

Abstract
Transforming growth factor beta 1 (TGF-β1) is implicated in osteoarthritis. We therefore studied the role of TGF-β1 signaling in the development of osteoarthritis in a developmental stage-dependent manner. Three different mouse models were investigated. First, the Tgf-β receptor II (Tgfbr2) was specifically removed from the mature cartilage of joints. Tgfbr2-deficient mice were grown to 12 months of age and were then euthanized for collection of knee and temporomandibular joints. Second, Tgfbr2-deficient mice were subjected to destabilization of the medial meniscus (DMM) surgery. Knee joints were then collected from the mice at 8 and 16 weeks after the surgery. Third, wild-type mice were subjected to DMM at the age of 8 weeks. Immediately after the surgery, these mice were treated with the Tgfbr2 inhibitor losartan for 8 weeks and then euthanized for collection of knee joints. All joints were characterized for evidences of articular cartilage degeneration. Initiation or acceleration of articular cartilage degeneration was not observed by the genetic inactivation of Tgfbr2 in the joints at the age of 12 months. In fact, the removal of Tgfbr2 and treatment with losartan both delayed the progression of articular cartilage degeneration induced by DMM compared with control littermates. Therefore, we conclude that inhibition of Tgf-β1 signaling protects adult knee joints in mice against the development of osteoarthritis.
Transforming growth factor beta 1 (TGF-β1) is considered an anabolic factor to articular chondrocytes, based largely on results from in vitro and ex vivo experiments in which TGF-β1 can stimulate chondrocytes to synthesize and release extracellular matrix molecules, including proteoglycans and type II collagen.1, 2 In addition, results from other studies indicate that the genetic inactivation of Smad3 or disruption of the interaction of Tgf-β1 with its receptor, Tgf-β receptor type II (Tgfbr2), during early development results in osteoarthritis (OA)-like knee joints in mice.3, 4, 5 Moreover, a human genetic study reports that a two-nucleotide deletion, 741-742del AT (nonsense mutation), in SMAD3 causes early-onset OA in a human family.6 These investigations indicate that TGF-β1 is, at least, required for the normal development of a joint.
The lack of TGF-β1 signaling during early development can cause a normal joint to develop into an osteoarthritic joint. However, observations from studies of adult mice as opposed to developing mice suggest that the increase in the activity of TGF-β1 signaling may initiate and accelerate articular cartilage degeneration in adult joints. First, studies in animal models by Itayem et al7, 8 suggest that intra-articular injection of TGF-β1 into adult rat knee joints causes early onset of OA. Second, a study by Bakker et al9 reports that the constitutive overexpression of active TGF-β1 in adult mouse knee joints results in OA associated with increase in the production of proteoglycans in articular cartilage and hyperplasia of synovium and chondro-osteophyte formation. Note that the enhanced production of extracellular matrix molecules is not necessarily beneficial or physiologic to adult articular cartilage. For instance, one of the earliest pathologic signs in articular cartilage degeneration is the overproduction of proteoglycans in mouse models of OA.10, 11 Thus, the overproduction of the proteoglycans could disrupt the homeostasis of adult articular cartilage. Third, the above-mentioned human genetic study reports that a nucleotide change, 859C>T or 782C>T in SMAD3, increases the level of TGF-β1 and activity of the TGF-β1 signaling pathway in two human families associated with early-onset OA.6 In addition, data from a human genetic association study suggest that an increase in the expression of SMAD3 is a risk factor for the development of OA.12 This is in agreement with the observation from studies showing that the level of TGF-β1 is significantly higher in human osteoarthritic tissues than in healthy articular cartilage.13, 14 Fourth, we found that the protein level of Tgf-β1 was significantly increased in the articular chondrocyte of adult knee joints in two mouse models of OA, collagen type XI gene-deficient mice and destabilization of the medial meniscus (DMM).15 On the basis of results from all of the aforementioned studies, a question remains: what is the exact role of TGF-β1 in the development of OA? We hypothesized that TGF-β1 signaling in the development of OA acts in a developmental stage-dependent manner. In this scenario, TGF-β1 is required for the development of articular cartilage; however, once a joint is formed, TGF-β1 is no longer needed. Therefore, induction of TGF-β1 in an adult joint causes articular cartilage degeneration, which eventually leads to OA.
To support our hypothesis, we evaluated the articular cartilage of knee joints for evidence of changes in structural characteristics and protein expression of genes in three different conditions of adult mice. First, Tgfbr2 was specifically removed from the articular cartilage of knee and temporomandibular (TM) joints of mice at the age of 8 weeks. The mice were grown to the age of 12 months, at which point knee and TM joints were collected. Second, adult mice (8 weeks old) without Tgfbr2 in the articular cartilage of their knee joints were subjected to DMM surgery (known to induce OA) and were euthanized at 8 and 16 weeks after DMM for the collection of knee joints. Third, adult wild-type C57BL/6 mice were subjected to DMM and then treated with a Tgfbr2 inhibitor, losartan. The mice were euthanized at 8 weeks after DMM for collection of knee joints. The articular cartilage of joints from the mice and their corresponding controls were analyzed.
Materials and Methods
Inducible Expression of CreERT2 in Articular Cartilages of Knee Joints and Condyles of Mandibles of TM Joints in Adult Mice
All of the animal experimental procedures were performed after approval from the Harvard Medical School Institutional Animal Care Committee. AgcCreERT2+/− mice were bred with Rosa26 lacZ reporter (Rosa26floxlacZ/floxlacZ) mice. Mice containing both AgcCreERT2 and Rosa26 lacZ were identified by PCR. A pair of PCR primers for AgcCreERT2+/− was forward 5′-TAACTACCTGTTTTGCCGGG-3′ and reverse 5′-GTCTGCCAGGTTGGTCAGTAA-3′. A pair of PCR primers for Rosa26+/floxlacZ was forward 5′-AAAGTCGCTCTGAGTTGTTAT-3′ and reverse 5′-GCGAAGAGTTTGTCCTCAACC-3′. The mice were injected intraperitoneally with tamoxifen. The mice were euthanized for the collection of knee joints and mandibles of TM joints. X-Gal staining of the joints was performed.
Efficiency of Tgfbr2 Removal in AgcCreERT2+/−;Tgfbr2f/f Mice
Three of the AgcCreERT2+/−;Tgfbr2f/f mice at the age of 8 weeks were injected intraperitoneally daily with tamoxifen at 2 mg/10 g body weight for 5 consecutive days and another three of the AgcCreERT2+/−;Tgfbr2f/f mice were injected with sunflower seed oil. The mice were then euthanized, and articular cartilages of knee joints were collected for isolation of the genomic DNAs. For the detection of the exon 2 of Tgfbr2, PCR was performed with two forward primers, Tgfbr2-F 5′-TAAACAAGGTCCGGAGCCCA-3′ and Tgfbr2-F4 5′-GGATGGCAAAGAGATAACCCA-3′ and one reverse primer, Tgfbr2-R 5′-AGAGTGAAGCCGTGGTAGGTGAGCTT-3′ (Figure 1).
Figure 1.
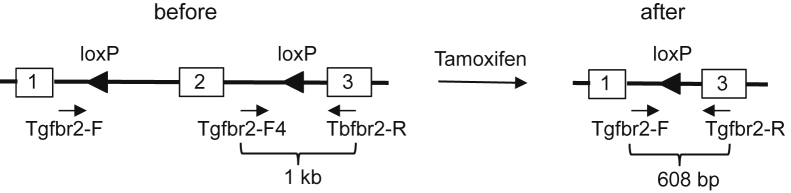
Genotyping strategy for the removal of the exon 2 of Tgfbr2.
For the quantitative measurement of the exon 2 of Tgfbr2, real-time PCR was performed. For an internal positive control, the following primers that target cartilage oligomeric matrix protein were used: forward 5′-ACCCACAACAGGCACATT-3′ and reverse 5′-TCAGTCATAGGAAGCAGG-3′ to generate a 142-bp PCR product. For the detection of the Tgfbr2 gene, the following primers for the exon 2 of Tgfbr2 were used: forward 5′-AACAGTGATGTCATGGCCAG-3′ and reverse 5′-CAGACTTCATGCGGCTTCTC-3′ to generate a 155-bp PCR product. PCR was performed with 25 μL of 1× PCR Master Mix (Life Technologies, Grand Island, NY) that contained each primer at 200 nmol/L and 0.5 μL of genomic DNA from the articular chondrocyte of mouse knee joint. Real-time PCR reaction was performed at 95°C for 3 minutes, followed by 50 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a final extension at 72°C for 4 minutes. Each sample was tested in triplicate.
Histologic Examination of Mouse Knee and TM Joints by Safranin O/Fast Green
At the age of 8 weeks, eight mice from AgcCreERT2+/−;Tgfbr2f/f mice were injected intraperitoneally with tamoxifen (see section above) and another eight AgcCreERT2+/−;Tgfbr2f/f mice were injected intraperitoneally with sunflower seed oil. At the age of 6 months, four mice from each group were euthanized for the collection of knee and TM joints. The remaining mice were kept alive to the age of 12 months and euthanized for the collection of knee and TM joints. Four wild-type littermates were also sacrificed at each age for the collection of knee and TM joints. For knee joints, the samples were fixed in 4% paraformaldehyde for 6 hours at room temperature, decalcified, and processed for paraffin embedding. For TM joints, mouse heads were cut along the midsagittal plane. The right half of the mouse heads were fixed in 4% paraformaldehyde for 6 hours at room temperature, decalcified, and embedded. The samples were then sectioned by serial sectioning at a 6-μm thickness from lateral to medial direction for the knee joints and in an anteroposterior direction for the TM joints. Every 10th section was collected for Safranin O/Fast Green staining.16
Immunohistostaining of Type X Collagen
Eight paraffin sections, distributed throughout each joint, from mice at the age of 12 months (n = 4) were selected for immunohistostaining. Paraffin sections from the growth plate of tibia of C57BL/6 mice at the age of 1 month were used as positive control. The sections were incubated with a rabbit polyclonal antibody (dilution 1:200) against type X collagen (Abcam, Cambridge, MA). After overnight incubation at 4°C, the sections were washed and treated with a biotinylated secondary antibody. Color development was performed with a peroxidase substrate (Vector Laboratories, Burlingame, CA) after treatment of the sections with a mixture of avidin and biotinylated horseradish peroxidase (Vector Laboratories). Staining without primary antibody was performed as negative control.
Destabilization of the Medial Meniscus
Two groups of mice, AgcCreERT2+/−;Tgfbr2−/− (tamoxifen treated), AgcCreERT2+/−;Tgfbr2+/+ (oil treated), at the age of 10 to 12 weeks were generated (see Histologic Examination of Mouse Knee and TM Joints by Safranin O/Fast Green). The mice were subjected either to DMM or sham surgery. Briefly, after the mice were anesthetized intraperitoneally with 90 mg ketamine/kg body weight and 10 mg Xylazine/kg mouse body weight, the right knees were prepared for aseptic surgery. The joint capsule immediately medial to the patellar tendon was opened. The medial meniscotibial ligament was sectioned. The joint capsule was then closed with 8-0 Vicryl (Ethicon, West Somerville, NJ) suture and the subcutaneous layer was closed with 7-0 suture. The skin was closed by the application of tissue adhesive. Sham surgery in which the ligament was visualized but not transected in mice was performed as a negative control.
Evaluation of Articular Cartilage Conditions by a Scoring System
The pathologic condition of articular cartilages was evaluated by a scoring system designed to assess the histology of OA in mouse joints; the system is recommended by the Osteoarthritis Research Society International histopathology initiative.11 The score 0 is for normal mouse articular cartilage and 6 is the maximal score, for vertical clefts/erosion to the calcified cartilage extending >75% of the articular surface.
Immunohistostaining for p-Smad2/3
Paraffin sections of randomly selected four mice from each experimental group were used for protein expression of phospho (p)Smad2/3. The experimental procedure was as described in Immunohistostaining of Type X Collagen with the exception of a primary rabbit polyclonal antibody (dilution 1:500) against p-Smad2/3 (Cell Signaling Technology, Danvers, MA). For each mouse, 10 to 12 paraffin sections were used for the staining. Thus, there were 40 to 48 (4 × 10 or 12) paraffin sections in each experimental group.
Losartan Treatment in DMM Mice
Four groups of C57BL/6 mice were selected as follows: sham with or without losartan (n = 7) and DMM with or without losartan (n = 7). Mice were subjected to DMM or sham surgery. Immediately after the surgery, one group of sham and one group of DMM mice were treated with losartan orally at the concentration of 600 mg/1000 mL drinking water for 8 weeks. The bottle was changed weekly, and the leftover losartan-water was measured to calculate daily water intake. The average intake of losartan was 2.5 mg/10 g mouse body weight daily. Mice without losartan treatment served as controls. Mice at 8 weeks after the surgery were euthanized for collection of knee joints. Structural characteristics of the knee joints was examined by Safranin O/Fast Green staining. The pathologic condition of the joints was evaluated by the scoring system.
Statistical Analysis
Ten to 12 paraffin sections, which were evenly distributed from an entire joint, were examined and scored. The score from the section of the worst condition was selected to represent that joint. Because six to eight mice were in an experimental group, six to eight scores were obtained for each group. An average score was then calculated from six to eight scores for each group.
We used t-tests with a significance level of 0.05 to determine whether a significant difference between any two scores was present. To determine sample size in this study, we performed a pilot study on the effect of the Tgfbr2 deficiency and losartan on mice with DMM. From the results, we concluded that a sample size of minimum six is required to achieve the specified confidence interval (95%) with at least 50% reduction of the score in the treatment group. We also used the t-test in the efficiency of the Tgfbr2 removal and immunohistostaining for p-Smad2/3 experiments.
Results
No Structural Characteristic Changes after Genetic Inactivation of Tgfbr2 in Articular Cartilage of Adult Mouse Joints
We first used a specific mouse strain, Cre-recombinase and the modified estrogen receptor (CreERT2) driven by the aggrecan promoter, AgcCreERT2. CreERT2 was highly inducible in the articular chondrocytes of knee joints (n = 4) in AgcCreERT2 mice at the age of 2 months (Figure 2, A and B). We noticed that the induced CreERT2 appeared in chondrocytes above the tidemark of the articular cartilage. This is consistent with previous findings.17 We also examined the efficiency of the ablation of Tgfbr2 by AgcCreERT2 in the articular chondrocyte of adult mouse knee joints. After several rounds of crossing AgcCreERT2 mice with floxed Tgfbr2 mice, we obtained compound mutant mice, heterozygous CreERT2 driven by the aggrecan promoter and homozygous floxed Tgfbr2 (AgcCreERT2+/−;Tgfbr2f/f). We found that the exon 2 genomic DNA of Tgfbr2 floxed by loxP sites was deleted in 86% articular chondrocytes of adult mouse knee joints (Figure 2C). The loss of the exon 2 resulted in a pre-mature stop codon immediately after the exon 1 of Tgfbr2. This result indicated that Tgfbr2 was deleted in most articular chondrocytes of adult knee joints in AgcCreERT2+/−;Tgfbr2f/f mice.
Figure 2.

The removal of the exon 2 of Tgfbr2 in articular chondrocytes of mouse knee joints. A: Before the Cre-recombination, a size of 1-kb PCR product is generated by the primers Tgfbr2-F4 and Tgfbr2-R and after the Cre-recombination, a size of 608-bp PCR product is generated by the primers Tgfbr2-F and Tgfbr2-R. B: Cells with intense X-gal staining (blue color) appear above the tidemark (dashed line) of the articular cartilage of knee joints in AgcCreERT2+/−,Rosa26+/floxlacZ mice at 2 months of age. A counterstain was performed with Safranin O staining of proteoglycan. C: Every articular chondrocyte in AgcCreERT2+/−;Tgfbr2+/+ littermates contains the exon 2 of Tgfbr2. However, <14% of articular chondrocytes with the exon 2 of Tgfbr2 are observed in the conditionally Tgfbr2-knockout mice. Significant difference was found in the number of the exon 2 of the Tgfbr2 cells between the two groups, ∗∗P < 0.01 (t-test).
On X-Gal staining, cells above the tidemark were considered positive staining cells (or aggrecan-producing cells), whereas cells below the tidemark were classified as negative staining cells. That may be, at least, part of the reason that 14% of cells contain the exon 2 of Tgfbr2 because the cells do not express aggrecan.
We then examined structural characteristic conditions of the articular cartilage of knee joints from AgcCreERT2+/−;Tgfbr2+/+, AgcCreERT2+/−;Tgfbr2−/− mice and their wild-type littermates at the ages of 6 and 12 months. Knee joints from four mice in each group were used for histology analysis. By Safranin O/Fast Green staining, we characterized the cartilage by four structural characteristic appearances, chondrocyte clustering, absence of proteoglycan staining, fibrillation, and missing cartilage. Without any of those phenotypes, we considered the articular cartilage to be normal. We found that there was no structural characteristic differences between AgcCreERT2+/−;Tgfbr2+/+ and the wild-type littermates, indicating no structural characteristic effect by the insertion of the CreERT2 in the mouse genome, consistent with earlier findings.17 We did not observe structural characteristic differences between AgcCreERT2+/−;Tgfbr2−/− and AgcCreERT2+/−;Tgfbr2+/+ mice (Figure 3), which suggested that the removal of Tgfbr2 from the articular cartilage after mice maturity (adult mice) did not cause any overt structural characteristic changes in knee joints of the mice.
Figure 3.

Structural characteristics of articular cartilage in mouse knee joints. No overt observable structural characteristic changes were found in the articular cartilage of knee joints in the Tgfbr2-deficient mice at the ages of 6 and 12 months (Safranin O/Fast Green). Scale bar = 100 μm.
Data from in vivo experiments indicated that the inhibition of activity of TGF-β1 signaling results in pre-mature hypertrophy of articular chondrocytes in mouse joints during early development (before maturity at the age of 6 to 8 weeks). Type X collagen is one of the markers for chondrocyte hypertrophy. We examined the protein expression of type X collagen in the articular cartilage of knee joints of mice at 12 months of age. In these mice, Tgfbr2 had been removed from the articular cartilage at the age of 2 months. Ten months after Tgfbr2 removal, we did not detect the protein expression of type X collagen in the articular cartilage of knee joints in these mice (Figure 4).
Figure 4.
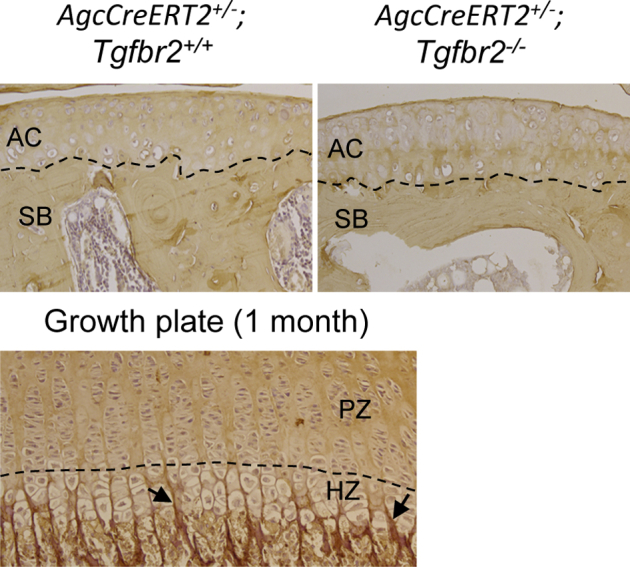
Immunohistostaining for collagen type X. Collagen type X is present in the extracellular matrix of the HZ of growth plates in mice at 1 month of age (arrowheads). The collagen type X is absent in the PZ of the growth plate. No positive staining of the collagen type X was found in the AC of both Tgfbr2-deficient mice and their AgcCreERT2+/−;Tgfbr2+/+ littermates at 12 months of age. The dashed line in the growth plate indicates the boundary between PZ and HZ. The dashed lines in ACs indicate the boundary between the articular cartilage and subchondral bone. AC, articular cartilage; HZ, hypertrophic zone; PZ, proliferative zone; SB, subchondral bone.
Although an independent study reports that the expression of CreERT2 is inducible in the articular chondrocyte of adult knee joints in AgcCreERT2 mice,17 it is unknown whether CreERt2 is also inducible in the condylar cartilage of adult TM joints. By crossing AgcCreERT2+/− mice with Rosa26floxlacZ/floxlacZ mice, we found that CreERT2 was highly expressed in the condylar cartilage of TM joints in mice at the age of 1 month and 6 months (Figure 5). This result indicated that AgcCreERT2 mouse strain could also be used to remove a gene of interest, such as Tgfbr2, in the condylar cartilage of adult TM joints.
Figure 5.

X-Gal staining in the condylar cartilage of TM joints in mice. Intensive X-gal staining cells (blue color) are observed in the bottom layer of condylar cartilage in TM joints of AgcCreERT2+/−,Rosa26+/floxlacZ mice at the ages of 1 (B and F) and 6 (D and H) months. The black dashed lines in F and H indicates the border between the condylar cartilage and subchondral bone. No X-gal staining cells are in the top layer of the condylar cartilage in TM joints, which is consistent with the structural characteristics of the condylar cartilage of TM joints by Safranin O/Fast Green staining (I and J). X-Gal staining cells are absent in the cartilage of TM joints in the control mice (A, C, E, and G). TM, temporomandibular.
We then examined the structural characteristics of TM joints from AgcCreERT2+/−;Tgfbr2+/+, AgcCreERT2+/−;Tgfbr2−/− mice and their wild-type littermates at the ages of 6 and 12 months. No structural characteristic differences were found in TM joints among the mice (Figure 6). This result was consistent with our observation that in adult knee joints in which the Tgfbr2 was removed from the adult articular cartilage, no overt structural characteristic changes were observed in the joints.
Figure 6.

Structural characteristics of condylar cartilage in mouse TM joints. No overt structural characteristic changes are seen in the condylar cartilage of TM joints in the Tgfbr2-deficient mice at the ages of 6 and 12 months. Safranin O/Fast Green. Scale bar = 100 μm. TM, temporomandibular.
Genetic inactivation of Tgfbr2 in articular cartilage of adult mouse knee joints results in a chondroprotective effect against the development of OA.
We first determined whether the removal of Tgfbr2 from the articular cartilage of adult knee joints could attenuate the progression of cartilage degeneration. We performed DMM surgery on knee joints of adult AgcCreERT2+/−;Tgfbr2−/− mice and their corresponding controls. We found significant disparities in the progressive process of the articular cartilage degeneration in knee joints between AgcCreERT2+/−;Tgfbr−/− and AgcCreERT2+/−;Tgfbr2+/+ mice at 8 and 16 weeks after DMM surgery (Figure 7A). The progression toward OA was dramatically delayed in the AgcCreERT2+/−;Tgfbr2−/− mice after the surgery. No abnormal structural characteristic changes were found in the sham surgery groups.
Figure 7.

Structural characteristic evaluation of the articular cartilage of knee joints from the Tgfbr2-deficent mice after DMM surgery. A: Fibrillation (arrow) in the articular cartilage of AgcCreERT2+/−;Tgfbr2+/+ littermates at 8 weeks after the surgery. However, only localized absence of the proteoglycans is observed in AgcCreERT2+/−;Tgfbr2−/− mice at 8 weeks after the surgery. At 16 weeks after the surgery, a complete loss of the articular cartilage (lesion reaching the tidemark and beyond) is evident in both femoral and tibia condyles of the AgcCreERT2+/−;Tgfbr2+/+ littermates. The missing cartilage is extended >75% of the surface area. In the AgcCreERT2+/−;Tgfbr2−/− mice, the fibrillation reaching to the tidemark is evident, and the missing cartilage is only extended <25% of the surface area. A delay in the degenerative process is seen in Tgfbr2-deficent mice after surgery. B: The mean scores were 2.19 ± 0.79 in AgcCreERT2+/−;Tgfbr2+/+ mice and 0.88 ± 0.22 in AgcCreERT2+/−;Tgfbr2−/− mice at 8 weeks after DMM surgery. C: The mean scores were 4.17 ± 0.70 in AgcCreERT2+/−;Tgfbr2+/+ mice and 2.25 ± 0.66 in AgcCreERT2+/−;Tgfbr2−/− mice at 16 weeks after DMM surgery. Data are expressed as means ± SD. n = 8 mice. ∗P < 0.05 (t-test). Scale bar = 100 μm. DMM, destabilization of the medial meniscus.
The condition of the articular cartilage was also evaluated with the scoring system designed to assess the histology of the articular cartilage in DMM model of OA (Figure 7, B and C). Mice at 8 weeks after sham surgery were used as a normal control (score = 0). At 8 weeks after DMM surgery, the average scores for AgcCreERT2+/−;Tgfbr2−/− mice and AgcCreERT2+/−;Tgfbr2+/+ littermates were 0.88 and 2.19, respectively. At 16 weeks after surgery, the average scores were 2.25 for AgcCreERT2+/−;Tgfbr2−/− littermates and 4.17 for AgcCreERT2+/−;Tgfbr2+/+ littermates. The scores were significantly different between the two groups at both time points (P < 0.05). In addition, we noticed the appearance of osteophytes in AgcCreERT2+/−;Tgfbr2+/+ littermates at 16 weeks. However, no evidence of osteophyte formation was found in AgcCreERT2+/−;Tgfbr2−/− littermates. We did not observe any abnormal structural characteristics in the subchondral bone and synovial tissue in both AgcCreERT2+/−;Tgfbr2−/− and AgcCreERT2+/−;Tgfbr2+/+ mice.
We then examined the protein expression of p-Smad2/3 in the articular cartilage of knee joints from mice at 8 weeks after DMM. The positive staining cells were counted in each paraffin section and were added to determine the total number of the positive cells from all of the sections in each group for quantitation of the percentage of cells expressing p-Smad2/3 protein. p-Smad2/3 protein-expressing cells were detected at approximately 2% in the articular cartilage of knee joints from all of the Tgfbr2-deficient mice (AgcCreERT2+/−;Tgfbr2−/−). However, p-Smad2/3 protein-positive cells were present at 17% in the articular cartilage of knee joints from the AgcCreERT2+/−;Tgfbr2+/+ littermates. The number of the positive staining cells was significantly different between the two groups (P < 0.05). The location of the p-Smad2/3+ cells was randomly scattered in the superficial layer of the articular cartilage (Figure 8).
Figure 8.
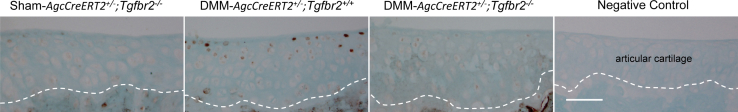
Immunohistostaining of p-Smad2/3 in the articular cartilage of the Tgfbr2-deficent mice. p-Smad2/3 protein-positive staining cells (17%) are present in the articular cartilage of AgcCreERT2+/−;Tgfbr2+/+ mice after DMM surgery. However, the p-Smad2/3+ staining cells are only detected at approximately 2% in the Tgfbr2-defecient mice. The white dashed line separates the articular cartilage from the subchondral bone. The number of the positive staining cells is significantly different between the two groups (P < 0.05). Scale bar = 50 μm. DMM, destabilization of the medial meniscus.
Delayed Progression of Surgically Induced Articular Cartilage Degeneration by the Oral Administration of Losartan in Adult Mice
In earlier experiments we found that the appearance of fibrillation at the superficial layer of articular cartilage was a hallmark of cartilage degeneration in mouse knee joints at 8 weeks after DMM.18 Indeed, we found fibrillation in knee joints of mice at 8 weeks after DMM without the treatment of losartan (Figure 9A). However, no fibrillation was observed in any of the mice at 8 weeks after DMM with the treatment of losartan. The degradation of proteoglycans was the only structural characteristic change that was seen in the DMM mice. No structural characteristic changes were found in mice undergoing sham surgery with or without the treatment of losartan. This result indicates that losartan could delay the progression of articular cartilage degeneration in the mouse model of OA.
Figure 9.

Structural characteristic evaluation of the articular cartilage of knee joints from losartan-treated mice at 8 weeks after DMM surgery. A: The structural characteristic conditions of articular cartilages in mice are similar to Tgfbr2-deficient mice. Fibrillation (arrow) in the articular cartilage of mice without treatment of losartan; no fibrillation is observed in mice with treatment of losartan (Safranin O/Fast Green). B: The mean scores were 2.14 ± 0.58 in mice without the treatment of losartan and 0.64 ± 0.23 in mice with the treatment of losartan. C: Immunostaining for p-Smad2/3 protein is similar to Tgfbr2-deficient mice. The p-Smad2/3 positive-staining cells (16%) are seen in the articular cartilage of mice without treatment of losartan after DMM, whereas the p-Smad2/3 positive-staining cells are detected at approximately 1% in the losartan-treated mice after DMM surgery. The white dashed line separates the articular cartilage from the subchondral bone. Data are expressed as means ± SD. n = 8 mice. n = 7 mice. ∗P < 0.05 (t-test). Scale bars: 100 μm (A); 50 μm (B). DMM, destabilization of the medial meniscus.
We evaluated the condition of articular cartilage of mouse knee joints by a scoring system (Figure 9B). A significant difference was found between the two groups, 0.64 in the losartan-treated group and 2.14 in the nontreated group (P < 0.05). This indicates that losartan is able to delay the progression of articular cartilage degeneration, induced by DMM, in adult mouse knee joints.
We also examined the protein expression of p-Smad2/3 in the articular cartilage of knee joints from mice treated with losartan. The expression profile of p-Smad2/3 in the knee joints was similar to what we observed in the articular cartilage of Tgfbr2-deficient mice. The protein expression of p-Smad2/3 was detected in 1% of cells in the articular cartilage of knee joints from DMM mice treated with losartan, whereas the protein of p-Smad2/3 was present in 16% of cells in the articular cartilage of knee joints from DMM mice without the treatment of losartan (Figure 9C). The number of the positive staining cells was significantly different between the two groups (P < 0.05). Again, the positive staining cells were randomly scattered in the superficial layer of the articular cartilage.
Discussion
TGF-β1 in the Development of Articular Cartilage
Results from three independent research groups indicate that Tgf-β1 is required for the development of articular cartilage of knee joints in mice.3, 4, 5 Tgf-β1/Smad2/3 may play an important role in the control of chondrocyte hypertrophic differentiation in articular cartilage. The complete removal of Smad3 in mouse germline cells results in a high number of hypertrophic chondrocytes in the basal layer of articular cartilage of knee joints in 1-month-old mice. The deficiency of Smad3 may enhance bone morphogenetic protein signaling and deregulate p38, which leads to chondrocyte hypertrophy.19, 20 Results from other in vitro studies indicate that TGF-β1 can inhibit chondrocyte hypertrophy by regulating the expression of some cartilage matrix proteins and metalloproteases.21, 22 In addition, the lack of the Tgf-β1/Smad2/3 signaling may activate runt-related transcription factor 2 (Runx2)-inducible expression of matrix metalloprotease 13 (MMP-13), which leads to the degeneration of articular cartilage.23 Clearly, TGF-β1 is a critical factor in the development of articular cartilage. However, no information suggests that this is also the case in adult (mature) articular cartilage.
TGF-β1 in the Maintenance of Articular Cartilage
The possible role of TGF-β1 in the maintenance of the mature articular cartilage remains unclear. Whether TGF-β1 is one of the key molecules in the maintenance of articular cartilages in adult joints remains unresolved. Tgf-β3 is present in the articular cartilage of adult mouse knee joints,24 whereas activin receptor-like kinase 1 (Alk1) and Alk5 are present in the articular cartilage of adult mouse knee joints.25 As mice age, the ratio of Alk1 to Alk5 increases, suggesting either a dramatic increase in Alk1 or a dramatic decrease in Alk5. A decrease in Alk5 would cause induction of cartilage-degrading enzymes, such as MMP-13, in aged cartilage. The causal relation between TGF-β signaling and aging articular cartilage needs to be explored further.
To understand the role of Tgf-β1 in the maintenance of mature articular cartilage, Tgfbr2 was conditionally knocked out in the articular cartilage of mice at the age of 8 weeks in this study. (Complete maturation of laboratory mice typically occurs at the age of 6 to 8 weeks.) Mice were then euthanized at the age of 12 months. No overt structural characteristic changes in the articular cartilage of the knee or the condylar cartilage of the TM joints of the Tgfbr2-deficient mice were observed. The protein expression of type X collagen, one of the molecular markers for chondrocyte hypertrophy, in the articular cartilage of knee joints of Tgfbr2-deficient mice was also examined. The presence of type X collagen was not detected in the cartilage, suggesting that the removal of Tgfbr2 from the mature articular cartilage may not have any effect on chondrocyte hypertrophic differentiation. In addition, the presence of Tgf-β1 was not detected in mature articular cartilage of mouse knee joints. These results suggest an insignificant role of Tgf-β1 in the maintenance of mature articular cartilage.
TGF-β1 in the Degenerative Process of Adult Articular Cartilage
Results from numerous independent studies and data from our previous investigation demonstrate that the protein expression of TGF-β1 is dramatically increased in human osteoarthritic cartilages and in the articular cartilage of mouse models of OA.13, 14, 15 Because data from in vitro experiments indicate that TGF-β1 can stimulate chondrocytes to synthesize extracellular matrix molecules, such as type II collagen and proteoglycans, it is suggested that the increased expression of TGF-β1 may partly counter articular cartilage degeneration. However, other investigations find that the up-regulated expression of TGF-β1 causes articular cartilage degeneration. First, mature articular cartilage is a relatively quiescent tissue. A study by Verzijl et al26 indicates that the half-life of type II collagen in humans is 117 years. The long half-life of type II collagen indicates a slow turnover of collagen in mature articular cartilage. This also suggests that it may not be needed for chondrocytes to continually produce type II collagen in mature articular cartilage. Thus, the TGF-β1–stimulated overproduction of type II collagen or/and proteoglycans in mature articular cartilage, in fact, may disrupt the homeostasis of the tissue, which eventually leads to cartilage degeneration. Second, an independent research group found that TGF-β1 induces a serine protease, high temperature requirement A1 (HTRA1) in human chondrocytes.27 Results from our in vitro experiments confirmed this observation.15 Furthermore, we found increased expression of Tgf-β1, p-Smad2/3, and HtrA1 in articular chondrocytes of knee joints in mouse models of OA. We also observed that increased expression of p-Smad2/3 and HtrA1 were co-localized in the chondrocyte. In addition, TGF-β1–induced expression of HTRA1 was inhibited by the ALK-5 inhibitor SB431542 in human and mouse chondrocytes, suggesting that the Tgf-β1 canonical signaling is activated to induce HtrA1 in articular chondrocytes of the mouse models of OA.
To determine whether the inhibition of Tgf-β1 signaling could attenuate the progression of articular cartilage degeneration, we conditionally deleted Tgfbr2 in the mature articular cartilage of knee joints of mice and then subjected the mice to DMM surgery. We found that the knee joints in the Tgfbr2-deficient mice were protected from degradation. To validate this observation, we treated wild-type mice with the Tgfbr2 inhibitor losartan28 immediately after DMM surgery. We also found that the knee joints in the losartan-treated mice were protected from being degraded. On the basis of these results, we conclude that inhibiting the activity of TGF-β1 signaling in mature articular cartilage can, in fact, delay the progression of articular cartilage degeneration.
Mechanical stress is believed to be the initial insult to articular cartilage during the development of OA. Induction of TGF-β1 is one of the responses to mechanical stress in chondrocytes. Our study demonstrated that normal mechanical loading of defective joints or an overloading of normal joints could stimulate chondrocytes to synthesize and release Tgf-β1 in mouse models of OA.15 A study by Lee et al29 reported that mechanical injury of bovine cartilage explants causes a significant increase in TGF-β1 gene expression. Another recent study by Madej et al30 showed that excessive mechanical stress can activate Tgf-β1 signaling via p-Smad2/3 in bovine articular cartilage. On the basis of the established and reported data, we propose a molecular pathway (Figure 10) underlying articular cartilage degeneration as follows: excessive mechanical stress can stimulate chondrocytes and other joint tissues to synthesize and release TGF-β1. The active TGF-β1 binds to its cognate receptor, TGFBR2, which induces expression of HTRA1 in chondrocytes.15, 27 Consequences of induction of HTRA1 are degradation of the pericellular matrix of chondrocytes and enhanced exposure of chondrocytes to type II collagen.31 Interaction of chondrocytes with type II collagen results in enhanced signaling through a cell surface receptor tyrosine kinase, discoidin domain receptor 2, for native type II collagen.32 This induces the expression of MMP-13 and expression of discoidin domain receptor 2 itself.33 MMP-13 degradation of type II collagen and aggrecan results in type II collagen and aggrecan fragments, which in turn may activate signals that further increase the synthesis of MMP-13.34 The end result is a feedback amplification loop that causes irreversible articular cartilage degeneration.
Figure 10.

A schematic illustration of the molecular pathways underlying articular cartilage degeneration. DDR2, discoidin domain receptor 2; MMP, matrix metalloprotease; TGF, transforming growth factor.
It is likely that the proposed molecular pathway is not the only sequential chain of the molecular events responsible for the articular cartilage degeneration, leading to OA. Other signaling pathways, such as Toll-like receptor signaling and Wnt/β-Catenin signaling, may also be involved in the initiation and progression of articular cartilage degeneration.35, 36 Furthermore, evidence is increasing that inflammation, such as cytokines released from the synovium or chondrocytes, plays a role in the articular cartilage destruction.37, 38 Other factors, such as chemokines,39 angiogenic factors, and neuropeptides,40 also play roles in the development of OA. Furthermore, many other genes, discovered by epigenetic approaches, are involved in the pathogenesis of OA.41, 42 Although it is a formidable challenge to determine how exactly all these factors cause OA, the molecular understanding of the complex genetic networks responsible for the articular cartilage destruction will provide invaluable information for the development of disease-modifying osteoarthritis drugs.
In summary, TGF-β1 is a potential pathogenic factor in the development of OA in adult joints. Therefore, inhibition of activity of TGF-β1, not induction of TGF-β1, should be considered in the prevention and treatment of OA.
Acknowledgments
The AgcCreERT2+/− mouse strain was provided by Drs. Stephen P. Henry and Benoit de Crombrugghe (MD Anderson Cancer Center, Houston, TX).
Footnotes
Supported in part by NIH grant R01 AR051989 (L.X. and L.Y.).
Disclosures: None declared.
Contributor Information
Yefu Li, Email: yefu_li@hms.harvard.edu.
Lin Xu, Email: lin_xu@hms.harvard.edu.
References
- 1.Galéra P., Vivien D., Pronost S., Bonaventure J., Rédini F., Loyau G., Pujol J.P. Transforming growth factor-beta 1 (TGF-beta 1) up-regulation of collagen type II in primary cultures of rabbit articular chondrocytes (RAC) involves increased mRNA levels without affecting mRNA stability and procollagen processing. J Cell Physiol. 1992;153:596–606. doi: 10.1002/jcp.1041530322. [DOI] [PubMed] [Google Scholar]
- 2.van Beuningen H.M., van der Kraan P.M., Arntz O.J., van den Berg W.B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab Invest. 1994;71:279–290. [PubMed] [Google Scholar]
- 3.Serra R., Johnson M., Filvaroff E.H., LaBorde J., Sheehan D.M., Derynck R., Moses H.L. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang X., Chen L., Xu X., Li C., Huang C., Deng C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153:35–46. doi: 10.1083/jcb.153.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen J., Li J., Wang B., Jin H., Wang M., Zhang Y., Yang Y., Im H.J., O'Keefe R., Chen D. Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013;65:3107–3119. doi: 10.1002/art.38122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van de Laar I.M., Oldenburg R.A., Pals G., Roos-Hesselink J.W., de Graaf B.M., Verhagen J.M., Hoedemaekers Y.M., Willemsen R., Severijnen L.A., Venselaar H., Vriend G., Pattynama P.M., Collée M., Majoor-Krakauer D., Poldermans D., Frohn-Mulder I.M., Micha D., Timmermans J., Hilhorst-Hofstee Y., Bierma-Zeinstra S.M., Willems P.J., Kros J.M., Oei E.H., Oostra B.A., Wessels M.W., Bertoli-Avella A.M. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 7.Itayem R., Mengarelli-Widholm S., Hulth A., Reinholt F.P. Ultrastructural studies on the effect of transforming growth factor-β1 on rat articular cartilage. APMIS. 1997;105:221–228. doi: 10.1111/j.1699-0463.1997.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 8.Itayem R., Mengarelli-WidholmI S., Reinholt F. The lone-term effect of a short course of transforming growth factorβ1 on rat articular cartilage. APMIS. 1999;107:183–192. doi: 10.1111/j.1699-0463.1999.tb01543.x. [DOI] [PubMed] [Google Scholar]
- 9.Bakker A.C., van de Loo F.A., van Beuningen H.M., Sime P., van Lent P.L., van der Kraan P.M., Richards C.D., van den Berg W.B. Overexpression of active TGF-beta-1 in the murine knee joint: evidence for synovial-layer-dependent chondro-osteophyte formation. Osteoarthritis Cartilage. 2001;9:128–136. doi: 10.1053/joca.2000.0368. [DOI] [PubMed] [Google Scholar]
- 10.Xu L., Flahiff C.M., Waldman B.A., Wu D., Olsen B.R., Setton L.A., Li Y. Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of chondrodysplasia gene (cho) Arthritis Rheum. 2003;48:2509–2518. doi: 10.1002/art.11233. [DOI] [PubMed] [Google Scholar]
- 11.Xu L., Polur I., Lim C., Servais J.M., Dobeck J., Li Y., Olsen B.R. Early-onset osteoarthritis of mouse temporomandibular joint induced by partial discectomy. Osteoarthritis Cartilage. 2009;17:917–922. doi: 10.1016/j.joca.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raine E.V., Reynard L.N., van de Laar I.M., Bertoli-Avella A.M., Loughlin J. Identification and analysis of a SMAD3 cis-acting eQTL operating in primary osteoarthritis and in the aneurysms and osteoarthritis syndrome. Osteoarthritis Cartilage. 2014;22:698–705. doi: 10.1016/j.joca.2014.02.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlaak J.F., Pfers I., Meyer Zum Büschenfelde K.H., Märker-Hermann E. Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin Exp Rheumatol. 1996;14:155–162. [PubMed] [Google Scholar]
- 14.Kawamura I., Maeda S., Imamura K., Setoguchi T., Yokouchi M., Ishidou Y., Komiya S. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J Biol Chem. 2012;287:29101–29113. doi: 10.1074/jbc.M112.349415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L., Golshirazian I., Asbury B.J., Li Y. Induction of high temperature requirement A1, a serine protease, by TGF-beta1 in articular chondrocytes of mouse models of OA. Histol Histopathol. 2014;29:609–618. doi: 10.14670/HH-29.10.609. [DOI] [PubMed] [Google Scholar]
- 16.Kiernan J.A. ed 3. Butterworth-Heinemann; Oxford, UK: 1999. Histological & Histochemical Methods. Theory & Practice. [Google Scholar]
- 17.Henry S.P., Liang S., Akdemir K.C., de Crombrugghe B. The postnatal role of Sox9 in cartilage. J Bone Miner Res. 2012;27:2511–2525. doi: 10.1002/jbmr.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L., Servais J., Polur I., Kim D., Lee P., Chung K., Li Y. Attenuation of osteoarthritis progression by reduction of the discoidin domain receptor 2 in mice. Arthritis Rheum. 2010;62:2736–2744. doi: 10.1002/art.27582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T.F., Darowish M., Zuscik M.J., Chen D., Schwarz E.M., Rosier R.N., Drissi H., O'Keefe R.J. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li T.F., Gao L., Sheu T.J., Sampson E.R., Flick L.M., Konttinen Y.T., Chen D., Schwarz E.M., Zuscik M.J., Jonason J.H., O'Keefe R.J. Aberrant hypertrophy in Smad3-deficient murine chondrocytes is rescued by restoring transforming growth factor beta-activated kinase 1/activating transcription factor 2 signaling: a potential clinical implication for osteoarthritis. Arthritis Rheum. 2010;62:2359–2369. doi: 10.1002/art.27537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballock R.T., Heydemann A., Wakefield L.M., Flanders K.C., Roberts A.B., Sporn M.B. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytes: regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- 22.Dreier R. Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res Ther. 2010;12:216. doi: 10.1186/ar3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C.G., Thuillier D., Chin E.N., Alliston T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum. 2012;64:3278–3289. doi: 10.1002/art.34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaney Davidson E.N., Vitters E.L., van der Kraan P.M., van den Berg W.B. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Ann Rheum Dis. 2006;65:1414–1421. doi: 10.1136/ard.2005.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaney Davidson E.N., Remst D.F., Vitters E.L., van Beuningen H.M., Blom A.B., Goumans M.J., van den Berg W.B., van der Kraan P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J Immunol. 2009;182:7937–7945. doi: 10.4049/jimmunol.0803991. [DOI] [PubMed] [Google Scholar]
- 26.Verzijl N., DeGroot J., Thorpe S.R., Bank R.A., Shaw J.N., Lyons T.J., Bijlsma J.W., Lafeber F.P., Baynes J.W., TeKoppele J.M. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 27.Urano T., Narusawa K., Kobayashi S., Shiraki M., Horie-Inoue K., Sasaki N., Hosoi T., Ouchi Y., Nakamura T., Inoue S. Association of HTRA1 promoter polymorphism with spinal disc degeneration in Japanese women. J Bone Miner Metab. 2010;28:220–226. doi: 10.1007/s00774-009-0124-0. [DOI] [PubMed] [Google Scholar]
- 28.Guo Z.X., Qiu M.C. [Losartan downregulates the expression of transforming growth factor-β type I and type II receptors in kidney of diabetic rat] Zhonghua Nei Ke Za Zhi. 2003;42:403–408. Chinese. [PubMed] [Google Scholar]
- 29.Lee J.H., Fitzgerald J.B., Dimicco M.A., Grodzinsky A.J. Mechanical injury of cartilage explants causes specific time-dependent changes in chondrocyte gene expression. Arthritis Rheum. 2005;52:2386–2395. doi: 10.1002/art.21215. [DOI] [PubMed] [Google Scholar]
- 30.Madej W., van Caam A., Blaney Davidson E.N., van der Kraan P.M., Buma P. Physiological and excessive mechanical compression of articular cartilage activates Smad2/3P signaling. Osteoarthritis Cartilage. 2014;22:1018–1025. doi: 10.1016/j.joca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 31.Polur I., Lee P.L., Servais J.M., Xu L., Li Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol. 2010;25:599–608. doi: 10.14670/hh-25.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leitinger B., Steplewski A., Fertala A. The D2 period of collagen II contains a specific binding site for the human discoidin domain receptor, DDR2. J Mol Biol. 2004;344:993–1003. doi: 10.1016/j.jmb.2004.09.089. [DOI] [PubMed] [Google Scholar]
- 33.Xu L., Peng H., Wu D., Hu K., Goldring M.B., Olsen B.R., Li Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem. 2005;280:548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- 34.Fichter M., Körner U., Schömburg J., Jennings L., Cole A.A., Mollenhauer J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J Orthop Res. 2006;24:63–70. doi: 10.1002/jor.20001. [DOI] [PubMed] [Google Scholar]
- 35.Miclea R.L., Siebelt M., Finos L., Goeman J.J., Löwik C.W., Oostdijk W., Weinans H., Wit J.M., Robanus-Maandag E.C., Karperien M. Inhibition of Gsk3β in cartilage induces osteoarthritic features through activation of the canonical Wnt signaling pathway. Osteoarthritis Cartilage. 2011;19:1363–1372. doi: 10.1016/j.joca.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Hou Y., Lin H., Zhu L., Liu Z., Hu F., Shi J., Yang T., Shi X., Zhu M., Godley B.F., Wang Q., Li Z., Zhao Y. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA1 expression. Arthritis Rheum. 2013;65:2835–2846. doi: 10.1002/art.38124. [DOI] [PubMed] [Google Scholar]
- 37.Loeser R.F. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006;54:1357–1360. doi: 10.1002/art.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scanzello C.R., Goldring S.R. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51:249–257. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vergunst C.E., van de Sande M.G., Lebre M.C., Tak P.P. The role of chemokines in rheumatoid arthritis and osteoarthritis. Scand J Rheumatol. 2005;34:415–425. doi: 10.1080/03009740500439159. [DOI] [PubMed] [Google Scholar]
- 40.Mapp P.I., Walsh D.A. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat Rev Rheumatol. 2012;8:390–398. doi: 10.1038/nrrheum.2012.80. [DOI] [PubMed] [Google Scholar]
- 41.Goldring M.B., Marcu K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol Med. 2012;18:109–118. doi: 10.1016/j.molmed.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M., Wang J. Epigenetics and osteoarthritis. Genes Dis. 2015;2:69–75. doi: 10.1016/j.gendis.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]