

Abstract
Immune cells have important roles during disease and are known to contribute to secondary, inflammation-induced injury after traumatic brain injury. To delineate the functional role of macrophages during traumatic brain injury, we depleted macrophages using transgenic CD11b-DTR mice and subjected them to controlled cortical impact. We found that macrophage depletion had no effect on lesion size assessed by T2-weighted MRI scans twenty-eight days after injury. Macrophage depletion resulted in a robust increase in proinflammatory gene expression in both the ipsilateral and contralateral hemispheres after controlled cortical impact. Interestingly, this sizeable increase in inflammation did not affect lesion development. We also showed that macrophage depletion resulted in increased proinflammatory gene expression in the brain and kidney in the absence of injury. These data demonstrate that depletion of macrophages in CD11b-DTR mice can significantly modulate the inflammatory response during brain injury without affecting lesion formation. These data also reveal a potentially confounding inflammatory effect in CD11b-DTR mice that must be considered when analyzing the effects of macrophage depletion in disease models.
Keywords: macrophage depletion, CD11b-DTR, traumatic brain injury, inflammation
1. INTRODUCTION
Immune cells have critical roles in the pathophysiology of traumatic brain injury (TBI), and immune cell-mediated damage is a major mechanism of secondary injury that occurs during the inflammatory response to TBI. The early inflammatory phase of TBI involves a coordinated response by numerous cell types including neurons, glia, and vascular cells, as well as recruited immune cells. Through both the activation of both resident and peripherally recruited immune cells, the site of brain injury is inundated with proinflammatory mediators that can exacerbate primary injury by inducing neuron death, blood-brain barrier breakdown, and edema formation.
Many proinflammatory mediators are known to exacerbate tissue damage, and the presence of certain proinflammatory cytokines is associated with poor outcome in patients with TBI (Ferreira et al., 2014). Consistent with these findings, genetic deletion or inhibition of many proinflammatory mediators like IL-1β, NOX, and TLRs are protective during brain injury (Ahmad et al., 2013; Clausen et al., 2011; Dohi et al., 2010; Tehranian et al., 2002). In contrast, deletion or inhibition of other proinflammatory cytokines like TNF-α and IL-6 have been shown to exacerbate injury and impair neurological function (Clausen et al., 2011; Quintana et al., 2007; Scherbel et al., 1999). It is evident that many of these cytokines have complex and variable, time-dependent effects that can alter the pathophysiology of TBI. Whether or not specific signaling pathways and cell types in the inflammatory response are inherently protective, detrimental, or a more variegated balance between both extremes is not well understood.
Macrophages and neutrophils are major secretors of inflammatory cytokines and are thought to significantly contribute to secondary injury through the production of proinflammatory mediators. Resident and peripheral immune cells can be recruited to injured tissue very rapidly, and neutrophils and monocytes have been shown to infiltrate the brain within minutes after injury (Roth et al., 2014). Immune cell expansion continues over the course of several days, and the early, innate immune cell response to TBI peaks around 1–3 days after the initial primary injury (Jin et al., 2012). Inhibiting leukocyte recruitment through genetic deletion of MCP1, its cognate receptor CCR2, or by blocking intercellular adhesion molecule 1 (ICAM-1) has been shown to have modest neuroprotective effects during traumatic brain injury (Hsieh et al., 2014; Knoblach and Faden, 2002; Semple et al., 2010). More recently, techniques to target specific immune cell types have been employed, and it has been shown that depletion of neutrophils using anti-Gr-1 antibody decreases lesion volume and edema after TBI (Kenne et al., 2012).
Macrophages are phenotypically heterogeneous and fall within a spectrum of classically activated macrophage (CAM) and alternatively activated macrophage (AAM) phenotypes, sometimes referred to as M1 and M2, respectively. CAMs are generally proinflammatory and promote inflammation by secreting various proinflammatory mediators, whereas AAMs are often thought to be involved in wound healing and are thought to have reparative and protective roles in many diseases. Microglia also exhibit phenotypic changes in response to chemokine and cytokine stimulation resulting in gene expression profiles that are analogous to the CAM and AAM phenotypes (Ponomarev et al., 2007). The presence of both CAM and AAM phenotypes have been identified after TBI, although their functional roles and how they affect the inflammatory process is largely unknown.
Attenuating the production of inflammatory cytokines through direct targeting of macrophage populations during TBI may have therapeutic potential to reduce the damaging effects of inflammation. Depletion models using a transgenic diphtheria toxin receptor or HSV-thymidine kinase under the control of the CD11b promoter (CD11b-DTR, CD11b-HSV-TK), as well as clodronate liposomes, have been utilized to deplete macrophages in order to gain a better understanding of their roles during the inflammatory and reparative phases of tissue injury (Cailhier et al., 2005; Heppner et al., 2005). Macrophage depletion has been shown to have neuroprotective effects during spinal cord injury and also suppresses inflammation in a cortical freeze injury model (Amankulor et al., 2009; Popovich et al., 1999). In contrast, macrophage depletion during stroke, and also during brain development, has been reported to have detrimental effects (Gliem et al., 2012; Ueno et al., 2013).
Although macrophages are thought to have a pathological role during TBI, strategies to specifically target macrophages in order to inhibit or modulate their phenotype are limited. In the current study, we examined the role of macrophages during TBI using transgenic CD11b-DTR mice. Here we determined the effect that macrophage depletion has on lesion development and the inflammatory response during controlled cortical impact. We also evaluated neuroinflammatory responses to macrophage depletion in the absence of injury to further characterize this model of macrophage depletion.
2. RESULTS
2.1. Depletion of macrophages during traumatic brain injury
To determine the role that macrophages have during the pathophysiology of traumatic brain injury, we used a transgenically expressed diphtheria toxin receptor with expression restricted to CD11b+ cells (CD11b-DTR). CD11b-DTR mice were injected with either PBS or diphtheria toxin (DT, 20 ng/g) 24 hours prior to controlled cortical impact, and then again 24 hours after injury. Previous reports have shown that similar doses of DT selectively deplete macrophages and circulating monocytes without affecting lymphocytes or neutrophils (Cailhier et al., 2005). To verify that macrophages were sufficiently depleted during injury, we isolated and analyzed peritoneal cells from wild type control (WT) and CD11b-DTR mice 48 hours after controlled cortical impact. Flow cytometric analysis of peritoneal cells showed that administration of diphtheria toxin resulted in a nearly complete depletion of CD45+CD11b+ leukocytes (Figure 1A) and F4/80+ macrophages (Figure 1B). To determine if DT administration depletes resident microglia in the absence of injury, we performed immunohistochemical analysis of brain sections in WT and CD11b-DTR mice using the microglia- and macrophage-specific Iba1 antibody (Figure 1C). Iba1 staining revealed no significant differences in the number of microglia in cortical and subcortical regions of mice that received DT in the absence of TBI (Table 1). We also analyzed the number of microglia and macrophages in the lesion 48 hr after TBI (Figure 2). There was an approximate 2-fold increase in the number of microglia in the TBI lesion of both WT and CD11b-DTR mice compared to uninjured mice (see Table 2). However, no significant decreases in Iba1+ microglia and macrophages were detected at the border of the TBI lesion in CD11b-DTR mice compared to controls (Figure 2).
Figure 1. Administration of diphtheria toxin depletes macrophages in CD11b-DTR mice during traumatic brain injury.

Flow cytometric analysis of (A) CD11b+ cells, and (B) F4/80+ macrophages from isolated peritoneal cells 48 hours after TBI in CD11b-DTR mice treated with PBS or diphtheria toxin (DT). (C) Analysis of microglia after DT administration in the absence of TBI. Representative photomicrographs of cortical brain sections stained with microglia- and macrophage- immunoreactive Iba1 antibody. N = 4 per group, ****P < 0.0001.
Table 1.
| Anatomical Region | Iba1+ cells/field
|
||
|---|---|---|---|
| WT
|
CD11b-DTR
|
||
| Mean ± S.E. | Mean ± S.E. | P-value | |
| Cortex | |||
| Primary Motor | 19 ± 1.5 | 17 ± 0.4 | 0.410 |
| Secondary Motor | 18 ± 0.6 | 19 ± 1.1 | 0.261 |
| Primary somatosensory | 18 ± 0.6 | 18 ± 1.1 | 0.692 |
| Secondary somatosensory | 19 ± 1.4 | 20 ± 0.9 | 0.707 |
| Basal ganglia | |||
| Medial | 16 ± 1.1 | 14 ± 0.8 | 0.291 |
| Lateral | 15 ± 0.8 | 13 ± 0.5 | 0.070 |
Values represent mean ± S.E. of Iba1+ cells observed in different anatomical regions within the cerebrum. Cells were counted in a 40X field.
Figure 2. Immunohistochemical analysis of macrophages and microglia 48 h after traumatic brain injury.

A, Representative photomicrographs of TBI lesions from WT and CD11b-DTR mice stained with the Iba1 antibody. (B) Quantification of Iba1 immunoreactive cells in the TBI lesion. N = 5 per group.
2.2. Effect of macrophage depletion on lesion size after traumatic brain injury
In order to determine the effect that macrophages have on brain lesion formation during traumatic brain injury, we depleted macrophages and analyzed the lesion size after controlled cortical impact. We first performed T2-weighted MRIs 48 hours after cortical impact, but a poor T2 signal at this time point made it difficult to delineate the lesion margins. We therefore measured the lesion size 28 days after cortical impact when the lesion is easily delineated and quantified, which is consistent with other reports using MRI (Skardelly et al., 2011). After 28 days, the lesions were detected by T2-weighted MRI and quantified in both control and CD11b+-cell depleted mice, although no significant differences were detected between the control and macrophage depleted groups (Figure 3).
Figure 3. Lesion size in macrophage depleted mice after traumatic brain injury.
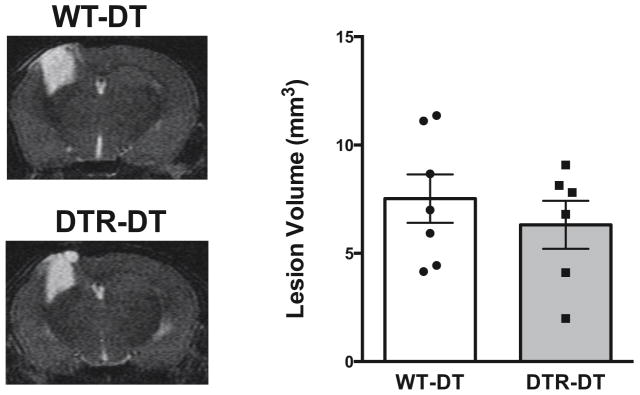
Lesion volume was measured in wild type control (WT-DT) and macrophage depleted mice (DTR-DT) by analyzing T2-weighted MRI scans 28 days after controlled cortical impact. N = 6–7 per group.
2.3. Depletion of macrophages increases proinflammatory gene expression after traumatic brain injury
Immune cells are major secretors of proinflammatory cytokines and can have a role in inflammation-mediated damage. To examine the effect that macrophage depletion has on inflammatory signaling, we analyzed the gene expression of proinflammatory cytokines and chemokines 48 hours after controlled cortical impact. In control mice, controlled cortical impact resulted in a significant increase in the expression of proinflammatory mediators in the ispilateral hemisphere compared to the unaffected contralateral hemisphere. Depletion of macrophages in CD11b-DTR mice resulted in a significant increase in the expression of CAM proinflammatory cytokines in the ipsilateral hemisphere compared to PBS treated controls (Figure 4A). Macrophage depletion had differential effects on the expression of AAM markers (Figure 4B). An increase in the gene expression of several CAM and AAM markers was also detected in the contralateral hemisphere of macrophage-depleted mice when compared to controls.
Figure 4. Analysis of inflammatory gene expression in macrophage depleted mice after traumatic brain injury.
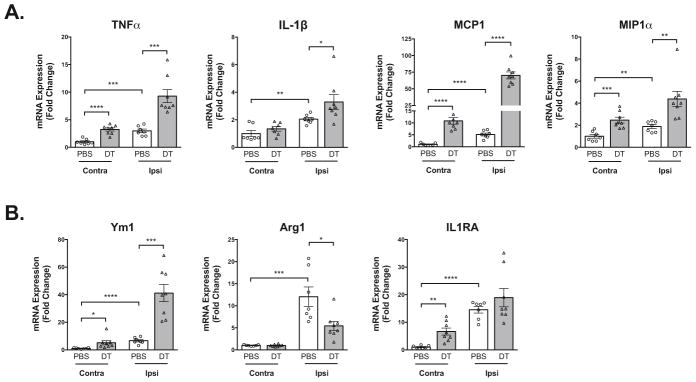
Gene expression of proinflammatory CAM markers (A) and AAM markers (B) was analyzed by qRT-PCR in contralateral (Contra) and ipsilateral (Ipsi) brain hemispheres 48 hours after controlled cortical impact. N = 7–8 per group, *P < 0.05, **P < 0. 01, ***P < 0.001, ****P < 0.0001.
2.4. Macrophage depletion induces inflammation in the brain in the absence of injury
To determine if the increases in inflammatory gene expression in the contralateral hemisphere were due to transmission of proinflammatory responses in the ipsilateral hemisphere or rather due to the macrophage depletion, we analyzed the effect that macrophage depletion has on inflammatory gene expression in the absence of injury. Control and CD11b-DTR mice received PBS or diphtheria toxin at the same dose and treatment timeline that was used during the controlled cortical impact studies. We found that depletion of macrophages resulted in a significant increase in proinflammatory gene expression in the brain even in the absence of injury. The major proinflammatory cytokines TNF-α, IL-1β, and IL-6, and chemokines MCP1 and Mip1α were significantly increased after macrophage depletion (Figure 5). In addition, several AAM markers Ym1, IL-4 and IL-1RA were also increased in response to macrophage depletion.
Figure 5. Induction of brain inflammation by macrophage depletion in the absence of traumatic brain injury.
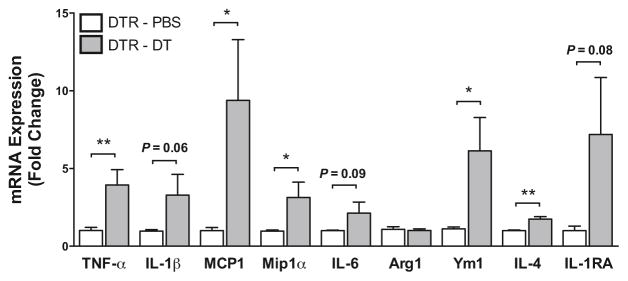
Gene expression of proinflammatory CAM and AAM markers was analyzed in the left cerebral (contralateral) hemisphere by qRT-PCR 72 hours after macrophage depletion. N = 7 per group. *P < 0.05, **P < 0. 01.
To verify that brain inflammation was due to the depletion of macrophages, we treated wild type mice with PBS or diphtheria toxin and analyzed inflammatory markers in the brain. Upon administration of diphtheria toxin, we found no significant increases in proinflammatory gene expression or AAM markers (Figure 6). This suggests that increased inflammation in the uninjured CD11b-DTR mice was due to depletion of macrophages rather than a complication from the diphtheria toxin.
Figure 6. Diphtheria toxin treatment does not induce inflammatory gene expression in the brain.
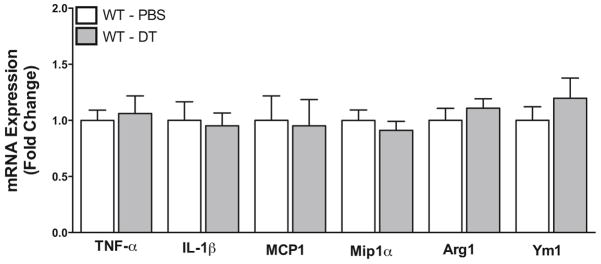
Gene expression of proinflammatory CAM and AAM markers was analyzed in the left cerebral (contralateral) hemisphere by qRT-PCR 72 hours after administration of PBS or diphtheria toxin (DT) in wild type (WT) mice. N = 9 per group
2.5. Macrophage depletion selectively induces inflammation in different organ systems
CD11b-DTR mice have been widely used to study the role of macrophages in a range of diseases including liver and kidney injury (Duffield et al., 2005; Lu et al., 2012). To determine the effect of macrophage depletion in other tissues, we depleted macrophages and analyzed inflammatory gene expression. In the kidney, we found a significant increase in the expression of proinflammatory cytokines TNF-α and IL-6, and the chemokine MCP1. In contrast, IL-1β and several AAM markers were unchanged (Figure 7A). We also analyzed the liver for changes in inflammation, but no significant differences were detected suggesting that macrophage depletion induces inflammation in a tissue-specific manner (Figure 7B).
Figure 7. Effect of macrophage depletion on inflammatory gene expression in the kidney and liver.

Inflammatory gene expression was analyzed by qRT-PCR 72 hours after CD11b+-cell depletion in (A) kidney, and (B) liver. N = 5 per group, *P < 0.05, **P < 0. 01.
3. DISCUSSION
In the present study, we examined the role of macrophages during the pathophysiology of traumatic brain injury. Here we found that depletion of macrophages using CD11b-DTR transgenic mice had no effect on lesion size after controlled cortical impact, but significantly increased inflammatory gene expression in the brain. Significant upregulation of proinflammatory cytokines and chemokines was present in both ipsilateral and contralateral brain hemispheres of macrophage-depleted mice after brain injury. In addition, macrophage depletion resulted in an increase in inflammatory gene expression in the brain in the absence of injury. Further analysis revealed that macrophage depletion also selectively induced inflammatory gene expression in the kidney, but not the liver.
Macrophages are known to have important roles during various diseases, and several molecular and genetic macrophage depletion models have been employed in order to understand their roles. Here we found that depletion of macrophages in CD11b-DTR mice had no effect on the lesion size after traumatic brain injury. While this may suggest that macrophages have a lesser role in mediating secondary injury and lesion development after TBI, it is difficult to draw this conclusion from these data because of the preexisting inflammation that was detected as a result of macrophage depletion. In addition, although prolonged proinflammatory macrophage responses are thought to exacerbate injury by preventing the resolution of inflammation, macrophages also have important phagocytic and reparative functions that are necessary for proper healing. Therefore, the lack of effect on lesion size may reflect inhibition of detrimental, but also reparative functions of macrophages.
Inhibiting the recruitment of blood-borne monocytes and macrophages by blocking chemokines and adhesion molecules has been shown to be protective during TBI, as well as in other models with inflammatory injury including stroke, myocardial infarction, and hypertensive cardiac remodeling. Interestingly and similar to our results, depletion of macrophages in these other models either had no effect or resulted in increased injury (Duffield et al., 2005; Gliem et al., 2012; van Amerongen et al., 2007; Zandbergen et al., 2009). These findings may again reflect a critical role for macrophages at specific stages after injury. It is also possible that blood-borne derived macrophages have more proinflammatory and detrimental roles in secondary injury, and inhibiting the influx of these cells rather than depleting them may suppress inflammation while still allowing for critical reparative functions by resident tissue macrophages. Likewise, in the heart it was shown that monocyte-derived CCR2+ macrophages are largely involved in promoting inflammation and have increased inflammasome activation (Epelman et al., 2014).
Depending on the phase of the injury, macrophages can have different CAM and AAM phenotypes, which have different roles during the inflammatory response. In a liver injury model, it was shown that depletion of macrophages during injury progression had opposite effects when compared to depletion during the recovery phase (Duffield et al., 2005). Similarly, inhibition of macrophages during different inflammatory phases after myocardial infarction can also have differential effects (Nahrendorf et al., 2007). Therefore, a better understanding of specific macrophage phenotypes during the inflammatory response to TBI is necessary in order to determine if time-dependent depletion of macrophages would be a more viable approach to suppress specific inflammatory processes.
Since macrophages secrete numerous proinflammatory cytokines, we hypothesized that depletion of macrophages would suppress inflammatory signaling after TBI. Surprisingly, we found that there were large increases in proinflammatory gene expression in both ipsilateral and contralateral brain hemispheres after TBI. This contrasts a previous report, which found that macrophage depletion suppressed inflammation after a cortical freeze injury model (Amankulor et al., 2009). These results may reflect differences in the brain injury model, and it was not reported whether macrophage depletion induced inflammation in the uninjured hemisphere.
A concerning and relevant finding of this study was that depletion of macrophages using the CD11b-DTR model induces brain inflammation in the absence of injury. This is a potentially confounding effect that makes interpretation of results from disease models difficult. Since many proinflammatory cytokines are known to exacerbate tissue injury, a preexisting inflammatory response resulting from macrophage depletion could significantly alter the pathophysiology of disease. Interestingly, despite a robust increase in inflammatory cytokines and chemokines in macrophage-depleted mice, there was no difference in the lesion size. This is surprising because proinflammatory cytokines and chemokines are known to be involved in the pathophysiology of TBI and are thought to promote injury, and numerous studies have shown that enhanced proinflammatory signaling can exacerbate brain injury. Consistent with our findings, another recently published report also detected inflammation in the brain after depleting microglia using the transgenic CD11b-HSVTK model (Bennett and Brody, 2014). In this previous report it was also found that microglia depletion did not prevent axon degeneration in a concussive brain injury model (Bennett and Brody, 2014).
In addition to the brain, we found that macrophage depletion also increased inflammatory gene expression in the kidney, but not the liver. These findings are consistent with a previous report showing that diphtheria toxin resulted in depletion of macrophages in the kidney, but not the liver (Cailhier et al., 2005). Therefore, the differences in inflammatory gene expression in specific tissues may be due to the extent of macrophage depletion in these tissues. Although cell type depletion models are a powerful tool in the analysis of the roles of macrophages, dendritic cells, neutrophils, and other immune cells, the ramifications of global depletion can be significant. For example, depletion of CD11c+ cells using CD11c-DTR mice results in neutrophilia (Tittel et al., 2012), which can have important roles in the pathophysiology of many diseases.
In this study, we found that DT administration did not significantly deplete microglia in uninjured mice. Similarly, we did not detect any significant differences in the number of microglia/macrophages in the brain in CD11b-DTR mice even after TBI. A previous report has shown that diphtheria toxin crosses the blood-brain barrier after intraperitoneal administration and effectively induces demyelination in an oligodendrocyte-specific, cre-inducible DTR model (Buch et al., 2005). Futhermore, subcutaneous administration of DT was found to significantly reduce microglia numbers in neonatal CD11b-DTR mice, although to a much lesser extent than macrophages (Ueno et al., 2013). More complex analysis of brain leukocytes using flow cytometry would be necessary to fully determine the extent of microglia and macrophage depletion, and further analysis of microglia populations may reveal the effect that DT administration has on microglia phenotype in CD11b-DTR mice.
In summary, we have demonstrated that depletion of macrophages increased the inflammatory response to TBI without significantly affecting lesion formation. We also found that macrophage depletion using CD11b-DTR mice induced inflammation in the brain and kidney, revealing a confounding effect of this commonly used model.
4. EXPERIMENTAL PROCEDURE
4.1. Animals and Treatments
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23) and were approved by the University Committee on Use and Care of Animals of the University of Michigan. Male CD11b-DTR and littermate wild type mice on a C57BL/6 background were maintained on standard laboratory chow (5001, LabDiet) and water ad libitum. For CD11b+ cell depletion and diphtheria toxin treatment, mice weighing 20–25 g were injected with diphtheria toxin (20 ng/g body weight, Sigma-Aldrich) 24 hr before and 24 hr after controlled cortical impact and then once every subsequent week for long term treatment.
4.2. Controlled Cortical Impact
Mice were anesthetized with isoflurane (5% induction, 1.5% maintenance) and core body temperature was maintained at 37°C using a heating pad controlled by rectal thermometer. The head was stabilized in a stereotaxic frame and a craniectomy (3.5 mm diameter) was made in the right parietal bone using a micromotor drill. Cortical impact was performed using a head impactor (TBI-0310, Precision systems and Instrumentation) fitted with a 3 mm impact tip. The impact velocity was set to 3.65 m/s with a compression depth of 1.00 mm and a dwell time of 500 msec. After cortical impact, the incision was sutured and the animals were allowed to recover.
4.3. Evaluation of Lesion Volume
The lesion volume was analyzed by T2-weighted MRI using a 7.0T Varian Unity Inova MR scanner (183-mm horizontal bore; Varian, Palo Alto, CA), with the body temperature maintained at 37°C by forced heated air. A double-tuned volume radiofrequency coil was used to scan the head region of the mice. Axial T2-weighted images were acquired using a fast spin-echo sequence with the following parameters: repetition time (TR)/effective echo time (TE), 4000/60 ms; echo spacing, 15 ms; number of echoes, 8; field of view (FOV), 20×20 mm; matrix, 256×128; slice thickness, 0.5 mm; number of slices, 25; and number of scans, 1 (total scan time ~2.5 min.). The lesion volumes were quantified by a blinded observer using NIH ImageJ software (version 1.43).
4.4. Gene Expression Analysis
Relative mRNA expression was determined using quantitative reverse transcription–polymerase chain reaction. Total RNA was extracted from whole cerebral hemispheres using TRIzol reagent and purified using an RNeasy Mini Kit (Qiagen) with an on-column DNase digestion. RNA (1 ug) was reverse transcribed to cDNA with an Applied Biosystems kit and quantitative reverse transcription–polymerase chain reaction was performed using a 7900HT fast real-time PCR system (Applied Biosystems). The relative mRNA expression was quantified by the comparative method and normalized to HPRT.
4.5. Flow cytometry
Peritoneal cells were isolated from the peritoneal cavity by peritoneal lavage using PBS. Cells were incubated in Fc Block for 10 min and then stained with indicated antibodies for 30 min at 4°C. Stained cells were washed twice in PBS and fixed in 0.1% paraformaldehyde before analysis. Flow cytometric analysis was performed on a FACSCanto II Flow Cytometer (BD Biosciences) equipped with three lasers (405-nm violet laser, 488-nm blue laser, and 640-nm red laser) and analyzed with FlowJo software (Treestar).
4.6. Statistics
A Shapiro-Wilk normality test was used to determine if data were normally distributed. For normally distributed data with equal variance, statistical comparison of mean values between groups was performed with the Student t test or by a two-way ANOVA with a Bonferroni post-test, and values are presented as mean ± SEM. For normally distributed data with unequal variance, a Welch’s t test was used. Data that were not normally distributed were analyzed with the nonparametric Mann-Whitney test. All statistical analysis of data was performed in GraphPad Prism (version 6; GraphPad Software, Inc). P < 0.05 was considered significant.
HIGHLIGHTS.
Macrophage depletion increases inflammation after traumatic brain injury
Enhanced inflammation in macrophage depleted mice does not affect lesion size
Macrophage depletion increases inflammation in select tissues in the absence of injury
Acknowledgments
This study was supported by National Institutes of Health grant HL112610 and American Heart Association Grant-in-Aid 12GRNT11890006 to R.M.M, and National Institutes of Health fellowship NS077780 to R.A.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, Cuzzocrea S. Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS One. 2013;8:e57208. doi: 10.1371/journal.pone.0057208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci. 2009;29:10299–308. doi: 10.1523/JNEUROSCI.2500-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RE, Brody DL. Acute Reduction of Microglia Does Not Alter Axonal Injury In a Mouse Model of Repetitive Concussive Traumatic Brain Injury. J Neurotrauma. 2014 doi: 10.1089/neu.2013.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2:419–26. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, Savill J, Hughes J, Lang RA. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol. 2005;174:2336–42. doi: 10.4049/jimmunol.174.4.2336. [DOI] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Israelsson C, Hedin J, Ebendal T, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2011;34:110–23. doi: 10.1111/j.1460-9568.2011.07723.x. [DOI] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. 2010;7:41. doi: 10.1186/1742-2094-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira LC, Regner A, Miotto KD, Moura SD, Ikuta N, Vargas AE, Chies JA, Simon D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014:1–6. doi: 10.3109/02699052.2014.916818. [DOI] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71:743–52. doi: 10.1002/ana.23529. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–52. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen M, Charo IF, Huang EJ, Liu J, Nakamura MC. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma. 2014 doi: 10.1089/neu.2013.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7:e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblach SM, Faden AI. Administration of either anti-intercellular adhesion molecule-1 or a nonspecific control antibody improves recovery after traumatic brain injury in the rat. J Neurotrauma. 2002;19:1039–50. doi: 10.1089/089771502760341956. [DOI] [PubMed] [Google Scholar]
- Lu L, Faubel S, He Z, Andres Hernando A, Jani A, Kedl R, Edelstein CL. Depletion of macrophages and dendritic cells in ischemic acute kidney injury. Am J Nephrol. 2012;35:181–90. doi: 10.1159/000335582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–65. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Quintana A, Molinero A, Florit S, Manso Y, Comes G, Carrasco J, Giralt M, Borup R, Nielsen FC, Campbell IL, Penkowa M, Hidalgo J. Diverging mechanisms for TNF-alpha receptors in normal mouse brains and in functional recovery after injury: From gene to behavior. J Neurosci Res. 2007;85:2668–85. doi: 10.1002/jnr.21126. [DOI] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–8. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–6. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab. 2010;30:769–82. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skardelly M, Gaber K, Burdack S, Scheidt F, Hilbig H, Boltze J, Forschler A, Schwarz S, Schwarz J, Meixensberger J, Schuhmann MU. Long-term benefit of human fetal neuronal progenitor cell transplantation in a clinically adapted model after traumatic brain injury. J Neurotrauma. 2011;28:401–14. doi: 10.1089/neu.2010.1526. [DOI] [PubMed] [Google Scholar]
- Tehranian R, Andell-Jonsson S, Beni SM, Yatsiv I, Shohami E, Bartfai T, Lundkvist J, Iverfeldt K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–51. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- Tittel AP, Heuser C, Ohliger C, Llanto C, Yona S, Hammerling GJ, Engel DR, Garbi N, Kurts C. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat Methods. 2012;9:385–90. doi: 10.1038/nmeth.1905. [DOI] [PubMed] [Google Scholar]
- Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–51. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–29. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandbergen HR, Sharma UC, Gupta S, Verjans JW, van den Borne S, Pokharel S, van Brakel T, Duijvestijn A, van Rooijen N, Maessen JG, Reutelingsperger C, Pinto YM, Narula J, Hofstra L. Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. J Cardiovasc Pharmacol Ther. 2009;14:68–75. doi: 10.1177/1074248408329860. [DOI] [PubMed] [Google Scholar]