

Abstract
Impaired cardiovascular function during acute myocardial infarction (MI) is partly associated with recruitment of activated polymorphonuclear neutrophils. The protective role of arjunolic acid (AA; 2:3:23-Trihydroxy olean-12-en-28-oic acid) is studied in the modulation of neutrophil functions in vitro by measuring the reactive oxygen species (ROS) generation. Neutrophils were isolated from normal and acute MI mice to find out the efficacy of AA in reducing oxidative stress. Stimulation of neutrophils with phorbol-12-myristate-13-acetate (PMA) resulted in an oxidative burst of superoxide anion (O2•−) and enhanced release of lysosomal enzymes. The treatment of neutrophils with PMA induced phosphorylation of Ser345 on p47phox, a cytosolic component of NADPH oxidase. Furthermore, we observed activated ERK induced phosphorylation of Ser345 in MI neutrophils. Treatment with AA significantly inhibited the phosphorylation of P47phox and ERK in the stimulated controls and MI neutrophils. Oxidative phosphorylation activities in MI cells were lower than in control, while the glycolysis rates were elevated in MI cells compared to the control. In addition, we observed AA decreased intracellular oxidative stress and reduced the levels of O2•− in neutrophils. This study therefore identifies targets for AA in activated neutrophils mediated by the MAPK pathway on p47phox involved in ROS generation.
1. Introduction
Neutrophils (Activated) have been implicated in the pathogenesis of several disease processes, including myocardial injury mediated by severe ischemia (Hendryk et al., 2010), reperfusion (Saxena et al., 2013), angina (Keresztes et al., 2013) and myocardial infarction (MI) (Kaya et al., 2013). Upon activation, polymorphonuclear neutrophils (PMN) not only generate superoxide anion (O2•−) and hydrogen peroxide (H2O2) but also secrete myeloperoxidase (MPO), a hemoprotein expressed abundantly in neutrophils, and to a lesser extent in monocytes and macrophages (Miriyala et al., 2013a, Miriyala et al., 2013b) as part of oxidative repertoire. MPO, in presence of H2O2, is anatomically poised to catalytically consume endothelial-derived nitric oxide (NO), thereby impairing NO-dependent signaling in smooth muscle cells and subsequent vascular relaxation (Baldus et al., 2004). The neutrophil respiratory burst by cytokines is associated with partial phosphorylation of the NADPH oxidase component p47phox and MAPKs (El-Benna et al., 2009).
The dynamic functions of leukocytes require a combined metabolic mechanism to encounter energetic demand which is likely to involve both glycolysis and mitochondrial oxidative phosphorylation. Mitochondrial function is inversely exaggerated by oxidative stress. Leakage of reactive oxygen species may lead to damage of the mitochondrial membrane potential, proteins and DNA which leads to inhibition of the oxidative phosphorylation. Discovering the role of bioenergetics and mitochondrial dysfunction in neutrophils is often examined by using freshly isolated cells.
We and others have demonstrated the protective role of arjunolic acid (AA; 2:3:23-Trihydroxy olean-12-en-28-oic acid) (Fig 1) on platelet aggregation (Sumitra et al., 2001), hepatotoxicity (Manna et al., 2007), nephrotoxicity (Sherif, 2014) and diabetes (Manna and Sil, 2012). Its antioxidant property coupled with metal chelating property defends different organs from drug-induced organ pathophysiology. The cytoprotection of AA, at least in part, results from the detoxification of reactive oxygen species (ROS). The underlying mechanism is still unknown. In this present work, we identified that AA could inhibit ROS activation through phosphorylation of p47phox and extracellular regulated kinase (ERK) in activated neutrophils. Furthermore, we observed that AA could inhibit the oxidative phosphorylation (OXPHOS) in mitochondria, which is the predominant mode for O2 consumption in cells, and act as the primary source of ROS in cells due to leaked electrons.
Figure 1.
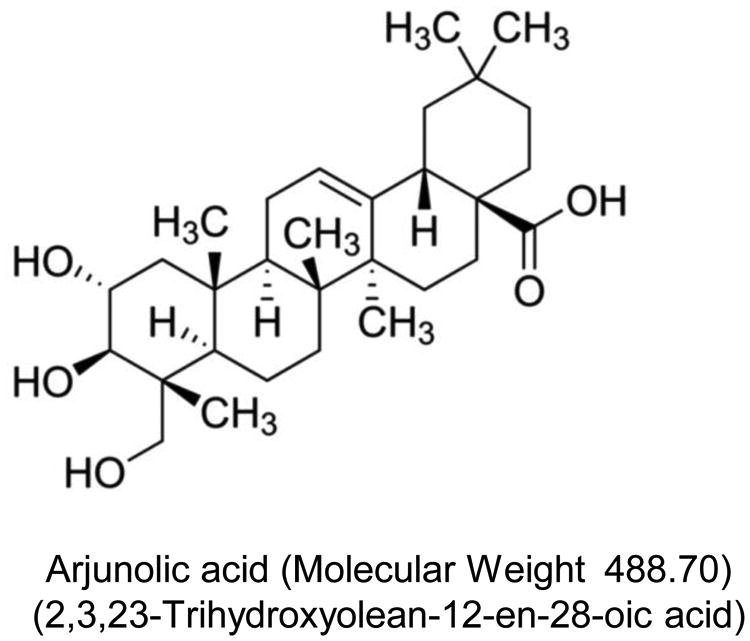
Structure of AA.
2. Materials and Methods
This study conforms to the guiding principles of the Institutional Animal Ethics Committee (IAEC), Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication No. 85-23, revised 1996).
2.1 Chemicals
The following chemicals were purchased from Sigma (St. Louis, MO, USA): AA (Cat# SMB00119; Lot# 122228133V), Phorbol 12-myristate 13 acetate (PMA), cytochrome C, superoxide dismutase, phenol red, dextran, O-dianisidine hydrochloride, ficollhistopaque (1077), glycerol, hexadecyltrimethylammonium bromide, Hank's balanced salt solution (HBSS), horse radish peroxidase, Triton ×100, MitoTEMPO, Folin-Ciocalteau reagent, hemoglobin, hydrogen peroxide, p-nitro phenol phosphate. All other chemicals used were of analytical grade.
2.2 Experimental Animals
Male mice weighing 30-35g were inbred in a pathogen free facility and were maintained in environmentally controlled rooms with 12h light/ dark cycle. The animals received commercial chow diet and water ad libitum.
2.3 Development of The Infarct Model
MI model was induced in the mice as described earlier (Miriyala et al., 2013b) with modifications to minimize the early mortality rate. As previously detailed, mice were anesthetized using endotracheal delivered isoflurane and were subjected to a left thoracotomy. After establishing positive pressure respiration, left intercostal thoracotomy was performed using aseptic technique and the third and fourth intercostal ribs were separated with a small retractor to expose the heart. The pericardium was opened carefully to avoid any injury to the heart. The left coronary artery (LCA) and its branches could be seen easily without any amplification or use of a surgical microscope. The pattern of the LCA was carefully examined in order to ligate the left anterior descending coronary artery. A 6-0 a traumatic proline silk suture was passed through the epicardial layer around the midway of the left anterior descending coronary artery. Following coronary occlusion, the thorax was closed in layers, the endotracheal tube was removed and the animals were brought back to normal respiration.
2.4 Isolation of Neutrophils From Mice
Blood was drawn from sham operated and infarct rats and neutrophils were separated according to Newman et al. (Newman et al., 1982). The neutrophils were suspended in HBSS and the cell concentration was determined using a hemocytometer.
2.5 Stimulation Studies
To ascertain the optimal concentration of AA on neutrophils isolated from mice, myeloperoxidase (MPO) was used as a sensitive marker and the results showed an inhibitory concentration (IC50) of 97.94 ± 8.33μM (data not shown). Hence, further studies were carried out with an optimal concentration of 120μM for AA. Neutrophils (1×106 cells /well) were allowed to adhere for 1h and were stimulated by the addition of PMA (100ng/ml) (Gurskaya et al., 1996) for 1h at 37°C. In the case of unstimulated cells and MI neutrophils, after 1h adherence, AA was added and the culture was terminated after 1h of AA treatment.
MPO was assayed in the cell lysate after extracting the enzyme in phosphate buffer containing hexadecyl trimethylammonium bromide (Seekamp et al., 1993). In H2O2 assay (Pick and Mizel, 1981), to the adherent neutrophils, 100μl (1μg/ml) of PMA, followed by 1000μl of HBSS containing horse radish peroxidase (19 units/ml) and phenol red (0.02%) were added. After 1h of incubation, 100μl of 1M NaOH was added and the color developed was read at 605nm.
For the assay of O2•− (Guthrie et al., 1984), the medium was removed and the adherent neutrophils were incubated for 1h in HBSS containing 80μM cytochrome C and 10μM PMA. The color developed in the supernatant was read at 580nm. The amount of O2•− released was measured by the amount of cytochrome C and was quantitated using a molar extinction coefficient of 21× 103 M-1cm-1.
For the lysosomal enzyme assays, after removing the cell free medium, the cells were lysed by adding 0.5ml of ice cold 0.1% Triton × 100 in 0.25M sucrose, subjected to repeated freezing and thawing and assayed for acid phosphatase (Barrett, 1977) and cathepsin D (Sapolsky et al., 1973).
2.6 Immunoblotting Studies
Neutrophils were lysed in buffer (10mM Tris–HCl pH 7.2, 1% Nonidet P-40, 158mM NaCl, 1mM EDTA, 50mM NaF, 1mM PMSF, 10μg/ml aprotinin, 10μg/ml leupeptin, 1mM sodium orthovanadate, 10mM sodium pyrophosphate), and lysates were centrifuged at 16,000 × g for 10min. Protein content in the lysate was determined (BCA protein assay; Pierce, Rockford, IL, USA); subsequent experiments were performed with the lysate with the indicated amount of protein. Immunoblotting (5μg protein/well) was performed with antibodies against ERK1 and 2/phospho-ERK Thr202/Tyr204, (Cell Signaling Technology, Danvers, MA, USA), p47phox Antibody, Anti-phospho-p47 phox (pSer345) antibody (Sigma), anti-gamma H2A.X (phospho S139) antibody (γH2Ax; abcam, Cambridge, MA, USA) and actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) as a loading control. Secondary antibodies were conjugated with Alexa680 (Molecular Probes, Carlsbad, CA, USA) or IRdye800 (Rockland Immunochemicals, Gilbertsville, PA, USA), and were detected and quantified using the Odyssey infrared imaging system (LI-COR, Lincoln, NE, USA). Untransferred proteins in the gel were stained with Coomassie blue as an additional method to confirm equal protein loading.
2.7 Mitochondrial Bioenergetics Measurements
Oxygen consumption was determined using the Seahorse Extracellular Flux (XF-96) analyzer (Seahorse Bioscience, Chicopee, MA). The XF-96 measures the concentration of oxygen and free protons in the medium above a monolayer of cells in real-time. Thus, the rates of oxygen consumption and proton production can be measured across several samples at a time. To allow comparison between experiments, data are presented as oxygen consumption rate (OCR) in pMoles/min/104 cells and the extracellular acidification rate (ECAR) in mpH/min/104 cells. Freshly isolated neutrophils were seeded at 125Kcells/well into gelatin-coated Seahorse Bioscience XF micro plates, cultured in the presence or absence of 2 g/L D-glucose, and then centrifuged to adhere to the bottom of the wells. OCR was measured four times and plotted as a function of cells under the basal condition followed by the sequential addition of Oligomycin (1 μg/ml, an inhibitor of mitochondrial ATP-synthase) was injected into the XF medium to estimate the OCR coupled to ATP synthesis and represented as ATP-linked. The residual OCR minus the non-mitochondrial OCR can be attributed to proton leak. FCCP (1μM), an uncoupler, is added to determine the maximal OCR, followed by Antimycin (1μM), an inhibitor of mitochondrial respiration, to determine nonmitochondrial sources of oxygen consumption. The ATP-linked OCR was calculated as the difference between the basal OCR and the OCR measured after the addition of oligomycin. The OCR maximal capacity was the direct rate measured after the addition of FCCP. Reserve capacity is a measure of the amount of ATP that can be produced under energetic demand and can be calculated as the difference between the maximum rate of respiration and the basal. The OCR values were normalized to total protein content in the corresponding wells and expressed as pmol/min/mg protein.
To calculate ECAR measurements, cells were washed and changed to assay media lacking glucose. Basal ECAR were measured four times and plotted as a function of cells under the basal condition followed by the sequential addition of glucose (25mM), oligomycin (1μg/ml) and 2-deoxyglucose (25mM), an inhibitor for the hexokinase. The rate of glycolysis was determined by the difference between the basal ECAR from the ECAR after the addition of glucose. Glycolytic reserve was determined by subtracting the ECAR following the addition of oligomycin from the ECAR following the addition of glucose.
2.8. Quantification of Intracellular Superoxide and Prooxidant
Dihydroethidium (DHE, Invitrogen), which exhibits blue fluorescence in the cytosol until oxidized, was used to estimate the levels of superoxide after AA treatment. To confirm the level of superoxide induced by AA, the cells were pretreated with 50mg PEG-SOD (Sigma) for 1 hour followed by AA treatment. Antimycin (Sigma) was used as a positive control because it has been shown to increase superoxide in all tested cell lines. The procedures for DHE were conducted using a FACScan protocol using a flow cytometry.
2.9 Statistical Analysis
Unless otherwise stated, results are expressed as mean ± standard deviation of the mean. In vitro studies were repeated a minimum of three times. One-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test or the unpaired Student's t-test were used to identify significant differences between groups. Statistical analysis was performed using Sigma-STAT software, version 3.5 (Systat Software Inc., San Jose, CA, USA). A P-value of less than 0.05 was considered significant.
3. Results
The neutrophil NADPH-oxidase can be activated by protein kinase C (PKC) agonists such as PMA, resulting in O2•− release. To investigate the effect of AA on PMA-induced NADPH-oxidase activity, O2•− and H2O2 production, we used neutrophils derived from the normal and acute MI mice. Treatment with AA showed a protective effect towards free radical generation on stimulated neutrophils (P<0.01; Fig 2). In comparison with PMA treated cells, AA cells demonstrated a significant decrease in PMA-induced MPO activation (P<0.01). Concomitant with the above results, there was a significant increase in cathepsin D and acid phosphatase in the stimulated neutrophils and AA significantly reduced the levels in the stimulated neutrophils (P<0.01; Fig 3). Consistent with these observations, neutrophils derived from MI mice were significantly inhibited upon AA treatment.
Figure 2.

Effect of AA on MI mice neutrophils ROS generation. All values are mean ± SD (n = 6). H2O2 level was expressed as μmoles of H2O2 liberated/0.5×106 cells. O2•− level was expressed as nmoles of O2- liberated /min/mg protein. MPO level was expressed as μmoles of H2O2 utilized/min/mg protein. NS = nonsignificant as compared to control; *P<0.01 as compared to control; #P<0.01 as compared to PMA stimulated cells; $P<0.01 as compared to MI cells.
Figure 3.

Effect of AA on MI mice neutrophils lysosomal release. All values are mean ± SD (n = 6). Acid phosphatase activity was expressed as μmoles of p-nitrophenol liberated /h/mg protein. Cathepsin D activity was expressed as μmoles of tyrosine liberated /h/mg protein. NS = nonsignificant as compared to control; *P<0.05 as compared to control; #P<0.05 as compared to PMA stimulated cells; $ P<0.05 as compared to MI cells.
We next examined whether MI-induced ROS production was associated with p47phox phosphorylation using specific anti-phosphoSer antibodies. Interestingly, Western blotting analysis indicated that stimulated and MI neutrophils induced a transient phosphorylation of p47phox on Ser345 (Fig 4a and 4b). As Ser345 is located in a sequence recognized and phosphorylated by MAPK, we examined p47phox phosphorylation at Ser345 by testing the effects of different MAPK inhibitors. As MI mediated ERK activation was observed in neutrophils (Fig 4b), we used PD98059 and U0126, which inhibits MEK1/2 (the upstream activator of ERK1/2), to analyze the role of these MAPK pathways in p47phox phosphorylation in neutrophils. In comparison with stimulated and MI neutrophils, AA treatment abrogated ERK activation (P<0.001; Fig 4b) in parallel with inhibition of p47phox phosphorylation on Ser345. Similarly MEK inhibition essentially abolished phosphorylation at Ser345. We found that PMA-induced p47phox phosphorylation precedes ROS production as both AA and PD98059 inhibited PMA-induced ROS production (P<0.01; Fig 4c). Regulation of intracellular Ca2+ homeostasis and production of ATP are crucial factors in mitochondria mediated ROS generation. We found that PMA plays a pivotal role in the mitochondria respiration and regulates the formation of ROS. Various respiratory complexes that leak electrons to oxygen (producing O2•−) are the precursors of most ROS. Incubation of cells with AA significantly reduced PMA-induced ROS production (Fig 4c) similar to MitoTEMPO, a mitochondria-targeted antioxidant with superoxide and alky scavenging properties. These results suggest that a considerable portion of MI and induced ROS production occurs at the levels of mitochondria.
Figure 4.
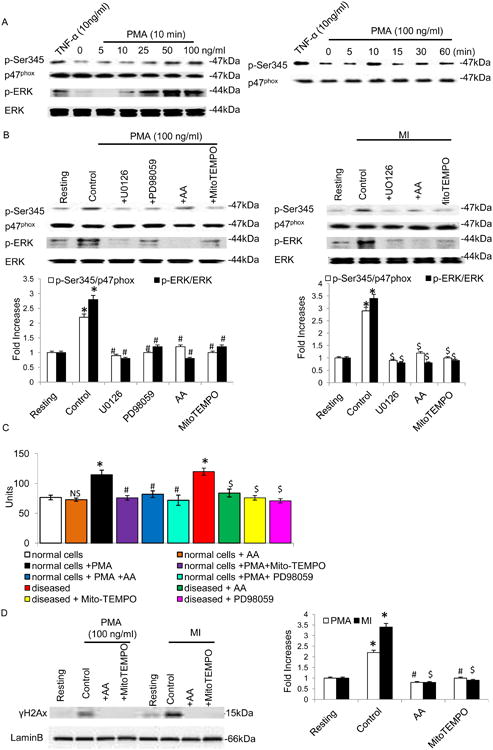
Effect of MAPK inhibitors on MI induced p47phox and MAPK activation. A) Effect of neutrophil agonists on the phosphorylation of Ser345. Neutrophils (1 × 106 cells/ml) were incubated with TNF-α (10ng/ml) for 20 minutes, and different concentrations of PMA for different time periods. (B and D) Stimulated or MI Neutrophils (1 × 106 cells/ml) were incubated with PD98059 (50μM) or UO126 (10μM) for 10 minutes. Cells were then lysed, and proteins were analyzed with SDS-PAGE and immunoblotting with anti–phosphorERK1/2, anti-phospho-Ser345 antibody and anti-gamma H2A.X (phospho S139) antibody. Data are representative of 4 experiments. C) Effect of AA on MI mice neutrophils ROS generation. All values are mean ± SD (n = 6). O2•− level was expressed as nmoles of O2- liberated /min/mg protein. NS = nonsignificant as compared to control; *P<0.01 as compared to control; #P<0.01 as compared to PMA stimulated cells; $P<0.01 as compared to MI cells.
To test the significance of DNA damage in MI, neutrophils were manifested by an increase in γH2AX levels, which are indicative of DNA fragmentation. The results, shown in Fig 4d, demonstrate that treatment with PMA and the MI neutrophils led to increased γH2AX levels, while the AA treatment showed a rescue effect. Next, we used a XF96 analyzer to determine the effect of PMA on respiration in the intact neutrophils. Adding PMA to these cells immediately increased oxygen consumption as compared to AA cells (Fig 5a). OXPHOS in mitochondria are the primary source of ROS in cells due to leaked electrons. This apparent increase in respiration was reversible by oligomycin, which blocks ATP synthase (Fig 5a). We examined OXPHOS levels in these cells from stimulated and MI. The rate of oxygen consumption per cell was significantly lower in stimulated and MI cells (Fig 5b). The most noticeable difference in the O2 consumption was found in the presence of carbonilcyanide p-trifluromethoxyphenylhydrazone (FCCP). FCCP is an inner membrane pore opener that resets the proton gradient between mitochondrial matrix and interspace, resulting in continuous transport of protons and consuming O2 at the maximum potential. Remarkably, while the FCCP treatment increased O2 consumption in AA, the treatment showed no effect on the O2 consumption in the MI neutrophils (P<0.01; Fig 5b). The antimycin A, an inhibitor of complex III, further reduced but did not completely abolish oxygen consumption in MI cells. The result indicated that the low basal OXPHOS activity in MI cells was due to unusually low OXPHOS potential. We examined glycolysis in these cells by measuring extracellular acidification (ECAR), and the pattern exactly opposite to that of oxygen consumption rate (OCR) was observed for glycolysis rates between AA and MI neutrophils. The result was supported by measuring cellular lactate concentration, an indicator for glycolysis, which was also significantly higher in MI than in AA cells (Fig 5d). The reverse relation of oxidative phosphorylation and glycolysis rates indicated changes in the metabolism for energy generation in activated neutrophils.
Figure 5.
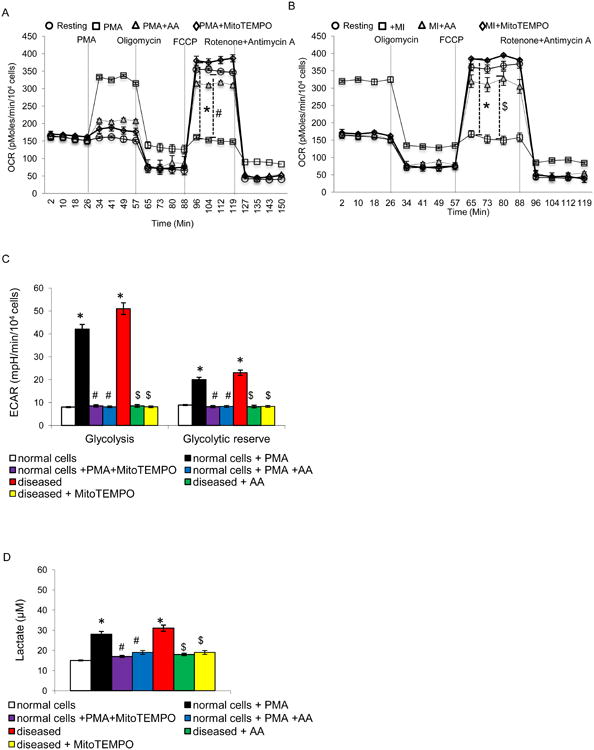

Mitochondrial activities in stimulated and MI neutrophils. (A and B) oxygen consumption rate (OCR, pmol/min/104 cells) determined with Seahorse analyzer for OXPHOS activity. (C) Extracellular acidification rate (ECAR) by Seahorse for glycolysis activity. (D) Cellular lactate concentrations. (E) Superoxide radicals produced in the cells were analyzed using a dihydroethidium fluorescence probe. Antimycin was added to induce cellular superoxide radicals and PEG-SOD was used to catalyze the produced superoxide radicals. All values are mean ± SD (n = 4). *P<0.05 as compared to control; #P<0.05 as compared to PMA stimulated cells; $P<0.05 as compared to MI cells.
To explore the free radical scavenging property of AA, superoxide anion was measured using dihydroethidium, a fluorescent cholesterol analog (Fig 5e). The levels of superoxide radicals in MI and PMA treated cells were consistently and significantly higher than the levels in AA treated cells. Antimycin A, an inhibitor of ETC complex III, is able to inhibit mtOXPHOS, leading to the generation of high levels of superoxide radicals. In turn, the radicals can be removed by adding PEG-SOD.
4. Discussion
In the present study, we show that acute MI induced NADPH oxidase activation in mice neutrophils. The extensive release of ROS is linked to the pathogenesis of tissue damage following ischemia in stroke and MI (Schnabel and Blankenberg, 2007). Inflammatory response in myocardial tissue with an early phase of neutrophil accumulation might exacerbate tissue injury through the release of free radicals and proteolytic enzymes (Mayadas et al., 2014). Preliminary studies were carried out using PMA stimulated mice neutrophils and AA was found to significantly inhibit ROS species generation and lysosomal enzymes release at a concentration of 120μM. The concentration of 120μM did not seem to be very high since earlier experiments showed that AA effectively inhibited platelet aggregation above 140μM (Sumitra et al., 2001). Antioxidant effects of AA are thought to be mainly attributed to the scavenging of oxygen-derived free radicals (Hemalatha et al., 2010).
It was shown recently that the neutrophils characteristically invaded the myocardial tissue during ischemia and they were observed to be the major source of free radicals (Shao et al., 2012). MPO primarily generated highly reactive intermediates such as tyrosyl radicals, which could rapidly consume NO at near diffusion-limited rates (Carr et al., 2000). Thus, NO consumption by MPO in complex physiological fluids was further accelerated, rather than reduced, by biological reductants. MPO-dependent NO consumption was preceded by the oxidation of MPO by H2O2. Since the majority of PMN-derived superoxide spontaneously or enzymatically dismutated to H2O2, the leukocyte and vessel wall NADPH oxidases, as well as other oxidoreductases, could further contribute to NO catabolism by providing the H2O2 required for activating MPO (Carr et al., 2000). The results of this study showing AA significantly reducing O2•− H2O2 and MPO levels in both stimulated and MI neutrophils were also in agreement with our earlier reports wherein elevated levels of ROS and MPO were significantly inhibited by AA in vivo (Sumitra et al., 2001). In experimental models, interventions that depleted neutrophils or inhibited their function played a central role in ventricular remodeling (Askari et al., 2003). Any disturbance in this equilibrium in favor of free radicals caused an increase in oxidative stress and initiated subcellular changes leading to ischemic cardiomyopathy (Rajasekaran et al., 2007) and heart failure (Shao et al., 2012). An increased activity of lysosomal enzymes such as acid phosphatase and cathepsin D was observed in MI neutrophils, which was abrogated upon AA treatment. These proteases mediate extracellular matrix degradation, myofibroblast trans-differentiation and myocardium extracellular matrix protein (e.g. collagen, fibronectin) expression (Chen et al., 2013). Elevated lysosomal enzymes levels have been shown to contribute directly to cardiac rupture/stability in human MI (Hilfiker-Kleiner et al., 2007).
Activation of NADPH oxidases (Nox) is initiated by phosphorylation of p47phox by kinases, including PKC and MAPK, and hence p47phox is called the organizer subunit of Nox (Ago et al., 2003). Activation of the NADPH oxidase and increased oxidative stress has been shown to play a key role in experimental and human heart failure (Heymes et al., 2003, Doerries et al., 2007). Furthermore, p47phox is critical for oxidase activation in intact cells and the Src homology 3 domain has been implicated in the assembly and maintenance of the NADPH oxidase activity in PMNs (Dang et al., 2006). Neutrophils isolated from MI mice showed a stronger oxidative burst than the controls. In addition, p47phox-Ser345 phosphorylation and the MAPKs (ERK1/2) that mediate this process was upregulated. As Ser345 is located in a sequence recognized and phosphorylated by MAPK, we tested the AA and MAPK inhibitors and found that it abrogated p47phox –Ser345 phosphorylation in stimulated and MI neutrophils.
A hallmark of mitochondrial dysfunction is increased production of the oxygen radical O2•− a normal byproduct of OXPHOS that is formed at increased rates when the electron transport chain is defective. In this report, we found that MI directly affects mitochondrial functions in neutrophils. With regards to changes in the respiration of cells, PMA increased their oxygen consumption, whereas AA and Mito-TEMPO showed a protective effect. Thus, AA showed a protective effect on induced and MI neutrophils. The present findings suggest that interference with signaling cascades might be acutely reversible by administration of an antioxidant agent such as AA and that inhibition of oxidative burst from neutrophils might have therapeutic potential in acute infarction and ischemic cardiomyopathy.
5. Conclusions
In conclusion, owing to the action of hydrogen donation, metal ion chelating and the resonance stabilization of carboxyl radical, AA could regulate OXPHOS in the mitochondria and subsequently inhibit ROS generation through abrogated p47phox-Ser345 phosphorylation in stimulated and MI neutrophils. To the best of our knowledge, this is the first report showing the protective effect of AA on p47phox phosphorylation and mitochondrial bioenergetics, which is imperative for the production of ROS and important from a drug discovery perspective.
Acknowledgments
The authors wish to thank Ms. Talicia A Tarver who assisted in the proof-reading of the manuscript. This project was supported by grants from NIH (R25RR026019 and R25OD010954); Beginning Grant-in-Aid (0950118G); and a Scientist Development Grant (10SDG4190036) from the American Heart Association
Footnotes
Conflict of interest statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ago T, Kuribayashi F, Hiroaki H, Takeya R, Ito T, Kohda D, Sumimoto H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proceedings of the National Academy of Sciences. 2003;100:4474–4479. doi: 10.1073/pnas.0735712100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askari AT, Brennan ML, Zhou X, Drinko J, Morehead A, Thomas JD, Topol EJ, Hazen SL, Penn MS. Myeloperoxidase and Plasminogen Activator Inhibitor 1 Play a Central Role in Ventricular Remodeling after Myocardial Infarction. J Exp Med. 2003;197:615–624. doi: 10.1084/jem.20021426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldus S, Heitzer T, Eiserich JP, Lau D, Mollnau H, Ortak M, Petri S, Goldmann B, Duchstein HJ, Berger J, Helmchen U, Freeman BA, Meinertz T, Münzel T. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radical Biology and Medicine. 2004;37:902–911. doi: 10.1016/j.freeradbiomed.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Barrett AJ, KMF . Lysosomal enzymes. Amsterdan: Elsevier North Holland Biomedical Press; 1977. Editor. [Google Scholar]
- Carr AC, McCall MR, Frei B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species: Reaction Pathways and Antioxidant Protection. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1716–1723. doi: 10.1161/01.atv.20.7.1716. [DOI] [PubMed] [Google Scholar]
- Chen H, Wang J, Xiang MX, Lin Y, He A, Jin CN, Guan J, Sukhova GK, Libby P, Wang JA, Shi GP. Cathepsin S-mediated fibroblast trans-differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovascular Research. 2013;100:84–94. doi: 10.1093/cvr/cvt158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang PMC, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo MA, El-Benna J. A specific p47phox - serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. The Journal of Clinical Investigation. 2006;116:2033–2043. doi: 10.1172/JCI27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, Sorrentino S, Manes C, Schieffer B, Drexler H, Landmesser U. Critical Role of the NAD(P)H Oxidase Subunit p47phox for Left Ventricular Remodeling/Dysfunction and Survival After Myocardial Infarction. Circulation Research. 2007;100:894–903. doi: 10.1161/01.RES.0000261657.76299.ff. [DOI] [PubMed] [Google Scholar]
- El-Benna J, Dang PMC, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NG, Diatchenko L, Chenchik A, Siebert PD, Khaspekov GL, Lukyanov KA, Vagner LL, Ermolaeva OD, Lukyanov SA, Sverdlov ED. Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Anal Biochem. 1996;240:90–97. doi: 10.1006/abio.1996.0334. [DOI] [PubMed] [Google Scholar]
- Guthrie LA, McPhail LC, Henson PM, Johnston RB., Jr Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med. 1984;160:1656–1671. doi: 10.1084/jem.160.6.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemalatha T, Pulavendran S, Balachandran C, Manohar BM, Puvanakrishnan R. Arjunolic acid: a novel phytomedicine with multifunctional therapeutic applications. Indian J Exp Biol. 2010;48:238–247. [PubMed] [Google Scholar]
- Hendryk S, Czuba Z, Jedrzejewska-Szypulka H, Bazowski P, Dolezych H, Krol W. Increase in activity of neutrophils and proinflammatory mediators in rats following acute and prolonged focal cerebral ischemia and reperfusion. Acta Neurochir Suppl. 2010;106:29–35. doi: 10.1007/978-3-211-98811-4_4. [DOI] [PubMed] [Google Scholar]
- Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. Journal of the American College of Cardiology. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand JL, Desjardins F, Ansari A, Struman I, Nguyen NQN, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H. A Cathepsin D-Cleaved 16 kDa Form of Prolactin Mediates Postpartum Cardiomyopathy. Cell. 2007;128:589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- Kaya MG, Akpek M, Lam YY, Yarlioglues M, Celik T, Gunebakmaz O, Duran M, Ulucan S, Keser A, Oguzhan A, Gibson MC. Prognostic value of neutrophil/lymphocyte ratio in patients with ST-elevated myocardial infarction undergoing primary coronary intervention: A prospective, multicenter study. International Journal of Cardiology. 2013;168:1154–1159. doi: 10.1016/j.ijcard.2012.11.074. [DOI] [PubMed] [Google Scholar]
- Keresztes M, Horváth T, Ocsovszki I, Földesi I, Serfőző G, Boda K, Ungi I. ACTH- and Cortisol-Associated Neutrophil Modulation in Coronary Artery Disease Patients Undergoing Stent Implantation. PLoS ONE. 2013;8:e71902. doi: 10.1371/journal.pone.0071902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna P, Sil PC. Arjunolic acid: beneficial role in type 1 diabetes and its associated organ pathophysiology. Free Radical Research. 2012;46:815–830. doi: 10.3109/10715762.2012.683431. [DOI] [PubMed] [Google Scholar]
- Manna P, Sinha M, Pal P, Sil PC. Arjunolic acid, a triterpenoid saponin, ameliorates arsenic-induced cyto-toxicity in hepatocytes. Chemico-Biological Interactions. 2007;170:187–200. doi: 10.1016/j.cbi.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Mayadas TN, Cullere X, Lowell CA. The Multifaceted Functions of Neutrophils. Annual Review of Pathology: Mechanisms of Disease. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miriyala S, Panchatcharam M, Landar A, Ramanujam M, Chatterjee S, Muthuswamy A. The Janus of Oxidative Stress Signaling in Different Pathophysiological Conditions. Oxidative Medicine and Cellular Longevity. 2013a;2013:2. doi: 10.1155/2013/708796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miriyala S, Panchatcharam M, Ramanujam M, Puvanakrishnan R. A Novel Tetrapeptide Derivative Exhibits In Vitro Inhibition of Neutrophil-Derived Reactive Oxygen Species and Lysosomal Enzymes Release. Oxidative Medicine and Cellular Longevity. 2013b;2013:7. doi: 10.1155/2013/853210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman SL, Henson JE, Henson PM. Phagocytosis of senescent neutrophils by human monocyte-derived macrophages and rabbit inflammatory macrophages. J Exp Med. 1982;156:430–442. doi: 10.1084/jem.156.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick E, Mizel D. Rapid microassays for the measurement of superoxide and hydrogen peroxide production by macrophages in culture using an automatic enzyme immunoassay reader. J Immunol Methods. 1981;46:211–226. doi: 10.1016/0022-1759(81)90138-1. [DOI] [PubMed] [Google Scholar]
- Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human αB-Crystallin Mutation Causes Oxido-Reductive Stress and Protein Aggregation Cardiomyopathy in Mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky AI, Altman RD, Howell DS. Cathepsin D activity in normal and osteoarthritic human cartilage. Fed Proc. 1973;32:1489–1493. [PubMed] [Google Scholar]
- Saxena P, Aggarwal S, Misso NL, Passage J, Newman MAJ, Thompson PJ, d'Udekem Y, Praporski S, Konstantinov IE. Remote ischaemic preconditioning down-regulates kinin receptor expression in neutrophils of patients undergoing heart surgery. Interactive CardioVascular and Thoracic Surgery. 2013;17:653–658. doi: 10.1093/icvts/ivt279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Blankenberg S. Oxidative Stress in Cardiovascular Disease: Successful Translation From Bench to Bedside? Circulation. 2007;116:1338–1340. doi: 10.1161/CIRCULATIONAHA.107.728394. [DOI] [PubMed] [Google Scholar]
- Seekamp A, Mulligan MS, Till GO, Ward PA. Requirements for neutrophil products and L-arginine in ischemia-reperfusion injury. Am J Pathol. 1993;142:1217–1226. [PMC free article] [PubMed] [Google Scholar]
- Shao D, Oka Si, Brady CD, Haendeler J, Eaton P, Sadoshima J. Redox modification of cell signaling in the cardiovascular system. Journal of Molecular and Cellular Cardiology. 2012;52:550–558. doi: 10.1016/j.yjmcc.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherif I. Amelioration of cisplatin-induced nephrotoxicity in rats by triterpenoid saponin of Terminalia arjuna. Clinical and Experimental Nephrology. 2014:1–7. doi: 10.1007/s10157-014-1056-0. [DOI] [PubMed] [Google Scholar]
- Sumitra M, Manikandan P, Kumar D, Arutselvan N, Balakrishna K, Manohar B, Puvanakrishnan R. Experimental myocardial necrosis in rats: Role of arjunolic acid on platelet aggregation, coagulation and antioxidant status. Molecular and Cellular Biochemistry. 2001;224:135–142. doi: 10.1023/a:1011927812753. [DOI] [PubMed] [Google Scholar]