y216/Y279 (Abcam), p-GSK-3β S9 (Cell Signaling Technology), pan-GSK-3β (BD Transduction Laboratory, San Diego, CA). Density of each band on Western blot was analyzed using Image J software. Each experiment was performed at least three times.
4-3. Histology and Immuno-fluorescent staining
Paraffin-embedded sections collected between bregma −3.14 and −4.80 from the formalin-fixed brain tissues were stained by with Nissl stain kit (IHCWORLD, Woodstock, MD) to detect Nissl bodies in cortex and hippocampal CA1 and dentate gyrus (DG) areas according to the manufacturer’s instruction. The sections were also used for immunofluorescent staining of certex for NeuN (Millipore, Bedford, MA), neurofilament SMI-31 (Abcam, Cambridge, MA) and p-tau S396. BX60 Olympus fluorescent/light microscope equipped with DP-70 camera (Olympus, Tokyo, Japan) was used for imaging of histological and immunofluorescent staining. All images were taken at 400x magnification from 6 rats per group in a blinded manner. The intensities of fluorescence were quantified from three randomly picked fields by Image-Pro Plus (MediaCybernetics, Bethesda, MD, USA).
4-4. Calpain activity assay
Analysis of calpain activity in cell free system was performed using purified active calpain 1 included in calpain activity assay kit (Abcam) according to the manufacturer’s instructions. Analysis of calpain activity in the brain tissue and primary neuron culture was performed using the same assay kit. Briefly, equal amounts of brain and neuronal lysate were incubated with substrate (Ac-LLY-AFC) and reaction buffer and calpain-mediated cleavage of substrate was analyzed by fluorometric analysis.
4-5. In vitro kinase assay for Cdk5, and GSK-3β
The brain tissues and neuron cells were lysed in lysis buffer (50mM Tris-HCl, pH 7.4, 0.5mM EDTA, 10.5M EGTA, 0.5mM activated sodium orthovanadate, 10mM sodium β-glycerol phosphate, 50mM sodium fluoride, 5mM sodium pyrophosphate, 1% Triton X-100, and protease inhibitor cocktail) and clarified by centrifugation. Equal amount of tissue and cell lysates were immunoprecipitated with anti-CDK5 antibody (Abcam) or anti-GSK-3β antibody (BD Transduction Laboratories) for 4 hrs at 4°C and then pulled down with Protein A/G sepharose beads (SantaCruz Biotech.). Following the washing of the immuno-precipitates, the resulted pellets were incubated with biotin-labeled Cdk5 substrate (Biotin-Ahx-PKTPKKAKKL; Enzo life sciences, Farmingdale, NY) or biotin-labeled GSK-3β substrate (Biotin-RRAAEELDSRAGSPQL; AnaSpec, San Jose, CA) in kinase assay buffer (5 mM MOPS, pH 7. 2, 2.5 mM β-glycerol-phosphate, 5 mM MgCl2, 1 mM EGTA, 0.4 mM EDTA, 100μM ATP, and 0.05mM dithiothreitol) for 15 minutes. The reactions were then stopped by adding 100 mM EDTA. Following the centrifugation, the supernatant containing phosphorylated biotin-labeled substrates were incubated in streptavidin coated 96 well plate (Pierce/Thermo Scientific, Rockford, IL). The phosphorylation of substrate peptides were detected by incubation with horse-radish-peroxidase (HRP)-conjugated anti-phospho-Thr antibody (Cell signaling) for analysis of Cdk5 activity and HRP-conjugated anti-phospho-Ser antibody for analysis of GSK-3β activity. The colorimetric analysis of HRP activities were assayed by using with using TMB HRP-substrate solution (Pierce/Thermo Scientific) and SpectraMax 190 ELISA reader (Molecular Device, Sunnyvale, CA).
4-6. Nitro tyrosine ELISA
Analysis of nitro tyrosine (N-Tyr) levels in the brain tissue was performed using OxiSelect ™ Nitrotyrosine ELISA Kit (Cell Biolabs, Inc., Cat# STA 305, San Diago, CA) according to the manufacturer’s instructions. Briefly, brain tissues (n=6) were homogenized (dounce homogenizer) in assay diluent provided with the kit and equal amounts (500μg) of brain lysates and nitrated BSA standards were added to an N-Tyr coated EIA plate and followed by incubation with anti-nitro tyrosine antibody. Following washing, the pates were incubated HRP conjugated secondary antibody and the levels of N-Tyr were measured by incubation with 3,3′,5,5′-tetramethylbenzidine (TMB) solution and colorimetric analysis at 450nm.
4-7. Synaptosomal preparation
Isolation of functional synaptosomes were prepared from normal healthy male adult Sprague-Dawley rats weighing 300–400 g by using Syn-PER synaptic protein isolation reagent (Thermo Scientific, IL). Briefly, the rat brain cortices were homogenized in 0.32M sucrose buffer by using a 7mL Dounce tissue grinder. The homogenate was centrifuged at 1200 × g for 10 minutes to remove cell debris, and the supernatant was centrifuged at 15,000 × g for 20 minutes. The resulted pellets containing synaptosomes were assayed for protein concentration.
4-8. Statistical Analysis
All values shown in the figures are expressed as mean ± SEM. The results were examined by one- and two-way ANOVA; then individual group means were compared with the Bonferroni test. A p value <0.05 was considered significant.
Highlight.
GSNO attenuates neurodegeneration in rat model of chronic cerebral hypoperfusion.
GSNO inhibits tau hyperphosphorylation via direct or indirect mechanisms.
GSNO inhibits GSK3β and calpain/p25/Cdk5 pathways via direct or indirect mechanisms.
GSNO attenuates iNOS expression and nitrotyrosine formation.
Acknowledgments
Je-Seong Won and Balasubramaniam Annamalai both contributed equal effort and are co-first authors for this manuscript. This work was supported by grants from NIH and VA (NS072511, BX001062, NS037766 and BX001072). We also acknowledge Ms. Joyce Bryan for their help in procurement of animals and supplies.
Abbreviations
- AD
Alzheimer’s disease
- CAA
cerebral amyloid angiopathy
- CCH
chronic cerebral hypoperfusion
- GSNO
S-nitrosoglutathione
- MCI
mild cognitive impairment
- N-Tyr
nitro tyrosine
- pBCCAO
permanent bilateral common carotid artery occlusion
- VaD
vascular dementia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal A, Khera A, Singh I, Sandhir R. S-nitrosoglutathione prevents blood-brain barrier disruption associated with increased matrix metalloproteinase-9 activity in experimental diabetes. J Neurochem. 2014 doi: 10.1111/jnc.12939. [DOI] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–8. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annamalai B, Won JS, Choi S, Singh I, Singh AK. Role of S-nitrosoglutathione mediated mechanisms in tau hyper-phosphorylation. Biochem Biophys Res Commun. 2015;458:214–9. doi: 10.1016/j.bbrc.2015.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Booden MA, Buss JE. S-Nitrosocysteine increases palmitate turnover on Ha-Ras in NIH 3T3 cells. J Biol Chem. 2000;275:22037–47. doi: 10.1074/jbc.M001813200. [DOI] [PubMed] [Google Scholar]
- Bentourkia M, Bol A, Ivanoiu A, Labar D, Sibomana M, Coppens A, Michel C, Cosnard G, De Volder AG. Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: effect of aging. J Neurol Sci. 2000;181:19–28. doi: 10.1016/s0022-510x(00)00396-8. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–6. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35:1119–21. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- Butler AR, Rhodes P. Chemistry, analysis, and biological roles of S-nitrosothiols. Anal Biochem. 1997;249:1–9. doi: 10.1006/abio.1997.2129. [DOI] [PubMed] [Google Scholar]
- Ceron PI, Cremonez DC, Bendhack LM, Tedesco AC. The relaxation induced by S-nitroso-glutathione and S-nitroso-N-acetylcysteine in rat aorta is not related to nitric oxide production. J Pharmacol Exp Ther. 2001;298:686–94. [PubMed] [Google Scholar]
- Chiueh CC. Neuroprotective properties of nitric oxide. Ann N Y Acad Sci. 1999;890:301–11. doi: 10.1111/j.1749-6632.1999.tb08007.x. [DOI] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004;377:249–55. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- de Belder AJ, MacAllister R, Radomski MW, Moncada S, Vallance PJ. Effects of S-nitroso-glutathione in the human forearm circulation: evidence for selective inhibition of platelet activation. Cardiovasc Res. 1994;28:691–4. doi: 10.1093/cvr/28.5.691. [DOI] [PubMed] [Google Scholar]
- de la Torre JC, Butler K, Kozlowski P, Fortin T, Saunders JK. Correlates between nuclear magnetic resonance spectroscopy, diffusion weighted imaging, and CA1 morphometry following chronic brain ischemia. J Neurosci Res. 1995;41:238–45. doi: 10.1002/jnr.490410211. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000;21:331–42. doi: 10.1016/s0197-4580(00)00111-1. [DOI] [PubMed] [Google Scholar]
- de la Torre JC, Pappas BA, Prevot V, Emmerling MR, Mantione K, Fortin T, Watson MD, Stefano GB. Hippocampal nitric oxide upregulation precedes memory loss and A beta 1–40 accumulation after chronic brain hypoperfusion in rats. Neurol Res. 2003;25:635–41. doi: 10.1179/016164103101201931. [DOI] [PubMed] [Google Scholar]
- de la Torre JC, Aliev G. Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab. 2005;25:663–72. doi: 10.1038/sj.jcbfm.9600057. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. How do heart disease and stroke become risk factors for Alzheimer’s disease? Neurol Res. 2006;28:637–44. doi: 10.1179/016164106X130362. [DOI] [PubMed] [Google Scholar]
- Ding R, Chen Y, Yang S, Deng X, Fu Z, Feng L, Cai Y, Du M, Zhou Y, Tang Y. Blood-brain barrier disruption induced by hemoglobin in vivo: Involvement of up-regulation of nitric oxide synthase and peroxynitrite formation. Brain Res. 2014;1571:25–38. doi: 10.1016/j.brainres.2014.04.042. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol (1985) 2006;100:739–43. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra P, Fernandez AP, Castro-Blanco S, Encinas JM, Serrano J, Bentura ML, Munoz P, Martinez-Murillo R, Rodrigo J. Expression of nitric oxide system in clinically evaluated cases of Alzheimer’s disease. Neurobiol Dis. 2004;15:287–305. doi: 10.1016/j.nbd.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–7. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Hamano T, Shirafuji N, Nakamoto Y. Chronic cerebral hypoperfusion induces tau hyperphosphorylation accompanied by axonal damage in adult mice. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 10:P789. [Google Scholar]
- Goni-Oliver P, Lucas JJ, Avila J, Hernandez F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–13. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Gordon-Krajcer W, Kozniewska E, Lazarewicz JW, Ksiezak-Reding H. Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem Res. 2007;32:729–37. doi: 10.1007/s11064-006-9199-3. [DOI] [PubMed] [Google Scholar]
- Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J Biol Chem. 2013;288:26473–9. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol. 2012;52:330–40. doi: 10.1016/j.yjmcc.2011.07.029. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Lee JM, Zipfel GJ, Wozniak DF. Characterizing learning deficits and hippocampal neuron loss following transient global cerebral ischemia in rats. Brain Res. 2005;1043:48–56. doi: 10.1016/j.brainres.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Heo S, Prakash RS, Voss MW, Erickson KI, Ouyang C, Sutton BP, Kramer AF. Resting hippocampal blood flow, spatial memory and aging. Brain Res. 2010;1315:119–27. doi: 10.1016/j.brainres.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Patterson SI, Smith DS, Skene JH. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature. 1993;366:562–5. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- Hogg N, Singh RJ, Konorev E, Joseph J, Kalyanaraman B. S-Nitrosoglutathione as a substrate for gamma-glutamyl transpeptidase. Biochem J. 1997;323(Pt 2):477–81. doi: 10.1042/bj3230477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB J. 2000;14:691–8. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ. Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;16:477–83. doi: 10.1161/01.hyp.16.5.477. [DOI] [PubMed] [Google Scholar]
- Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J Biochem. 1997;121:179–88. [PubMed] [Google Scholar]
- Ju TC, Chen SD, Liu CC, Yang DI. Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med. 2005;38:938–49. doi: 10.1016/j.freeradbiomed.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- Kawano T, Kunz A, Abe T, Girouard H, Anrather J, Zhou P, Iadecola C. iNOS-derived NO and nox2-derived superoxide confer tolerance to excitotoxic brain injury through peroxynitrite. J Cereb Blood Flow Metab. 2007;27:1453–62. doi: 10.1038/sj.jcbfm.9600449. [DOI] [PubMed] [Google Scholar]
- Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, Singh I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25:177–92. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- Khan M, Sakakima H, Dhammu TS, Shunmugavel A, Im YB, Gilg AG, Singh AK, Singh I. S-nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation. 2011;8:78. doi: 10.1186/1742-2094-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Sakakima H, Shunmugavel A, Gilg AG, Singh AK, Singh I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem. 2012;123(Suppl 2):86–97. doi: 10.1111/j.1471-4159.2012.07947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Matsuda F, Baarine M, Dhindsa TS, Singh I, Singh AK. Promoting endothelial function by S-nitrosoglutathione through the HIF-1alpha/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats. Drug Des Devel Ther. 2015;9:2233–47. doi: 10.2147/DDDT.S77115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Won JS, Singh AK, Sharma AK, Singh I. STAT3 regulation by S-nitrosylation: implication for inflammatory disease. Antioxid Redox Signal. 2014;20:2514–27. doi: 10.1089/ars.2013.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz LO, Schroeder P, Sies H. Peroxynitrite signaling: receptor tyrosine kinases and activation of stress-responsive pathways. Free Radic Biol Med. 2002;33:737–43. doi: 10.1016/s0891-5849(02)00892-4. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Roberts R. Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis. 2010;20:699–709. doi: 10.3233/JAD-2010-091555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-{beta} Am J Pathol. 2010;177:300–10. doi: 10.2353/ajpath.2010.090750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konigsmark BW. Contemporary research methods in neuroanatomy. Springer; Berlin Heidelberg New York: 1970. Methods for counting of neurons. [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, Konig S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71:833–44. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kutzing MK, Firestein BL. Altered uric acid levels and disease states. J Pharmacol Exp Ther. 2008;324:1–7. doi: 10.1124/jpet.107.129031. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6:17. doi: 10.1186/1750-1326-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford EJ, Brown AS, Wainwright RJ, de Belder AJ, Thomas MR, Smith RE, Radomski MW, Martin JF, Moncada S. Inhibition of platelet activity by S-nitrosoglutathione during coronary angioplasty. Lancet. 1994;344:1458–60. doi: 10.1016/s0140-6736(94)90287-9. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–4. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lee MS, Tsai LH. Cdk5: one of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis. 2003;5:127–37. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- Leenders KL, Perani D, Lammertsma AA, Heather JD, Buckingham P, Healy MJ, Gibbs JM, Wise RJ, Hatazawa J, Herold S, et al. Cerebral blood flow, blood volume and oxygen utilization. Normal values and effect of age. Brain. 1990;113(Pt 1):27–47. doi: 10.1093/brain/113.1.27. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Hoque A, Bates JN. Differentiation of L- and D-S-nitrosothiol recognition sites in vivo. J Cardiovasc Pharmacol. 2005;46:660–71. doi: 10.1097/01.fjc.0000181714.94827.5d. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Vassalli G, Pacher P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci (Landmark Ed) 2009;14:4809–14. doi: 10.2741/3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, Gaston B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature. 2001;413:171–4. doi: 10.1038/35093117. [DOI] [PubMed] [Google Scholar]
- Llorens-Martin M, Jurado J, Hernandez F, Avila J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein CJ. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc Res. 2007;75:240–6. doi: 10.1016/j.cardiores.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth HJ, Munch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–43. doi: 10.1016/s0006-8993(02)03280-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Cadenas S, Lamas S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med. 2011;51:17–29. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Masada T, Itano T, Fujisawa M, Miyamoto O, Tokuda M, Matsui H, Nagao S, Hatase O. Embryonic transplantation and ischemic memory deficit. Neurosci Res. 1997;27:249–55. doi: 10.1016/s0168-0102(97)01158-9. [DOI] [PubMed] [Google Scholar]
- Mayer B, Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Brunner F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J Biol Chem. 1998;273:3264–70. doi: 10.1074/jbc.273.6.3264. [DOI] [PubMed] [Google Scholar]
- McAndrew J, Patel RP, Jo H, Cornwell T, Lincoln T, Moellering D, White CR, Matalon S, Darley-Usmar V. The interplay of nitric oxide and peroxynitrite with signal transduction pathways: implications for disease. Semin Perinatol. 1997;21:351–66. doi: 10.1016/s0146-0005(97)80002-x. [DOI] [PubMed] [Google Scholar]
- Michetti M, Salamino F, Melloni E, Pontremoli S. Reversible inactivation of calpain isoforms by nitric oxide. Biochem Biophys Res Commun. 1995;207:1009–14. doi: 10.1006/bbrc.1995.1285. [DOI] [PubMed] [Google Scholar]
- Ooigawa H, Nawashiro H, Fukui S, Otani N, Osumi A, Toyooka T, Shima K. The fate of Nissl-stained dark neurons following traumatic brain injury in rats: difference between neocortex and hippocampus regarding survival rate. Acta Neuropathol. 2006;112:471–81. doi: 10.1007/s00401-006-0108-2. [DOI] [PubMed] [Google Scholar]
- Pannu R, Singh I. Pharmacological strategies for the regulation of inducible nitric oxide synthase: neurodegenerative versus neuroprotective mechanisms. Neurochem Int. 2006;49:170–82. doi: 10.1016/j.neuint.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–64. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- Pazos AJ, Green EJ, Busto R, McCabe PM, Baena RC, Ginsberg MD, Globus MY, Schneiderman N, Dietrich WD. Effects of combined postischemic hypothermia and delayed N-tert-butyl-alpha-pheylnitrone (PBN) administration on histopathologicaland behavioral deficits associated with transient global ischemia in rats. Brain Res. 1999;846:186–95. doi: 10.1016/s0006-8993(99)02010-7. [DOI] [PubMed] [Google Scholar]
- Prasad R, Giri S, Nath N, Singh I, Singh AK. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007;55:65–77. doi: 10.1002/glia.20436. [DOI] [PubMed] [Google Scholar]
- Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, Fulton DJ. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic Biol Med. 2012;52:1806–19. doi: 10.1016/j.freeradbiomed.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JF, Fu Y, Vana L, Kanaan NM, Binder LI. Tyrosine nitration within the proline-rich region of Tau in Alzheimer’s disease. Am J Pathol. 2011;178:2275–85. doi: 10.1016/j.ajpath.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samengo G, Avik A, Fedor B, Whittaker D, Myung KH, Wehling-Henricks M, Tidball JG. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell. 2012;11:1036–45. doi: 10.1111/acel.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–88. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Shunmugavel A, Khan M, Chou PC, Singh I. Spinal cord injury induced arrest in estrous cycle of rats is ameliorated by S-nitrosoglutathione: novel therapeutic agent to treat amenorrhea. J Sex Med. 2012;9:148–58. doi: 10.1111/j.1743-6109.2011.02526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997;17:2653–7. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89:444–8. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Sako K, Yura S, Yonemasu Y. Cerebral blood flow and histopathological changes following permanent bilateral carotid artery ligation in Wistar rats. Exp Brain Res. 1992;89:87–92. doi: 10.1007/BF00229004. [DOI] [PubMed] [Google Scholar]
- Wang X, Xing A, Xu C, Cai Q, Liu H, Li L. Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-beta oligomerization in rats. J Alzheimers Dis. 2010;21:813–22. doi: 10.3233/JAD-2010-100216. [DOI] [PubMed] [Google Scholar]
- Won JS, Kim J, Annamalai B, Shunmugavel A, Singh I, Singh AK. Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J Alzheimers Dis. 2013;34:621–35. doi: 10.3233/JAD-121786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Wang M, Zhang W, Bai M, Du Y, Zhang Z, Li Z, Miao J. Neuronal damage, central cholinergic dysfunction and oxidative damage correlate with cognitive deficits in rats with chronic cerebral hypoperfusion. Neurobiol Learn Mem. 2014;109:7–19. doi: 10.1016/j.nlm.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Zaman K, Carraro S, Doherty J, Henderson EM, Lendermon E, Liu L, Verghese G, Zigler M, Ross M, Park E, Palmer LA, Doctor A, Stamler JS, Gaston B. S-nitrosylating agents: a novel class of compounds that increase cystic fibrosis transmembrane conductance regulator expression and maturation in epithelial cells. Mol Pharmacol. 2006;70:1435–42. doi: 10.1124/mol.106.023242. [DOI] [PubMed] [Google Scholar]
- Zeigler MM, Doseff AI, Galloway MF, Opalek JM, Nowicki PT, Zweier JL, Sen CK, Marsh CB. Presentation of nitric oxide regulates monocyte survival through effects on caspase-9 and caspase-3 activation. J Biol Chem. 2003;278:12894–902. doi: 10.1074/jbc.M213125200. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Liu YH, Yin J, Li HL, Wang Q, Wang JZ. Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J. 2006;20:1431–42. doi: 10.1096/fj.05-5223com. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Gong CX. From chronic cerebral hypoperfusion to Alzheimer-like brain pathology and neurodegeneration. Cell Mol Neurobiol. 2015;35:101–10. doi: 10.1007/s10571-014-0127-9. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]