

Abstract
ProLuCID, a new algorithm for peptide identification using tandem mass spectrometry and protein sequence databases has been developed. This algorithm uses a three tier scoring scheme. First, a binomial probability is used as a preliminary scoring scheme to select candidate peptides. The binomial probability scores generated by ProLuCID minimize molecular weight bias and are independent of database size. A modified cross-correlation score is calculated for each candidate peptide identified by the binomial probability. This cross-correlation scoring function models the isotopic distributions of fragment ions of candidate peptides which ultimately results in higher sensitivity and specificity than that obtained with the SEQUEST XCorr. Finally, ProLuCID uses the distribution of XCorr values for all of the selected candidate peptides to compute a Z score for the peptide hit with the highest XCorr. The ProLuCID Z score combines the discriminative power of XCorr and DeltaCN, the standard parameters for assessing the quality of the peptide identification using SEQUEST, and displays significant improvement in specificity over ProLuCID XCorr alone. ProLuCID is also able to take advantage of high resolution MS/MS spectra leading to further improvements in specificity when compared to low resolution tandem MS data. A comparison of filtered data searched with SEQUEST and ProLuCID using the same false discovery rate as estimated by a target-decoy database strategy, shows that ProLuCID was able to identify as many as 25% more proteins than SEQUEST. ProLuCID is implemented in Java and can be easily installed on a single computer or a computer cluster.
Graphical Abstract

Introduction
In recent years, shotgun proteomics1,2 has emerged as a robust and sensitive method for identifying and quantifying proteins in a complex biological sample and is now a preferred method for large-scale proteomic analyses .3,4 The strategy is based on proteolytic digestion of complex protein mixtures into peptides followed by identification of the peptides using tandem mass spectrometry (MS/MS). Peptide identifications can be used to identify their corresponding proteins using an automated database search. Recent improvements in MS technologies allow the acquisition of hundreds of thousands of MS/MS spectra over the course of one LC/MS/MS analysis5-7, and a large-scale shotgun proteomics project typically generates hundreds of millions of MS/MS spectra. Each of these spectra has to be correlated with the amino acid sequence of a peptide and corresponding protein. The sensitivity and efficiency of the database search program used is of critical importance in any high-throughput protein identification experiment.
There are five basic types of algorithms used to assign tandem mass spectra to peptide sequences: (1) cross-correlation methods that correlate experimental spectra with theoretical spectra.8-10 (2) methods using unambiguous “peptide sequence tags” derived from spectra that are used to search known sequences.11-15 (3) peptide de novo sequencing16-24 (4) probability-based matching that calculates a score based on the statistical significance of a match between an observed peptide fragment and those calculated from a sequence library25-32 and (5) blind or unrestricted modification search and spectra alignment-based algorithms.21,33-37 Cross-correlation approaches and probability-based matching approaches are the two most commonly used database searching strategies in large scale shotgun proteomics experiments. Among these algorithms, SEQUEST8 and Mascot25 are the two most widely used database search engines. Studies have shown that cross-correlation-based intensity-modeling methods have higher sensitivity while probability-based methods have higher specificity.32,38
The advent and commercialization of the high-performance mass spectrometer enables routine, wide-spread high resolution high mass accuracy measurements of peptides in proteomics 39. Early studies using this hybrid instrument have demonstrated a number of advantages including high mass accuracy, high resolution, large space charge capacity, and high dynamic range 5,40. Venable et al., evaluated the use of the LTQ-Orbitrap for the quantification of stable isotope-labeled peptides and showed a 4-5 fold improvement in the number and quality of the peptide ratio measurements compared with similar analyses done on the LTQ41. In addition, the high mass accuracy generated by the LTQ-Orbitrap hybrid mass spectrometer can be used to improve the confidence of peptide identification and database search speed. One strategy for doing this is to obtain high mass resolution data for all precursor ions during the full MS scan in the Orbitrap mass analyzer and then collect low resolution MS/MS spectra on those precursor peptides in the linear ion trap. An alternative approach that takes advantage of the LTQ-Orbitrap is to collect both high resolution MS and MS/MS spectra in the Orbitrap mass analyzer for peptide identification.
Some database search programs utilize a two-step scoring scheme. The first step is a preliminary scoring (Sp) step that is used to select a fixed number of candidate peptides which are then analyzed using a more sophisticated second step of scoring. This Sp step is important for the speed of the identification process since the final scoring algorithms are usually slower, making them impractical for scoring every candidate sequence. One common method for Sp scoring is to use the number of shared peaks to select the final candidates. This is done by multiple algorithms, including the hypergeometric probability based PEP_PROBE 29, OMSSA 31 and the central limit theorem based PEP_PROBE 32. However, the “number of shared peaks” approach may not work well for a low quality spectrum, especially when the fragmentation is poor. Alternatively, the preliminary score (Sp) of SEQUEST is an empirically derived score that restricts the number of sequences analyzed in the correlation analysis. Sp sums the peak intensity of fragment ions matching the predicted sequence ions and accounts for the continuity of an ion series and the length of a peptide. The original score is:
| (1) |
where the first term in the product is the sum of ion abundances of all matched peaks, m is the number of matches, β is a ‘reward’ for each consecutive match of an ion series (for example, 0.075), ρ is a ‘reward’ for the presence of an ammonium ion (for example 0.15) and L is the number of all theoretical ions of an amino acid sequence. The final scoring uses one of the following two methods to measure closeness of fit between spectra and peptide sequences: the first method uses a shared peak model to generate a quantitative measure of the fit, while the second method uses fragment ion frequency to generate the probability the sequence and spectrum are the best fit 32. Because the final scoring is usually more sophisticated and sensitive than the preliminary scoring, the final scoring method would ideally be applied to each candidate peptide rather than a limited number of them.
It is well known that the results of an unfiltered database search includes a large number of false positive identifications from random hits to the database. Post-database search filtering programs, such as DTASelect 42,43, PeptideProphet, 44 and Search Engine Processor 45 are essential for the optimal separation of true peptide/protein hits from random hits. For a peptide to be successfully identified by a database search algorithm, it has to pass the following three tests: (1) it must be ranked high enough in the Sp scoring to be selected for the final scoring, (2) it must be assigned the top rank during the final scoring, and (3) its score or scores have to be high enough to pass the post-search filtering criteria 46. The major challenge to improvement of the overall performance of a database search algorithm is how to increase the sensitivity of searches while maintaining adequate discrimination between correct answers and false positives.
In this paper, we present ProLuCID, an MS/MS-based database search program with enhanced peptide identification sensitivity and specificity relative to SEQUEST. ProLuCID uses a three tiered scoring scheme to maximize the sensitivity of database searching. For its Sp scoring method, ProLuCID computes a binomial probability score for each candidate peptide with a calculated mass that matches a precursor mass within a user specified tolerance. Then, based on the binomial probability scores, it selects a user-specified number of candidate peptides for final scoring (default = 500) that are least likely to be random hits. For each candidate peptide selected for further analysis, ProLuCID calculates a modified cross-correlation score (XCorr) and then further generates another score (Z score) based on the distribution of the XCorr of all final candidate peptides for that spectra. This three-tiered scoring scheme gives ProLuCID significant higher sensitivity and specificity than SEQUEST. Here we show that for low mass accuracy MS/MS data, the cross-correlation-based Z score outperforms the binomial probability score in making correct spectral assignments, while the binomial probability score performs better with high mass accuracy tandem mass spectra MS/MS data.
The ProLuCID software and data used in this paper can be downloaded at http://fields.scripps.edu/downloads.php and http://fields.scripps.edu/published/ProLuCID respectively.
Experimental Section
Sample preparation
A variety of samples and instrument platforms were used to demonstrate the improvement in identification, regardless of sample complexity or instrument sensitivity. Samples of varying degrees of complexity were used in this study: a mixture of 17 known proteins, a human saliva sample, rat brain sample, human cell lysate, and a protein fractionated human cell lysate. The 17 protein mixture sample was used to assess the sensitivity and the specificity of ProLuCID and SEQUEST scores, while the other more complex samples were used to demonstrate the sensitivity improvement in protein identification with samples of medium to high complexity.
HEK 293 Cells
Standard HEK293 cell lysate was prepared from HEK cells grown in Dulbecco's modified Eagle's medium (D-MEM) with 10 % fetal bovine serum (FBS) supplemented with penicillin and streptomycin. Cells were grown (37 °C/5 % CO2) to approximately 80 % confluence in tissue culture flasks. Cells were washed twice with DPBS, scrapped from flasks, supplemented with protease inhibitor cocktail (Roche) and lysed by sonication. Protein concentration was determined by BCA assay. Standard samples were kept at -80 °C until use.
Protein Fractionation
HEK lysate was submitted to protein based fractionation by addition of organic solvent into ten protein fractions, effectively reducing the sample complexity. Protein pellets were washed with acetone and digested with trypsin. Dried pellets were dissolved in 8 M urea/100 mM Tris, pH 8.5. Proteins were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) and alkylated with 10 mM iodoacetamide (Sigma-Aldrich). Proteins were digested overnight at 37 °C in 2 M urea/100 mM Tris, pH 8.5, 1 mM CaCl2 with trypsin (Promega) in a ratio of 1:100 (enzyme:protein). Digestion was stopped with formic acid, 5 % final concentration. Debris was removed by centrifugation.
For the saliva and the rat brain samples, about 200 micrograms of proteins were solublized with 8 M urea/Invitrosol (Invitrogen, Calsbad, CA), reduced with 10 mM dithiothreitol, alkylated with 10 mM iodoacetomide, diluted with 4 volumes of 100 mM Tris-HCl, and then digested with trypsin overnight. After digestion, the pH was adjusted to ~ 2.5 using 90% formic acid. Sixty micrograms of protein digest from each sample was analyzed by MudPIT.
Multidimensional Protein Identification Technology
Digested proteins were pressure-loaded onto a fused silica capillary column packed with 3 cm of 5-μm Partisphere strong cation exchanger (SCX, Whatman, Clifton, NJ) and 3 cm of 5-μm Aqua C18 material (RP, Phenomenex, Ventura, CA) with a 2 μm filtered union (UpChurch Scientific, Oak Harbor, WA) attached to the SCX end. The column was washed with buffer containing 95% water, 5% acetonitrile, and 0.1% formic acid. After desalting, a 100-μm i.d. capillary with a 5-μm pulled tip packed with 10 cm 3-μm Aqua C18 material was attached to the filter union, and the entire split-column was placed inline with an Agilent 1100 quaternary HPLC (Agilent, Palo Alto, CA) and analyzed using a modified 12-step separation procedure described previously 2. Three buffer solution were used: 5% acetonitrile/0.1% formic acid (buffer A); 80% acetonitrile/0.1% formic acid (buffer B), and 500 mM ammonium acetate/5% acetonitrile/0.1% formic acid (buffer C). The first step consisted of a 100 min gradient from 0 to 100% buffer B, Steps 2-11 had the following profile: 3 min of 100% buffer A, 5 min of X% buffer C, a 100 min gradient from 15 to 45% buffer B. The 5 min buffer C percentage (X) were 5, 10, 15, 20, 25, 30, 35, 40, 55, and 75%, respectively, for steps 2-11. In the final step, the gradient contained 3 min of 100% buffer a, 20 min of 100% buffer C, a 10 min gradient from 0 to 15% buffer B, and a 107 min gradient from 15 to 100% buffer B. As peptides were eluted from the microcapillary column, they were electrosprayed directly into an LTQ or LTQ-Orbitrap mass spectrometer (Thermo-Fisher, Palo Alto, CA) with the application of a distal 2.4-kV spray voltage. A cycle of one full scan mass spectrum (400-1400 m/z) followed by 8 data dependent MS/MS spectra at a 35% normalized collision energy was repeated continuously throughout each step of the multidimensional separation.
Database Search
The data for the17 protein mix, the human saliva sample and the rat brain sample were searched against a database with sequences of the 17 proteins added to a S. pombe protein FASTA database (http://www.sanger.ac.uk/Projects/S_pombe/protein_download.shtml, release date of March 3, 2005), the IPI human protein FASTA database (version 3.06 release date of May 10, 2005), and the IPI rat protein FASTA database (version 3.08 release date of July 12, 2005), respectively. Each protein database was concatenated with reversed sequences of all the proteins to estimate false positive rate. ProLuCID database searches were performed with precursor ion mass tolerance of 3 amu for low accuracy data or between 5 and 50 ppm for FTMS data, while fragment ion mass tolerances were 0.4 amu for low-resolution data and 30 ppm for calculation of the high resolution probability score calculated for FT-MS/MS data. All searches considered a static modification of 57.0215 on cysteine due to carboxyamidomethylation. The database search was not restricted by enzymatic specificity. Each dataset was searched twice, once with SEQUEST and once with ProLuCID, and the search results were directly compared. Similar database searches (precursor ion mass tolerance 3 amu and no enzyme restriction) were done with SEQUEST, MASCOT, XTANDEM, OMSSA on the 17 protein mix dataset for sensitivity and specificity comparison. The raw and processed datasets are available at http://fields.scripps.edu/published/ProLuCID/.
Theory
ProLuCID utilizes a three tiered scoring scheme. It first selects candidate peptides (500 by default) for final scoring based on a binomial probability score. This binomial probability score is computed for each peptide in the protein database that has a calculated mass within the precursor mass ± user-defined mass tolerance. It then computes an XCorr and a Z score for each candidate peptide that is selected for final scoring. Previous studies have shown that the distribution of matching fragment ions between a set of candidate peptides and an experimental spectrum can be approximated by a Poisson distribution 20,29,31. As shown in figure 1, the number of fragment ions that match an experimental spectrum also fits a binomial distribution very well. The binomial distribution is the discrete probability distribution of the number of successes in a sequence of n independent yes/no experiments, each of which yields success with probability (p). Such a success/failure experiment is also called a Bernoulli experiment or Bernoulli trial. We consider the testing of each theoretical peak as a Bernoulli trial and compute the probability of a peptide with at least m random matches with the formula (1):
| (1) |
where n is the number of theoretical peaks of the candidate peptide tested, which is determined by the peptide length together with the miminum and maximum m/z in the spectrum; m is the number of theoretical peaks that match to a peak in the exprimental spectrum and is guaranteed not greater than n; p is the probability that any fragment ion matches a peak in the spectrum, as determined by the mass tolerance for a fragment ion match and the density and distribution of peaks in the experimental spectrum. The binomial probability score P(x>=m) is the probability of getting m or more matches when n theoretical peaks are tested. By design, the binomial probability score computed by ProLuCID is database independent and is solely dependent on characteristics of the spectrum and the peptide sequence.
Figure 1.

Distribution of number of fragment ion matched to a tandem mass spectrum of all candidate peptides (blue line) a protein database. The protein FASTA database contains amino acid sequences of the 17 proteins, all Pombe proteins and the reverse copy of each protein (10006 entries in total). The fit curve (pink line) is a binomial distribution B (22, 0.1391).
The second ProLuCID score is referred to as XCorr and is very similar to the SEQUEST XCorr. It is a cross-correlation of the experimental and theoretical spectra.
The correlation is processed and averaged to remove the periodic noise in the interval (-75 to 75). Unlike the SEQUEST cross-correlation procedure which assigns an intensity of 50 to the monoisotopic peak of each major peak series and an intensity of 25 to a window of 1 amu around the major peak, ProLuCID uses averagine 47 to model the isotopic distribution of each major ion peak based on it mass. Based on the averagine table, any isotopic peaks within the isotopic envelope that have at least 20% of the intensity of the base peak (i.e., the most intense peak) are assigned an intensity that is proportional to their theoretical intensity. In order to keep the ProLuCID XCorr comparable to the SEQUEST XCorr, we assign the intensity of the base peak in the isotopic envelope of each major fragment ion to 50 and the intensity of each minor peak (i.e., a ion, z ion, b loss of H2O, b loss of NH3 for CID spectra) to 10 as is done in SEQUEST.
In addition to the preliminary score and XCorr, ProLuCID computes a third score (Z score) for each final candidate peptide. For each spectrum, there should only be one correct answer and all the other candidate peptides are considered random hits. We have found that the distribution of XCorr's for the top 500 peptide hits to each spectrum is very close to a normal distribution with the true hit being an obvious outlier and statistically significantly different from the other final candidates. There are many ways to detect outliers from normal distributions and the Z score of Grubbs’ test 48 is the method implemented in ProLuCID. The Z score is calculated as the difference between the outlier and the mean divided by the standard deviation SD (eq. 2). A large Z score means that the XCorr of the top hit is significantly different from the other hits and the peptide is more likely to be a true hit.
| (2) |
X is the XCorr of the top hit, μ is the mean XCorr of all the final candidate peptides and n is the number of final candidate peptides.
Results and Discussion
Overview of ProLuCID
We have developed ProLuCID, a new database search algorithm for peptide identification that is highly flexible, efficient, and sensitive. ProLuCID is implemented in Java 1.6 and can be run on either a single CPU or multi-node computing cluster with 1.6 or later version Java. With Java multithreading technology, ProLuCID users can specify number of compute cores to be used to take advantage of multi-core architectures that come with most modern computers. It is can be used with protein FASTA databases or pre-processed databases for faster search speed. ProLuCID is also able to perform efficient and flexible differential modification searches and is capable of taking advantage of the high mass accuracy data generated by the latest instrumentation. These features of ProLuCID are described in greater detail below.
Using binomial probability as preliminary score (Sp to improve sensitivity
The goal of a tandem mass spectral database search is to identify the best peptide sequence match for a spectrum. The ProLuCID algorithm uses a three-tiered scoring scheme to assess the quality of a match between a spectrum and a peptide amino acid sequence from a protein database. First, ProLuCID uses a binomial probability score (Sp)to distinguish random matches and select peptide candidates for final cross-correlation scoring. Although the cross-correlation score provides higher sensitivity than the binomial probability score, it is computationally expensive (i.e., slow) and thus not practical for use in the initial scoring scheme. Instead, we select a user-defined number (500 by default) of candidate peptides for final scoring based on binomial probability scores. It is worth noting that the computation of exact binomial probability score is also a very slow process due to the computation of the factorials and exponentiations (see equation 1), thus ProLuCID uses an approximation method to compute the scores. A lookup table is calculated when the program starts and the approximate probability score can be retrieved based on the number of matched peaks, the number of peaks tried and the fraction of the region from the minimum m/z to maximum m/z in the tandem mass spectrum that are considered positive (the p in equation 1). In order to use the lookup table, the value of p is rounded and keeps only two significant digits so we can map any p to an integer between 1 and 100.. This approximation also make it feasible for ProLuCID to use the binomial probability score as a preliminary scoring method used for all candidate peptides within a given mass tolerance rather than just a final list of 100- 200 peptide candidates as is done in other database search programs. The advantage of using a more sophisticated scoring function as the preliminary scoring routine can be seen in figure 2. Based on the 17 protein mix dataset, the SEQUEST Sp score gives 5338 correct spectrum assignments the while ProLuCID binomial probability score gives 6353 correct spectrum assignments. Based on this result, we can conclude that the approximate binomial probability score displays better sensitivity than SEQUEST Sp score.
Figure 2.

Number of correct spectrum assignments by ProLuCID and SEQUEST XCorr and Sp scores. BC for both XCorr rank and Sp rank are correct; XC for XCorr rank is correct and Sp rank is incorrect; SPC for Sp rank is correct and XCorr rank is incorrect; FP for top hits on the reverse sequences of the 17 proteins. These results are based on a 6-step MudPIT with 75866 spectra. The ProLuCID XCorr outperforms SEQUEST XCorr in terms of number of correct spectrum assignments (7299 vs 6974); The ProLuCID Sp scores (binomial probability score) work better than SEQUEST Sp scores (6353 vs 5338); and ProLuCID XCorr gives more true hits the top rank than ProLuCID Sp (7299 vs 6353).
Matching the isotopic distribution of fragment ions
The second score generated by ProLuCID is a measure of cross-correlation between the experimental and theoretical spectra for a peptide and is referred to as XCorr. In SEQUEST, a theoretical spectrum is generated from predicted fragment ions for each peptide sequence (b- and y-ions for CID and c- and z-ions for ETD). In the theoretical spectrum, the main ion series products are assigned an abundance of 50, a window of 1 amu around the main fragment is assigned an intensity of 25, and water and ammonia losses are assigned an intensity of 10. The theoretical and normalized experimental spectra are then cross-correlated to obtain similarities between the spectra. In contrast to SEQUEST, ProLuCID models the isotope distribution of each fragment ion in order to generate a more realistic theoretical spectrum for cross-correlation. Theoretical isotopic abundance distributions for proteins and peptides were created using a look-up table of 150 averagine theoretical isotopic distributions with monoisotopic mass values for multiples of 500 Da up to 75,000 Da with all abundance distributions in the look-up table created by Mercury47. ProLuCID uses the averagine table to closely model the isotopic distribution of the fragment ions. This modification makes the distribution of ProLuCID XCorr of decoy hits closer to a normal distribution, and the score itself becomes more discriminative (Figure 3). Importantly, the benefits of modeling the isotope distribution are realized even for low-resolution LTQ data in which the charge states cannot be determined.
Figure 3.

Histogram of SEQUEST and ProLuCID XCorr scores, separated into true hitss and reverse hits, showing that the XCorr score generated by ProLuCID are more discriminative than those generated by SEQUEST, because ProLuCID closely models fragment ion isotopic distributions.
Since ProLuCID can be configured to output both the binomial probability score and XCorr for each candidate peptide, we can determine which score is more sensitive in identifying target peptides by comparing the number of true peptides (from the 17 protein mix) that are ranked as the top hit by each scoring scheme. From Figure 2, we can see that XCorr performs better than preliminary scoring using either the ProLuCID binomial probability score or the SEQUEST Sp score. The ProLuCID XCorr identifies more spectra correctly than the ProLuCID's binomial probability score (7299 vs 6353) while the ProLuCID probability score gets more correct spectral assignments than the SEQUEST Sp score (6353 vs 5201). Based on these results, we can conclude that ProLuCID's binomial probability is a better score than SEQUEST's Sp score, and ProLuCID's XCorr is a better score than ProLuCID's binomial probability score. The combination of binomial probability preliminary scoring and the modeling of the isotopic distribution of fragment ions make ProLuCID more sensitive than SEQUEST in terms of correct spectrum assignments (7299 vs 6974), regardless of the specificity of the scores.
Statistical Z score improves the specificity
In addition to the binomial probability and cross-correlation scores, ProLuCID outputs a Z score for each peptide hit. The Z score is a dimensionless score derived by subtracting the population mean from an individual raw score and then dividing the difference by the population standard deviation. It reveals how many units of the standard deviation a case is above or below the mean. Unlike XCorr, which is independent of database size and reflects the quality of the match between the experimental spectrum and the peptide sequence, the ProLuCID's Z score is database-dependent and reflects the quality of the match relative to near misses. A higher Z score indicates that the peptide hit is more likely to be a correct match to the spectrum.
Traditionally, filtering of database search results by DTASelect used threshold cutoffs for XCorr and DeltaCN, where DeltaCN is the difference between the top hit XCorr and the second best hit XCorr divided by the XCorr of the top hit. In the latest version of DTASelect (DTASelect2),43 these two measurements are combined using a discriminant function that dynamically sets the XCorr and DeltaCN values in order to achieve a user-specified false discovery rate. For either case, high confidence spectrum assignments generally have both high XCorr and high DeltaCN scores. Since the XCorr shows positive correlation across charge states (i.e. XCorr values increase for higher charge state spectra)49, different cutoffs are usually applied to assignments with different charge states. DeltaCN measures the difference between the best hit and the second best hit and has proven to be a very good measure for separating true hits from false . However, in some cases the sequence corresponding to the second highest XCorr might have very high sequence similarity to the top hit, making the DeltaCN value very small. Thus, even though the identification itself may be reliable, it would be discarded by DTASelect due to its similarity to the 2nd best hit. Tang et al 34 used the distance score which is defined as the difference between the highest score and the seventh highest score for each MS/MS spectrum. The distance score provides a measure of the separation between the highest scoring peptide and the pack of wrong peptides. The larger the distance score, the larger the probability that the highest scoring peptide is indeed a legitimate answer. The distributions of distance scores for correct peptides and incorrect peptides were found to be approximately Poisson. ProLuCID's Z score provides a statistical measurement to indicate how significant the difference between the best match and the rest of the matches to the same spectrum are. This measure provides an effective way of distinguishing the true hits from the random hits using a strong statistical foundation.
A common method for visualizing and comparing discrimination ability is the receiver operating characteristic (ROC) plot,50 in which one can read the false positive level that must be tolerated in order to obtain any given true positive level. In our case, we consider any identification that matches a peptide sequence from any of the 17 proteins as a true positive, and any identification that matches a peptide sequence from a reversed protein as a false positive. The ROC curves in figure 4A-4C clearly illustrate the improvement in sensitivity and specificity of the ProLuCID XCorr and Z score compared with the SEQUEST XCorr. Figure 4A is a typical ROC curve with the area under the curve being 0.89, 0.91 and 0.96 for the SEQUEST XCorr, ProLuCID XCorr and ProLuCID Z score, respectively. Based on these result, we can conclude that ProLuCID XCorr is a more discriminative score than SEQUEST XCorr and that the ProLuCID Z score shows significantly improved specificity over both SEQUEST and ProLuCID XCorr. Figure 4C is a modified ROC curve that plots the number of true hits against the false positive faction. These figures clearly show that ProLuCID XCorr and Z score have better sensitivity and specificity than SEQUEST XCorr and that the ProLuCID Z score shows better specificity than ProLuCID XCorr. We implemented Z score in SEQUEST and it show significant better specificity than SEQUEST XCorr (Figure 4).
Figure 4.
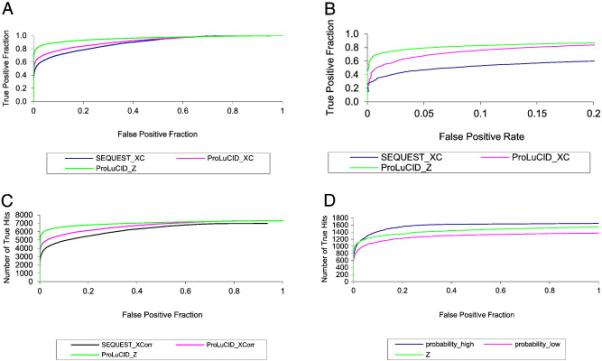
ROC curves of ProLuCID and SEQUEST scores. A. Typical ROC curves of SEQUEST XCorr, ProLuCID XCorr and ProLuCID Z score. B. Modified ROC curves, showing true positive fraction as a function of false positive rate. C. Plots of number of true hits against false positive fraction of SEQUEST XCorr, ProLuCID XCorr and ProLuCID Z score. D. Plots of number of true hits against false positive fraction of ProLuCID high mass accuracy probability score, low mass accuracy probability score and Z score.
It is also worth noting that the ProLuCID Z score distributions for charge +2 and charge +3 decoy hits as shown in figure 5 are very similar, indicating that the ProLuCID Z score is largely charge state independent. It is important for practical applications to know the true and false positive rates at given score thresholds. Figure 6 plots the false positive rate against ProLuCID Z scores. In this dataset, the spectrum assignment false positive rate is 10% at Z score 4.42, 5% at Z score 4.67 and 1% at Z score 5.28, respectively. Although the distribution of ProLuCID Z scores shows relatively small variation between different MudPIT runs, it is still dataset dependent to some degree.
Figure 5.

Histograms of ProLuCID Z scores of the true hits and decoy hits, showing good separation between the true hits and decoy hits, and that the distributions of the Z scores of the decoy hits of charge +2 and charge + 3 spectra are very similar.
Figure 6.

Plot of ProLuCID Z score as a function of false positive rate on the 17 protein mixture dataset.
Performance test with biological samples of medium and high complexity
In order to test the performance of ProLuCID on data from more complex samples, we performed 12-step MudPIT experiments with a human salivary sample and a rat brain whole cell lysate sample. Human saliva is a biological fluid with a medium level of complexity. In a large scale saliva protein cataloging project that combined results from over 200 MudPIT experiments, we previously identified about 1500 proteins with high confidence (<= 1% false positive rate). With a single 12-step MudPIT experiment using LTQ-Orbitrap, we identified 372 proteins with ProLuCID and 300 proteins with SEQUEST using the same DTASelect filtering criteria (at least two peptides per protein, each peptide has at least one tryptic terminus and 5% spectrum level false positive rate). From the results in table 1, we find that ProLuCID identifies more proteins than SEQUEST at similar false positive rate. On the more complex sample of rat brain whole cell lysate, we identified about 3345 proteins with ProLuCID compared with 2991 with SEQUEST (table 2), with false positive rates of 1.23% and 1.44% respectively. Thus, the improvements on the scoring methods used in ProLuCID versus SEQUEST leads to higher confidence in protein identifications. In table 3, we show that ProLuCID results show higher sequence coverage, peptide counts and spectrum counts than SEQUEST results.
Table 1.
Number of protein identified in the saliva sample with SEQUEST and ProLuCID after DTASelect filtering
| Search Program | Forward Hits | Decoy Hits | False Positive Rate |
|---|---|---|---|
| SEQUEST | 300 | 7 | 2.33% |
| ProLuCID | 372 | 7 | 1.89% |
Table 2.
Number of proteins identified in the rat brain sample with SEQUEST and ProLuCID after DTASelect filtering
| Search Program and filter options | Forward Hits | Decoy Hits | False Positive Rate |
|---|---|---|---|
| SEQUEST_XD | 2991 | 43 | 1.44% |
| ProLuCID_XD | 3330 | 51 | 1.53% |
| ProLuCID_Z | 3345 | 41 | 1.23% |
A twelve step MudPIT dataset with 139277 spectra was searched with SEQUEST and ProLuCID respectively and DTASelect2 was used to get the final protein lists. SEQUEST_XD for SEQUEST search and XCorr and DeltaCN for DTASelect filtering; ProLuCID_XD for ProLuCID search with XCorr and DeltaCN for DTASelect2 filtering; ProLuCID_Z for ProLuCID search and Z score only for DTASelect filtering (with additional –noxc –nodcn –sp options). DTASelect2 options –p 2 –y 1 –fp 0.05 were used for all three, and additional options (–noxc –nodcn –sp) for ProLuCID_Z to use Z score only for filtering. A protein was considered identified if it has at least two peptides pass the 5% PSM (peptide-spectrum-match) false positive rate filter and each peptide has at least one tryptical terminus.
Table 3.
Average number of peptide count, spectrum count and sequence coverage of 1000 proteins with highest sequence coverage identified in the rat brain sample with SEQUEST and ProLuCID after DTASelect2 filtering
| Algorithm | Peptide count | Spectrum count | Sequence Coverage |
|---|---|---|---|
| SEQUEST | 7.53 | 29.95 | 22.79% |
| ProLuCID | 8.50 | 39.67 | 24.65% |
Comparison with Comet and SEQUEST on Hela sample
We compared ProLuCID with Comet and SEQUEST by searching triplicate data from a Hela sample. The tandem mass spectra were searched against UniProt human database (downloaded on November 08, 2010). To estimate peptide probabilities and FDRs accurately, we used a target/decoy database containing the reversed sequences. The search space included fully tryptic peptide candidates that fell within the mass tolerance window with maximum three internal miscleavage constraints. Carbamidomethylation (+57.02146 Da) of cysteine was considered as a static modification. The validity of peptide/spectrum matches (PSMs) was assessed in DTASelect, using spectrum level FDR less than 1% and precursor delta mass threshold of 10 ppm.
Table 4 shows that ProLuCID identified more proteins than both SEQUEST and Comet based on all protein, peptide and spectrum average counts from triplicates. ProLuCID identified 12% more spectra than SEQUEST, and 5% more than Comet.
Table 4.
Search result comparison of ProLuCID, SEQUEST, and Comet. Triplicates of Hela sample were searched against the same protein database. Search results were filtered with the same DTASelect parameters. The results were averaged from triplicates.
| Algorithm | Protein count | Peptide count | Spectrum count |
|---|---|---|---|
| SEQUEST | 3798 | 28075 | 48217 |
| Comet | 3921 | 29810 | 51202 |
| ProLuCID | 3973 | 31165 | 53797 |
We also compared ProLuCID and Comet with half-tryptic (considering candidate peptides with at least one tryptic end) parameter. Comparing to fully tryptic search, the ProLuCID identified more spectra than Comet by even bigger difference. ProLuCID identified 20% more spectra and 19% more peptides than the Comet.
High Resolution MS and MS/MS database searches
The LTQ-Orbitrap hybrid mass spectrometer combines high resolution and mass accuracy with fast scan rates and the flexibility of two different mass analyzers which provides the user with the opportunity to operate the instrument in different modes. One mode uses the Orbitrap mass analyzer to collect all spectra for an experiment, including both high resolution full MS scans of precursor ions and high resolution tandem mass spectra after peptide fragmentation. The major advantages of this approach are the high mass accuracy of the precursor ion which restricts the number of candidate peptides that need to be considered by the database search algorithm, and the high mass accuracy of the fragment ions which could to lead to more confident peptide and protein identifications, as well as PTM localization. The disadvantage of this strategy, however, is the lower scan rate of the Orbitrap compared with the LTQ, which would result in the collection of fewer tandem mass spectra and likely fewer peptide and protein identifications. Alternatively, the LTQ-Orbitrap can be used so that the full MS scans are collected by the Orbitrap while the LTQ is used to obtain low resolution MS/MS spectra. In this approach, high mass accuracy is obtained for precursor ions while low mass accuracy is obtained for fragment ions. The advantage of this mode is that the high precursor mass accuracy can be used to reduce the false positive rate and/or speed up database search while a large number of tandem mass spectra are collected by the LTQ. Importantly, ProLuCID is capable of handling all of these possibilities and can search spectra with either high or low mass accuracy for both precursor and fragment ions, including deisotoped and decharged high-resolution MS/MS spectra 51
ProLuCID allows the user to specify the precursor and fragment ion mass tolerance from 1 ppm to 1000 ppm. When high precursor mass accuracy is specified, ProLuCID can be configured to use a very narrow precursor mass tolerance to reduce the number of candidate peptides and thus speed up the search. In this case, however, the mass spectrometer may select and record the non-monoisotopic peaks (i.e., peptide ions containing one or more 13C atoms) for MS/MS fragmentation which can prevent these spectra from being identified when searches are restricted to small m/z windows. To address this problem, ProLuCID selects candidate peptides by assuming the precursor can be either the M+0 (mono), M+1 (with one 13C) M+2 (with two 13C), etc., isotopic peak. The number of isotopic peaks considered by ProLuCID can be specified by the user in the ProLuCID search parameter file. This approach significantly reduces the number of candidate peptides and speeds up the database search without missing spectra obtained from the fragmentation of non-monoisotopic peaks.
Additionally, ProLuCID can use a preprocessed database in which the peptides are sorted by mass and can improve the computational efficiency by more than 1000~2000% over SEQUEST if stringent precursor mass tolerance (e.g., 5 ppm) is used. The search speed improvement can be more dramatic for differential modification searches and largely depends on the database size, precursor mass tolerance, enzyme restriction, etc.
Another advantage of high resolution full MS spectra is the ability to correctly assign charge states to the precursor ions. For low resolution data, the charge state of the precursor ions cannot easily be determined for spectra with charge states higher than +1. When the charge state of a multiply charged precursor ion cannot be determined, the spectrum is typically searched against the database twice, once assuming a +2 charge state and then again assuming a +3 charge state. In this approach, spectra with charge states higher than +3 are always incorrectly assigned. With high resolution Orbitrap data, charge states can be assigned to over 90% of MS/MS spectra using the in-house algorithm RawXtract. This eliminates the need to guess the charge state of precursor ions and enables peptides with charge states of +4 or higher to be identified (Figure 7). ProLuCID models +1 fragment ions for +1 and +2 spectra, +1 and +2 fragment ions for +3 spectra, and fragment ions of charge state from +1 to the floor of (z + 2)/2 for spectra with precursor charge state +4 or higher, where z is the precursor charge state.
Figure 7.

An example high precursor charge (+4) peptide spectrum identified by ProLuCID.
For high resolution MS/MS data, ProLuCID allows users to specify fragment ion mass tolerance in terms of parts-per-million (ppm., e.g. 20 ppm). We collected high resolution tandem mass spectra in a 4-step MudPIT experiment with the 17 protein mix sample. The same set of tandem mass spectra were searched as high resolution data using 30 ppm fragment mass tolerance and low resolution data using 0.4 amu fragment mass tolerance. From figure 4D, we can see that the ProLuCID binomial probability score for a high fragment mass accuracy search shows better sensitivity and specificity than the binomial probability score and the Z score for a low fragment mass accuracy search. It is worth noting that the Z score is computed based on an XCorr with low fragment ion mass accuracy.
ProLuCID takes a similar approach as SEQUEST for differential or variable modification searches. Users need to specify the type of modification and the maximum number of modifications to be considered. However, unlike SEQUEST in which the maximum number of modification types is set to 3 and each modification or mass shift can only occur to a maximum 3 amino acid residues, ProLuCID allows users to specify as many differential modification types as desired and each modification type or mass shift can be applied to as many residues as expected to be possible. This provides users the opportunity to search for unexpected modifications at a relatively low computational cost. Of course, for any given protein database, search times will increase as more modifications are considered.
Conclusions
ProLuCID achieves enhanced sensitivity and specificity by using a binomial probability score as a preliminary score, an improved XCorr, and the implementation of a novel Z score. ProLuCID Z score shows significantly higher sensitivity and specificity than SEQUEST XCorr. For high resolution (Orbitrap) MS/MS data, the ProLuCID probability score outperforms Z score, while Z score performs better than ProLuCID probability score for low mass accuracy (LTQ) MS/MS data. We show for typical shotgun proteomics experiments, using DTASelect with the same false positive rate filter, ProLuCID usually identifies about 10% ~ 25% more proteins than SEQUEST does. The overall confidence of the identified proteins is improved due to significant increases in peptide count, spectrum count and sequence coverage.
Supplementary Material
Significance.
The manuscript describes ProLuCID, a new algorithm for peptide identification using tandem mass spectrometry and protein sequence databases. This algorithm uses a three tier scoring scheme. First, a binomial probability is used as a preliminary scoring scheme to select candidate peptides. The binomial probability scores generated by ProLuCID minimize molecular weight bias and are independent of database size. A modified cross-correlation score is calculated for each candidate peptide identified by the binomial probability. This cross-correlation scoring function models the isotopic distributions of fragment ions of candidate peptides which ultimately results in higher sensitivity and specificity than that obtained with the SEQUEST XCorr. Finally, ProLuCID uses the distribution of XCorr values for all of the selected candidate peptides to compute a Z score for the peptide hit with the highest XCorr. The ProLuCID Z score combines the discriminative power of XCorr and DeltaCN, the standard parameters for assessing the quality of the peptide identification using SEQUEST, and displays significant improvement in specificity over ProLuCID XCorr alone. ProLuCID is also able to take advantage of high resolution MS/MS spectra leading to further improvements in specificity when compared to low resolution tandem MS data. A comparison of filtered data searched with SEQUEST and ProLuCID using the same false discovery rate as estimated by a target-decoy database strategy, shows that ProLuCID was able to identify as many as 25% more proteins than SEQUEST.
Table 5.
Search result comparison of ProLuCID and Comet with half tryptic parameter. Triplicates of Hela sample were searched against the same protein database. Search results were filtered with same DTASelect parameters. The results were averaged from triplicates.
| Algorithm | Protein count | Peptide count | Spectrum count |
|---|---|---|---|
| Comet | 3940 | 28400 | 48008 |
| ProLuCID | 4124 | 33710 | 57569 |
Acknowledgment
The authors thank Rovshan Sadygov, Akira Motoyama, Cristian Ruse and other members of the Yates laboratory for fruitful discussions. The work is supported by NIH Grant 5U01 DE016267-04, 5R01 HL079442-04, R01 MH067880, P41 GM103533 and NHLBI contract # HHSN268201000035C to J.R.Y.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Link AJ, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. doi:10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 2.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI. Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol Biol. 2006;367:87–120. doi: 10.1385/1-59745-275-0:87. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., 3rd Protein Analysis by Shotgun/Bottom-up Proteomics. Chem Rev. 2013 doi: 10.1021/cr3003533. doi:10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsen JV, et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Olsen JV, et al. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics. 2009;8:2759–2769. doi: 10.1074/mcp.M900375-MCP200. doi:10.1074/mcp.M900375-MCP200 M900375-MCP200 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Second TP, et al. Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Anal Chem. 2009;81:7757–7765. doi: 10.1021/ac901278y. doi:10.1021/ac901278y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eng JK, Mccormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 9.Diament BJ, Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. J Proteome Res. 2011;10:3871–3879. doi: 10.1021/pr101196n. doi:10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. J Proteome Res. 2008;7:4598–4602. doi: 10.1021/pr800420s. doi:10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- 11.Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 12.Sunyaev S, Liska AJ, Golod A, Shevchenko A. MultiTag: multiple error-tolerant sequence tag search for the sequence-similarity identification of proteins by mass spectrometry. Anal Chem. 2003;75:1307–1315. doi: 10.1021/ac026199a. [DOI] [PubMed] [Google Scholar]
- 13.Tabb DL, Saraf A, Yates JR., 3rd GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. doi:10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank A, Tanner S, Bafna V, Pevzner P. Peptide sequence tags for fast database search in mass-spectrometry. J Proteome Res. 2005;4:1287–1295. doi: 10.1021/pr050011x. doi:10.1021/pr050011x. [DOI] [PubMed] [Google Scholar]
- 15.Tanner S, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. doi:10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 16.Shevchenko A, Chernushevich I, Wilm M, Mann M. De Novo peptide sequencing by nanoelectrospray tandem mass spectrometry using triple quadrupole and quadrupole/time-of-flight instruments. Methods Mol Biol. 2000;146:1–16. doi: 10.1385/1-59259-045-4:1. doi:1-59259-045-4-1 [pii] 10.1385/1-59259-045-4:1. [DOI] [PubMed] [Google Scholar]
- 17.Chen T, Kao MY, Tepel M, Rush J, Church GM. A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 2001;8:325–337. doi: 10.1089/10665270152530872. doi:10.1089/10665270152530872. [DOI] [PubMed] [Google Scholar]
- 18.Johnson RS, Taylor JA. Searching sequence databases via de novo peptide sequencing by tandem mass spectrometry. Mol Biotechnol. 2002;22:301–315. doi: 10.1385/MB:22:3:301. doi:MB:22:3:301 [pii] 10.1385/MB:22:3:301. [DOI] [PubMed] [Google Scholar]
- 19.Ma B, et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. doi:10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 20.Lu B, Chen T. A suffix tree approach to the interpretation of tandem mass spectra: applications to peptides of non-specific digestion and post-translational modifications. Bioinformatics. 2003;19(Suppl 2):II113–II121. doi: 10.1093/bioinformatics/btg1068. [DOI] [PubMed] [Google Scholar]
- 21.Searle BC, et al. High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal Chem. 2004;76:2220–2230. doi: 10.1021/ac035258x. doi:10.1021/ac035258x. [DOI] [PubMed] [Google Scholar]
- 22.Chi H, et al. pNovo: de novo peptide sequencing and identification using HCD spectra. J Proteome Res. 2010;9:2713–2724. doi: 10.1021/pr100182k. doi:10.1021/pr100182k. [DOI] [PubMed] [Google Scholar]
- 23.Chi H, et al. pNovo+: De Novo Peptide Sequencing Using Complementary HCD and ETD Tandem Mass Spectra. J Proteome Res. 2013;12:615–625. doi: 10.1021/pr3006843. doi:10.1021/pr3006843. [DOI] [PubMed] [Google Scholar]
- 24.Frank A, Pevzner P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- 25.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 27.Fenyo D, Qin J, Chait BT. Protein identification using mass spectrometric information. Electrophoresis. 1998;19:998–1005. doi: 10.1002/elps.1150190615. doi:10.1002/elps.1150190615. [DOI] [PubMed] [Google Scholar]
- 28.Zhang N, Aebersold R, Schwikowski B. ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics. 2002;2:1406–1412. doi: 10.1002/1615-9861(200210)2:10<1406::AID-PROT1406>3.0.CO;2-9. doi:10.1002/1615-9861(200210)2:10<1406::AID PROT1406>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Sadygov RG, Yates JR., 3rd A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal Chem. 2003;75:3792–3798. doi: 10.1021/ac034157w. [DOI] [PubMed] [Google Scholar]
- 30.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. doi:10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 31.Geer LY, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 32.Sadygov R, Wohlschlegel J, Park SK, Xu T, Yates JR., 3rd Central limit theorem as an approximation for intensity-based scoring function. Anal Chem. 2006;78:89–95. doi: 10.1021/ac051206r. [DOI] [PubMed] [Google Scholar]
- 33.Searle BC, et al. Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J Proteome Res. 2005;4:546–554. doi: 10.1021/pr049781j. [DOI] [PubMed] [Google Scholar]
- 34.Tang WH, et al. Discovering known and unanticipated protein modifications using MS/MS database searching. Anal Chem. 2005;77:3931–3946. doi: 10.1021/ac0481046. [DOI] [PubMed] [Google Scholar]
- 35.Bandeira N, Tsur D, Frank A, Pevzner PA. Protein identification by spectral networks analysis. Proc Natl Acad Sci U S A. 2007;104:6140–6145. doi: 10.1073/pnas.0701130104. doi:0701130104 [pii] 10.1073/pnas.0701130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Na S, Bandeira N, Paek E. Fast multi-blind modification search through tandem mass spectrometry. Mol Cell Proteomics. 2012;11:M111 010199. doi: 10.1074/mcp.M111.010199. doi:10.1074/mcp.M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han X, He L, Xin L, Shan B, Ma B. PeaksPTM: Mass spectrometry-based identification of peptides with unspecified modifications. J Proteome Res. 2011;10:2930–2936. doi: 10.1021/pr200153k. doi:10.1021/pr200153k. [DOI] [PubMed] [Google Scholar]
- 38.Kapp EA, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. doi:10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 39.Hu Q, et al. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 40.Yates JR, Cociorva D, Liao L, Zabrouskov V. Performance of a linear ion trap-Orbitrap hybrid for peptide analysis. Anal Chem. 2006;78:493–500. doi: 10.1021/ac0514624. doi:10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
- 41.Venable JD, Wohlschlegel J, McClatchy DB, Park SK, Yates JR., 3rd Relative quantification of stable isotope labeled peptides using a linear ion trap-orbitrap hybrid mass spectrometer. Anal Chem. 2007;79:3056–3064. doi: 10.1021/ac062054i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cociorva D, D LT, Yates JR. Validation of tandem mass spectrometry database search results using DTASelect. Current protocols in bioinformatics / editoral board, Andreas D. Baxevanis ... [et al.] 2007 doi: 10.1002/0471250953.bi1304s16. Chapter 13, Unit 13 14, doi:10.1002/0471250953.bi1304s16. [DOI] [PubMed] [Google Scholar]
- 44.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 45.Carvalho PC, et al. Search engine processor: Filtering and organizing peptide spectrum matches. Proteomics. 2012;12:944–949. doi: 10.1002/pmic.201100529. doi:10.1002/pmic.201100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu B, Xu T, Park SK, Yates JR., 3rd Shotgun protein identification and quantification by mass spectrometry. Methods Mol Biol. 2009;564:261–288. doi: 10.1007/978-1-60761-157-8_15. doi:10.1007/978-1-60761-157-8_15. [DOI] [PubMed] [Google Scholar]
- 47.Rockwood AL, VanOrden SL, Smith RD. Ultrahigh resolution isotope distribution calculations. Rapid Communications in Mass Spectrometry. 1996;10:54–59. [Google Scholar]
- 48.Grubbs FE. Procedures for Detecting Outlying Observations in Samples. Technometrics. 1969;11:1. [Google Scholar]
- 49.MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., 3rd A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem. 2003;75:6912–6921. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 50.Swets JA. Measuring the accuracy of diagnostic systems. Science. 1988;240:1285–1293. doi: 10.1126/science.3287615. [DOI] [PubMed] [Google Scholar]
- 51.Carvalho PC, et al. YADA: a tool for taking the most out of high-resolution spectra. Bioinformatics. 2009;25:2734–2736. doi: 10.1093/bioinformatics/btp489. doi:10.1093/bioinformatics/btp489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.