

Abstract
Background
The opioid antagonists, naloxone/naltrexone, are involved in improving learning and memory, but their cellular and molecular mechanisms remain unknown. We investigated the effect of naloxone/naltrexone on hippocampal AMPAR trafficking, a molecular substrate of learning and memory, as a probable mechanism for the antagonists activity.
Methods
To measure naloxone/naltrexone-regulated AMPAR trafficking, pHluorin-GluA1 imaging and biochemical analyses were performed on primary hippocampal neurons. To establish the in vivo role of GluA1-S845 phosphorylation on the behavioral effect induced by inhibition of the endogenous μ-opioid receptor (MOR) by naltrexone, MOR knockout (MORKO) and GluA1-S845A mutant (in which Ser845 was mutated to Ala) mice were tested in a water maze after chronic naltrexone administration. Behavioral responses and GluA1 levels in the hippocampal postsynaptic density (PSD) in wild-type and GluA1-S845A mutants mice were compared using Western blot analysis.
Results
In vitro prolonged naloxone/naltrexone exposure significantly increased synaptic and extrasynaptic GluA1 membrane expression as well as GluA1-S845 phosphorylation. In the MORKO and GluA1-S845A mutant mice, naltrexone did not improve learning, which suggests that naltrexone acts via inhibition of endogenous MOR action and alteration of GluA1 phosphorylation. Naltrexone-treated wild-type mice had significantly increased phosphorylated GluA1-S845 and GluA1 levels in their hippocampal PSD on the third day of acquisition, which is the time when naltrexone significantly improved learning.
Conclusions
The beneficial effect of naltrexone on spatial learning and memory under normal conditions appears to be the result of increasing GluA1-S845 phosphorylation-dependent AMPAR trafficking. These results can be further explored in a mouse model of memory loss.
Keywords: Naltrexone, AMPA receptors, GluA1, GluA1-S845, Spatial Memory, GluA1-S845A mutants
Introduction
The endogenous opioid system modulates many functions, including learning and memory. For instance, intracerebral administration of β-endorphin or endomorphin-1/2 in rodents significantly impairs retention memory and learning in both inhibitory/passive avoidance and water maze tasks (1,2). Also, the μ-opioid receptor (MOR) agonist morphine, an exogenous opioid that mimics endorphin action, modulates learning and memory in both humans and animals (3,4) which is usually counteracted by an opioid antagonist, such as naloxone or naltrexone. More importantly, other investigations demonstrated that post-training administration of naloxone or naltrexone alone, which presumably inhibits endogenous opioids, ameliorates the retention of both passive and active avoidance conditioning (5,6,7) as well as acquisition on a radial arm maze in a novel spatial environment in rats (8). Moreover, experiments conducted on monkeys showed a significant naloxone-induced improvement of memory in animals with especially poor control scores, supporting the possibility that individuals with weak memory may be particularly responsive to the facilitating effects of naloxone/naltrexone (9). However, the molecular and cellular mechanisms by which naloxone/naltrexone promote enhanced memory still remain unknown.
One of the neurobiological modifications underlying different forms of learning has been shown to involve the recruitment and activation of additional functional excitatory glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs) on the neuronal postsynaptic membrane and increasing of dendritic spine size and density (10,11,12). GuA1, one of the four subunits constituting ionotropic AMPAR (13), plays a key role in activity-dependent synaptic trafficking of AMPARs, especially in the adult hippocampus, which is, among others, an essential structure for spatial learning and memory (14).
Chronic administration of morphine decreases the density of dendritic spines in vitro (15,16,17) and in vivo (18) as well as the amount of synaptic GluA1 through internalization in cultured hippocampal neurons (19). Also in a behavioral model of drug addiction, morphine can induce context-dependent sensitization through changes of AMPAR expression and phosphorylation in the postsynaptic density (PSD) of mice hippocampi (20). In contrast, treatment with naloxone or naltrexone or genetic deletion of MOR increases the density of dendritic spines (15). Thus, a probable mechanism by which naloxone and naltrexone improve learning and memory might be by affecting AMPAR trafficking through MOR inhibition.
In the present study, we first established that the facilitative effects of chronic naltrexone treatment on acquisition and retention in a water maze task are mediated by the blockade of endogenous MOR. Using cultured hippocampal neurons and following treatment with naloxone and naltrexone, we then observed induction of the temporal translocation of GluA1 from the intra-cellular compartment to the surface membrane with a parallel increase in GluA1-S845 phosphorylation, which has been shown to affect in AMPAR trafficking and synaptic delivery (21,22). The recruitment of AMPARs on the membrane surface and GluA1-S845 phosphorylation may be related to the memory improvements observed during acquisition which was demonstrated by comparing the behavioral responses and GluA1 levels in the PSD in both wild-type and GluA1-S845A mutant mice with chronic naltrexone administration.
Methods and Materials
Animals
Heterozygous GluA1-S845A mutant and homozygous C57BL/6J MORKO mice were generous gifts from Dr. Hey-Kyoung Lee (Johns Hopkins School of Medicine, Baltimore, Maryland) and Dr. John Pintar (Robert Wood Johnson Medical School, Piscataway, New Jersey), respectively. The homozygous mice in which the cre gene is absent were produced from the heterozygous GluA1-S845A line and identified by PCR. Wild-type, homozygous GluA1-S845A and MORKO mice were used for breeding and housed in a temperature-controlled (21–23°C) environment with a 12-hours light/dark cycle. Food and water were provided ad libitum. Experiments were conducted on two-months-old male mice (30–35 g). All animal procedures followed the National Institutes of Health (NIH) guidelines and were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Drug Treatment
Naltrexone hydrochloride was dissolved in 0.9% NaCl and was injected intraperitoneally at a concentration of 2 mg/kg twice/day. The mice were tested 30 minutes after injection.
Morris Water Maze
The task was carried out as previously described in (23).
For the acquisition of spatial reference memory from Days 1 to 6, each mouse was given 4 trials of a maximum of 60 seconds/day to find the hidden platform placed in the middle of the southwest quadrant. To initiate the trials, each animal was placed at randomized positions in the tank. During inter-trial intervals of 60 seconds, each mouse was allowed to stay on the platform until the next trial. The latency to find and climb on the platform, as well as the swimming distance, and speed, were recorded and averaged daily.
Retrieval of reference memory was performed on Day 7, 24 hours after the last training trial. Mice were given a 30-second retention (probe) test with the platform removed. The swim path and time spent in the platform (“training”) quadrant were recorded over 30 seconds. To test the extinction of the quadrant preference, interspersed probe trials were conducted, besides the Day 7, on Days 10 and 13 (4 and 7 days after the last training trial respectively). See Supplemental Methods and Materials for detailed description.
A video-computerized tracking system (ANY-maze, Stoelting Co, Wood Dale, Illinois) was used to record and analyze animal behavior.
Transfection of Hippocampal Cultures and Live-Cell Confocal Imaging
Dissociated neuronal cultures were prepared from the hippocampi of Sprague-Dawley rats (Harlan Laboratories, Indianapolis, Indiana) and C57BL/6J mice (Charles River Laboratories, Wilmington, Massachusetts) on postnatal Days 1 and 2 and maintained as previously described (24,25). For live-cell imaging experiments, neurons were plated at a density of 1 × 106 cells per dish onto a 35-mm glass-bottom Petri dish coated with poly-L-lysine (thickness of glass coverslip, 0.08 mm) (26). For all other biochemical studies, 3 × 106 cells were plated onto a poly-L-lysine-treated 60-mm Petri dish. From the day of plating, cultured neurons were counted as DIV1 (Day 1 in vitro). Neurons on DIV5–DIV9 were transfected with pHluorin-GluR1 (19) using a standard calcium phosphate coprecipitation method and were used for experiments between DIV18–DIV24. The averaged pHluorin-GluA1 fluorescence of the cells was based on the quantification of the pHluorin-GluA1 intensity of each individual neuron examined under a Leica DMIRE2 fluorescence microscope connected to a BD CARVII confocal imager and a Hamamatsu EM CCD camera. See detailed description in (19) and Supplemental Methods and Materials.
Surface Biotinylation on Hippocampal Cultured Neurons
After incubation with or without 10 μM naloxone or naltrexone for 1 day, the surface of cultured mouse and rat neurons (DIV18) were biotinylated according to the manufacturer’s instructions (Pierce Biotechnology, Rockford, Illinois). Fifty μL of each biotinylated sample were used to determine the total GluA1 input, and equal amounts of the remaining samples were immunoprecipitated and used for SDS-PAGE/Western blot analysis.
Subcellular Fractionation and Western Blot Analysis
The protocol used to isolate PSDs from the hippocampi was a modification of previously described procedures (27,28). Hippocampi from 6 mice were pooled, resuspended in a solution containing 0.32 M sucrose, 4 mM HEPES (pH7.4) with protease/phosphatase inhibitors (Sigma-Aldrich, St. Louis, Missouri) and homogenized. After several centrifugations, lysis and use of a sucrose gradient, a PSD pellet was obtained and resuspended in 50 mM HEPES, 2 mM EDTA pH7.4 with protease/phosphatase inhibitors. Proteins were then separated by SDS-PAGE and immunoblotted with antibodies against actin, phospho-GluA1-S845 (Millipore, Massachusetts) and GluA1 (Santa Cruz Biotechnology, California). See Supplemental Methods and Materials for detailed description.
Statistics
Data are presented as means ± SEMs. Statistical differences between values from 2 groups were determined using unpaired Student t tests, whereas statistical differences between 3 or more groups were analyzed using ANOVAs followed by Newman-Keuls’s post hoc comparisons. A difference of p < .05 was considered statistically significant.
Results
Effect of Chronic Naltrexone Administration on the Acquisition, Retrieval and Extinction of Spatial Reference Memory in Wild-type and MORKO mice
To determine the role of MOR in the mnemonic effect of naltrexone on spatial memory, wild-type and MORKO mice were both intraperitoneally injected with naltrexone at a dose of 2 mg/kg twice/day and tested in a standard Morris water maze task with a hidden platform for 6 consecutive days (Figures 1A–H). The dose of 2 mg/kg was chosen according early investigations that have encouraged the use of opioids antagonists as potential therapeutic agents in memory loss (6,7,8). Although naloxone and naltrexone act the same manner, naltrexone was used instead of naloxone in the behavioral studies because of its longer in vivo half-life. Three-way ANOVAs on swimming latency and distance to the hidden platform during the acquisition from the wild-type and MORKO mice revealed significant (in addition to significant main effects for “training day”, “treatment” and “genotype”) “treatment × genotype × training day” interactions (Figures 1B–C) (F5,620 = 10.31, p < .001; F5,620 = 2.62, p < .05). In the wild-type mice, chronic naltrexone administration induced faster learning as demonstrated by significant shorter latencies and distances on the second and third day of training compared to the saline-treated group (Figures 1B–C) (Days 2 and 3, p < .01, p < .001, respectively). However, there were no significant differences in the latency or distance to reach the platform between the saline- and naltrexone-treated MORKO mice (Figures 1B–C). Naltrexone does not improve the acquisition of spatial memory in MORKO mice.
Figure 1.

Effect of chronic naltrexone administration on the acquisition (A–D), retrieval and extinction (E–H) of spatial reference memory in MORKO and wild-type littermates in a water maze task. Points and whiskers are means ± SEMs. Chronic intraperitoneal treatment with naltrexone 2 mg/kg twice/day started 3 days (−2, −1, 0 on the timeline) before the first day of the training period of the task during which mice were given 4 trials/day to find the hidden platform for 6 consecutive days (1 to 6 on the timeline) (A). The naltrexone-treated wild-type mice (filled squares, n = 7) required less time (B) and distance (C) to find the submerged platform compared to saline-treated wild-type (open circles, n = 8), saline-treated MORKO (filled circles, n = 8) and naltrexone-treated MORKO (open inverted triangles, n = 9) mice. (D) There were no significant differences in the average swimming speed among the different groups during the 6 days of acquisition (n = 7–9/group).
After a training period during which mice learned the platform location from Day 1 to 6 (1 to 6 on timeline), three probe tests were performed on Days 7, 10 and 13 of the experiment (1, 4 and 7 days after the last day of acquisition) to examine the retrieval and extinction of spatial reference memory in MORKO and wild-type littermates. Chronic intraperitoneal treatment with naltrexone 2 mg/kg twice/day started 3 days (−2, −1, 0 on the timeline) before the first day of the acquisition (E). For each of the probe tests, the percentages of time spent (F) and distance swum (G) in the training quadrant were similar among the naltrexone-treated wild-type (filled squares, n = 8), saline-treated (filled circles, n = 8) and naltrexone-treated MORKO mice (open inverted triangles, n = 9). In contrast, the percentages of time spent (F) and distance swum (G) in the training quadrant were significantly lower in saline-treated wild-type mice (open circles, n = 8) on Day 13. Chronic treatment with naltrexone significantly delayed extinction in the wild-type mice. The absence of MOR in the knockout mice resulted in stable retrieval performances that were not affected by chronic treatment with naltrexone. (H) The average swimming speed during the probe tests was similar amoung the different groups (n = 8–9/group). Significant differences among groups were determined using three-way ANOVAs, followed by Newman-Keuls’s post hoc comparisons. *** p < .001, ** p < .01 and * p < .05 significant differences between saline- and naltrexone-treated wild-type mice, ¶ p < .05 and ¶¶ p < .01 significant differences between saline-treated wild-type and MORKO mice, £ p < .05 significant difference between saline-treated wild-type and naltrexone-treated MORKO mice.
Retrieval results in reactivating/utilizing the memory from acquisition and is induced by reexposure to the training context during the probe test. When the reexposure to the training context is performed several times without reinforcement (absence of platform in the training quadrant) the conditioned response (time and distance spent swimming into the training quadrant) gradually diminishes over time as an animal learns to uncouple the response from a stimulus (29,30,31). Thus 1, 4 and 7 days after the last day of acquisition, 3 probes trials were performed to test whether chronic naltrexone treatment would affect the long-term retrieval of the target quadrant for both wild-type and MORKO mice (Figures 1E–H). On Day 13, naltrexone treatment in the wild-type mice significantly increased the time spent and the distance swum in the target quadrant compared to the saline group (Figures 1F–G) (p < .05), while during the first probe on Day 7 the performances were similar. Naltrexone impaired extinction learning of the removed platform location. The specificity of naltrexone inhibition of MOR that might mediate the observed delay of extinction in the wild-type mice on Day 13 was verified in the MORKO mice in which the antagonist had no effect (Figures 1F–G). Moreover, control and drug-treated MORKO mice appear to have a slow rate of extinction similar to that of the naltrexone-treated wild-type mice.
Chronic Exposure to Naloxone/Naltrexone Increased both Synaptic and Extrasynaptic GluA1 Subunits as well as GluA1 Phosphorylation at Serine 845 (S845) on Cultured Hippocampal Neurons
To measure the temporal dynamics of opioid-antagonist-regulated AMPAR trafficking, a live-imaging system was used to acquire images of the same-labeled cultured hippocampal neuron before and after 10-μM naloxone exposure at various time points (3 hours and 1 and 3 days) (Figures 2A–B). Upon naloxone exposure, an increase in the fluorescence signal of the pHluorin-GluA1 was observed that reached a maximum level 1 day after treatment (Figure 2A). The average fluorescence of pHluorin-GluA1 from the entire image of naloxone-treated neurons significantly increased by 42.2% at 3 hours, 93.7% at 1 day and 43.6% at 3 days after drug application (compared to before naloxone treatment) (Figure 2B), indicating increased expression of GluA1 on the membrane surface following treatment with naloxone. Interestingly, quantitative analysis on adjacent dendrites and individual spines in Figure 2B showed that the enhancement of the surface fluorescence of pHluorin-GluA1 caused by naloxone treatment for 3 hours and 1 day was higher on adjacent dendrites (increase of 31% at 3 hours and 112.1% at 1 day) than on spines (increase of 10.8% at 3 hours and 93.3% at 1 day). On day 3, the adjacent spines express 62% less surface GluA1 compared to day 1 and 16% less than on spines: this can reflect on the one hand GluA1 trafficking from the extrasynaptic sites to the spines and on the other hand, AMPAR internalization.
Figure 2.

Chronic treatment with naloxone or naltrexone induced an increase in the synaptic and extra-synaptic surface GluA1. (A) Confocal images of primary rat hippocampal neurons expressing transfected pHluorin-GluA1 before, 3 hours (3 hrs), 1 day (1d) and 3 days (3d) after exposure to 10 μM naloxone (n = 15/group). Solid arrowheads denote that 10 μM naloxone progressively increased the fluorescence intensity of pHluorin-GluA1-labeled dendritic spines. Scale bar = 10 μm. (B) Mean pHluorin-GluA1 fluorescence from different regions (entire images, dendrites, and spines) of naloxone-treated neurons (n = 15/group). Naloxone significantly enhanced the amount of surface-expressed pHluorin-GluA1 on both the spines and dendrites in a time-dependent manner. (C) Surface biotinylation assay showed a significant increase in GluA1 on the wild-type mouse and rat cultured neuronal membranes after 1 day of exposure to 10-μM naltrexone (Naltr) or naloxone (Nalox) (n = 5–8/group). In MORKO cultured neuronal membranes, naltrexone or naloxone treatment had no observed effect on GluA1 quantity (n = 5/group). The surface-expressed GluA1 subunits were normalized to the untreated control (Ctl) (n = 5–8). On the Western blots of neurons treated with no drug (Ctl), 10 μM naltrexone (Naltr) or 10 μM naloxone (Nalox) for 1 day, GluA1 was detected in the total cell lysates (Input: Total) and in immunoprecipitated biotinylated proteins (IP: Surface). No significant change in the overall protein expression of AMPARs was noted during either naloxone or naltrexone treatment (Input: Total) (D) Time-lapse images were taken on rat hippocampal neurons expressing pHluorin-GluR1 before, and 1 day after, 10-μM naltrexone incubation (n = 5–7). Solid arrowheads indicate that 10 μM naltrexone increased the fluorescence intensity of pHluorin-GluA1 labeled dendritic spines. Scale bar = 10 μm. * p < .05, ** p < .01 and *** p < .001 relative to the untreated control.
Surface biotinylation experiments on cultured mouse and rat hippocampal neurons performed with 10 μM naltrexone showed a significant 1.7–3-fold increase of GluA1 expression, which was similar to that observed after 1 day of naloxone exposure (Figure 2C). An increased fluorescence signal of pHluorin-GluA1 on dendrites and spines monitored by live-imaging experiments on the same labeled neuron 1 day after treatment with 10 μM naltrexone confirmed the similarity of the effect of both naloxone and naltrexone on GluA1 surface expression (Figure 2D). The naltrexone effect is mediated by MOR inhibition as demonstrated by the absence of an increase in the cell-surface GluA1 in the naltrexone-treated primary hippocampal cultures derived from the MORKO mice (Figure 2C).
Because activation of Gi-coupled MOR can induce cAMP-dependent kinase (PKA) inactivation (32), we examined the effect of MOR inhibition by naloxone and naltrexone on the phosphorylation of GluA1 at S845, a substrate of PKA (33,34), which primes AMPARs for synaptic incorporation by trafficking AMPARs to extrasynaptic sites on the surface membrane (21,22). Cultured hippocampal neurons were incubated with 10 μM naloxone or naltrexone. The GluA1 complex was immunoprecipitated with an anti-GluA1 antibody followed by Western blot detection with phosphorylated GluA1-S845 (Figures 3A–B). The amount of phosphorylation at GluA1-S845 was significantly increased after 12 hours of naloxone (p < .001) or naltrexone (p < .01) incubation, reached a maximum level within 1 day (p < .001) and declined during the next 2 days (Figure 3B).
Figure 3.
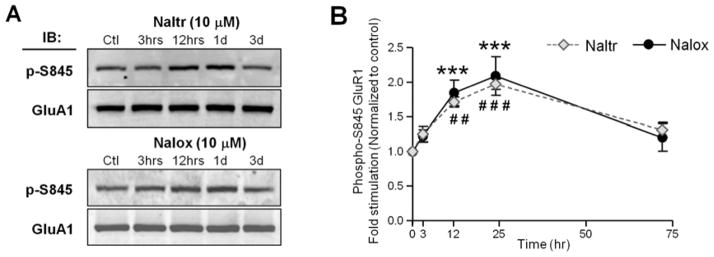
Chronic treatment with naloxone or naltrexone increased GluA1 phosphorylation at S845 on cultured hippocampal neurons. Points and whiskers are means ± SEMs. (A) The amount of GluA1-S845 phosphorylation (p-S845) and total GluA1 in the immunoprecipitated cells lysates from neurons treated with no drug (Ctl), 10 μM naltrexone (Naltr) or 10 μM naloxone (Nalox) for 3 hours (3 hrs), 1 day (1d) and 3 days (3d) were detected by Western blot analysis. The total amount of GluA1 was unchanged. (B) Densitometric quantifications of immunoblots (IB) in (A) on the amount of phosphorylated GluA1-S845 in the receptor complex (normalized to the untreated control, n = 7) revealed a significant increase of GluA1-S845 phosphorylation 12 and 24 hours (hrs) after naltrexone (n = 7) and naloxone (n = 5) treatments. # # p < .01 and # # # p < .01 relative to the data before naltrexone treatment, *** p < .001 relative to the data before naloxone treatment.
Effect of Chronic Naltrexone Injection on the Acquisition, Retrieval and Extinction of Spatial Reference Memory in Wild-type and GluA1-S845A Mutant mice
Because of the naloxone/naltrexone-induced increase in both the amount of extrasynaptic and synaptic membrane GluA1 levels and the increase in GluA1 phosphorylation at S845 in cultured hippocampal neurons, we hypothesize that naloxone/naltrexone would affect learning of spatial reference memory through the phosphorylation of S845 on GluA1. The use of GluA1-S845A mutant mice in which the mutation of Ser845 to Ala resulting in blunting its phosphorylation can reveal whether or not the phosphorylation of Ser845 plays a role in naltrexone-induced improvement of spatial reference memory acquisition. Wild-type and GluA1-S845A mutant mice were both intraperitoneally injected with naltrexone (2 mg/kg twice/day) and tested in the water maze (Figures 4A–G). Chronic naltrexone treatment in the GluA1-S845A mutants did not improve acquisition on the second and third day of training compared to the wild-type mice in which naltrexone facilitated learning (Figures 4A–B: three-way ANOVAs “treatment × genotype × training day”, latency F5,540 = 3.94, p < .01 and distance F5,540 = 2.33, p < .05).
Figure 4.

Effect of chronic naltrexone administration on the acquisition (A–C), retrieval and extinction (D–G) of spatial reference memory in GluA1-S845A mutants and wild-type littermates in a water maze task. Points and whiskers are means ± SEMs. All mice learned to find the submerged platform during the 6 days of acquisition (the design of the experiment is the same as described in figure 1A legend) but, when measuring the average escape latency (A) and distance (B), naltrexone-treated wild-types mice (filled squares) found the hidden platform significantly faster than the saline-treated wild-type mice (open circles), saline-treated GluA1-S845A mutants (open triangles), or naltrexone-treated GluA1-S845A mutants (filled diamonds) (n = 7/group). (C) The average swimming speed during the 6 days of acquisition was similar among the different groups (n = 7/group). The retrieval and extinction of spatial reference memory in GluA1-S845A mutants and wild-type littermates were measured in 3 probe tests performed on Days 7, 10 and 13 (1, 4 and 7 days after the last day of acquisition) (see description in figure 1E legend). The percentages of time spent (D) or distance swum (E) in the training quadrant were similar across the 3 probe tests for the naltrexone-treated wild-type mice (filled squares, n = 10). In contrast, the percentages of time spent (D) or distance swum (E) in the training quadrant decreased significantly over time for control mice (open circles, n = 10). Chronic treatment with naltrexone induced a slow extinction in the wild-type mice. In contrast, naltrexone-treated GluA1-S845A mutant mice (filled diamonds, n = 11) displayed retrieval impairment during the second and third probe tests compared to control wild-type mice (D, E). This result suggests that GluA1-S845 phosphorylation is involved in the naltrexone-induced delayed extinction in the wild-type mice. Saline-treated GluA1-S845A mutants (open triangles, n = 11) tended to retrieve better than the other groups, and their extinction rate was slower than that of the saline-treated wild-type mice. The dotted line on (D) and (E) corresponds to the level of random chance (25%). (F) The average swimming speed during the probe tests was similar among the groups (n = 10–11/group). (G) Track plots of wild-type and GluA1-S845A mutant mice treated with saline or naltrexone during the third probe (Day 13). The southwest quadrant (labeled SW on the figure) is the target quadrant where the platform was previously located during training. Significant differences among groups were determined using three-way ANOVAs, followed by Newman-Keuls’s post hoc comparisons. *** p < .001, ** p < .01 and * p < .05 significant differences between naltrexone- and saline-treated wild-type mice, # # p < .01 and # # # p < .001 significant differences between naltrexone- and saline-treated GluA1-S845A mutants, $$$ p < .001 significant difference between saline-treated wild-type mice and saline-treated GluA1-S845A mutants, && p < .01 significant difference between wild-type mice and GluA1-S845A mutants treated with naltrexone.
In regards to the extinction (Figures 4D–F), naltrexone differentially affected the performances depending on the genotype and the probe trial (Figures 4D–E: three-way ANOVAs “treatment × genotype × extinction day”, F2,76 = 3.31, p < .05, F2,76 = 3.50, p < .05). In GluA1-S845A mutants, the same reduction of the extinction rate induced by naltrexone in the wild-type mice was not observed, as shown by significantly lower retrieval scores compared to the saline-treated mutants during the first (p < .01), second (p < .001) and third (p < .01) probes or compared to the naltrexone-treated wild-type mice on Day 10 (p < .001). From Days 7 to 13, the GluA1-S845A mutant saline-treated mice displayed a slower extinction compared to the wild-type saline-treated mice (Figures 4D–E) (Day 13, p < .001).
In Vivo Increase in GluA1 Level and GluA1-S845 Phosphorylation at the PSD during the Acquisition of Spatial Reference Memory in Wild-type and GluA1-S845A Mutant mice
We postulate that the learning enhancement from treatment with naltrexone is the result of GluA1-S845 phosphorylation leading to an increase in GluA1 levels at the postsynaptic membrane. To confirm our hypothesis, the levels of phosphorylated GluR1-S845 and of AMPARs at the synapses were assessed by quantitative Western blot analysis. Hippocampi from mice in their home cage and from mice on different training days (Days 1, 3 and 5) (Figure 4A–C) were collected. The results from the Western blot analyses of the PSD fractions showed that naltrexone-treated mice exhibiting faster learning had significantly increased GluA1 (Figure 5A) (p < .01) and phosphorylated GluA1-S845 (Figure 5B) (p < .05) levels at the PSD on the third day of acquisition, which is when naltrexone significantly enhanced learning compared to the saline-treated mice. However, among the GluA1-S845 mutants, there was no significant change in the expression of GluA1 between saline and naltrexone treatment in the home cage, on Days 1, 3 and 5 of training, which is correlated with the absence of an effect from naltrexone treatment on the mutants’ performances during the acquisition. An effect of “time” on GluA1 protein levels (Figure 5A) and GluA1-S845 phosphorylation (Figure 5B) in saline-treated mice, as revealed by a two-way ANOVA (GluA1: F3,18 = 42.11, p < .0001; p-S845: F3,18 = 21.52, p < .0001), suggests that spatial reference learning without drug treatment requires an increase of postsynaptic GluA1-dependent neurotransmission.
Figure 5.

Chronic naltrexone treatment significantly increased GluA1 expression and GluA1-S845 phosphorylation during the third day of acquisition in the wild-type mice, but not in GluA1-S845A mutant mice. Bars and errors are means means ± SEMs. (A) Western blot analysis showing the difference between saline (S) and naltrexone (N) chronic treatment on the level of GluA1, phosphorylated GluA1-S845 (p-S845) and actin at the PSD of hippocampi extracted from wild-type mice (n = 6/group) in their home cage, and on different days of acquisition (Home cage, Day 1, Day 3, Day 5). (B, C) Densitometric quantifications of Western blots in (A). On the histograms, the ratios GluA1/actin (B) and p-S845/actin (C) are normalized to the saline-treated home cage group. (D) Western blot analysis showing the difference between saline (S) and naltrexone (N) chronic treatment in the level of GluA1, phosphorylated GluA1-S845 (p-S845) and actin at the PSD of hippocampi extracted from GluA1-S845A mutant mice (n = 6/group) in their home cage and on different days of acquisition (Home cage, Day 1, Day 3, Day 5). The Western blots confirmed the absence of phosphorylated GluA1-S845 staining in GluA1-S845A mutant mice. (E) Densitometric quantifications of Western blots in (D). On the histograms, the ratio GluA1/actin (E) is normalized to the saline-treated home cage group. Significant differences between saline- and naltrexone-treated mice were determined using two-way ANOVAs, followed by Newman-Keuls’s post hoc comparisons. * p < .05 and ** p < .01 significant differences between saline- and naltrexone-treated mice.
Discussion
Variations in the quantity of AMPARs have been suggested to be the basis of synaptic plasticity, whereby synapses become stronger or weaker by addition or removal of AMPARs, respectively (10,35,36,37). Because synaptic plasticity is disrupted in many neuropathologies (38), novel, effective drug therapies based on manipulations of the glutamatergic system through AMPARs offer an attractive alternative target to other therapies (39). However, before assessing the effects of naltrexone/naloxone on the progression of neuropathology, it is important to first establish the ability of naltrexone/naloxone to modify AMPAR trafficking in the context of normal cognition. This study is the first to demonstrate that naloxone/naltrexone alter postsynaptic AMPAR expression in the hippocampus and potentially other brain regions, to enhance learning and delay extinction.
We observed that naltrexone increases the speed of learning in a water maze task in wild-type mice through inhibition of endogenous MOR (Figures 1A–D). Although naltrexone does not appear to play a role in the retrieval process 24 hours after the last day of acquisition, the measured delayed extinction of the retrieved memory in naltrexone-treated wild-type mice also seems to indicate the blockade of endogenous opioids action on MOR by naltrexone as shown by a similar delayed extinction in the MORKO mice (Figure 1E–H). Moreover, the facilitative effect of 6-β-naltrexol, a MOR neutral antagonist (40,41), on learning in wild-type mice (Supplemental Figure S1) confirms that naltrexone acts as a neutral antagonist to induce its effect. The similarities of performances between the saline-treated wild-type and MORKO mice during the acquisition and the first probe are in line with previous studies in which the same genetic background and mutation were used (42,43,44).
Time-lapse imaging and biochemical experiments in cultured hippocampal neurons provided direct visual and quantitative evidence that naloxone/naltrexone caused a time-dependent increase of GluA1 insertion at both spines and dendrites (Figure 2). We also found that incubation with naloxone and naltrexone enhanced phosphorylated GluA1-S845, which could reflect either an activation of phosphorylation of GluA1-S845 or a reduction in the dephosphorylation of the receptor subunit (Figure 3). Increased GluA1-S845 phosphorylation has been reported to be a required step for the cell surface insertion of AMPARs on the extra- and perisynaptic sites in both cultured neurons and hippocampal slices ready for delivery to synapses (21,22,45). Our data suggest that naloxone/naltrexone can engage the surface insertion at the extrasynaptic sites of AMPARs through the phosphorylation of GluA1-S845. Naloxone and naltrexone have the highest affinity for MOR (46), which is predominantly expressed in the postsynaptic membrane of hippocampal neurons (47) and is colocalized with postsynaptic AMPARs (15). GluA1 internalization in primary hippocampal neurons promoted by prolonged application of morphine (19) demonstrates the inhibitory effect of opioids on AMPARs membrane expression through MOR activation. Thus, the probable blockade of endogenous opioids on postsynaptic MOR by naloxone/naltrexone would result in enhanced GluA1-S845 phosphorylation followed by an increase of AMPAR insertion at the extrasynaptic membrane. In the absence of MOR activity in cultured hippocampal neurons from MORKO mice, naloxone/naltrexone do not change the GluA1 surface expression compared to the control, which supports our hypothesis. In addition to demonstrating that naloxone and naltrexone treatment affects in vitro hippocampal synaptic plasticity regulation through AMPAR trafficking, the critical question as whether these mechanisms actually play an in vivo role in mediating spatial memory after opioid receptor inhibition was also addressed.
The lack of an effect induced by naltrexone during the acquisition in GluA1-S845A mutants indicates that GluA1-S845 phosphorylation is involved in the mechanism of naltrexone’s enhancement of learning in wild-type mice (Figures 4A–B). The blockade of endogenous opioids action on MOR by naltrexone disinhibits the effect of the basal signaling activity of MOR on the homeostatic AMPAR trafficking. Combined with learning-related neuronal activity, this would allow the insertion of a higher number of AMPARs and, thus, faster neurotransmission compared to the saline-treated mice. A significant increase in the amount of GluA1 as well as GluA1-S845 phosphorylation from the PSDs of the mice on the third day of acquisition (Figure 5A–B) confirms this hypothesis. Increased GluA1 correlates with the observed improvement in learning in wild-type naltrexone-treated mice compared to saline-treated mice (Figure 4A–C). Moreover, experiments on cultured hippocampal neurons showed that the significant increase in spine and protrusion density caused by naloxone treatment is blocked in cells transfected with dominant-negative mutant Ca2+/calmodulin-dependent protein kinase II (CaMKII) plasmids (48). In the cortex, the activation of CaMKII as well as other kinases has been shown to be involved in GluA1-S845 phosphorylation upregulation mechanisms (49,50). Thus, CaMKII may play a role in the mechanisms behind the increased GluA1 membrane expression by naltrexone. Similar performances between wild-type and GluA1-S845A mutant mice without drug treatment (Figure 4A–C) are consistent with a recent study reporting normal LTP in GluA1-S845A mutant mice (51). The authors of the study suggested that the absence of phosphorylation on S845 in the single mutant could be compensated by phosphorylation of S831 because double phosphomutants lacking both S845 and S831 sites expressed reduced and fast-decayed LTP (52). Because naltrexone did not improve the acquisition and change GluA1 levels in the GluA1-S845A mutants, GluA1-S831 phosphorylation is probably not required for learning facilitation by naltrexone. Then, GluA1-S845A mutation does not seem to affect the sensorimotor capabilities, escape motivation and non spatial associative learning in the cued learning version of the water and Barnes maze (Supplemental Figures S2 and S3, respectively). That is why the deficit in working memory (Supplemental Figure S4) in the control GluA1-S845A mutants cannot be clearly attributed to a difference in sensorimotor functions in the absence of GluA1-S845 phosphorylation.
Because drug-treated GluA1-S845A mutants display an overall lower retrieval performance compared to the other groups and more significantly to the saline-treated GluA1-S845A mutants during the interspersed probe trials (Figures 4D–E), naltrexone likely targets other molecular mechanisms important for retrieval and the maintenance of normal extinction. In this case, knowing that the mutation prohibits phosphorylation and dephosphorylation at the S845 site of GluA1 and thus decreases AMPAR trafficking, naltrexone could affect the phosphorylation of GluA1 at S831 or S818 to produce a faster extinction. Also, these poor retrieval performances induced by naltrexone in the mutants can reflect a lack of motivation, as suggested by a study on appetitive incentive learning which showed motivational deficits in absence of S831 site (53). Saline-treated GluA1-S845A mutants displayed enhanced performances during the first probe test and a resistance to extinction compared to the other groups. GluA1-S845 dephosphorylation is essential for activity-dependent internalization of AMPARs during long-term depression (LTD) and its absence could explain the specific deficit in LTD described in GluA1-S845A mutants (54,55,45). Several studies have suggested that LTD and AMPAR endocytosis underlie memory extinction (56,57). Thus, we hypothesize that in the GluA1-S845A mutants, the slow extinction learning in saline-treated mice may due to a deficit in LTD and that the retrieval deficits induced by naltrexone may result from LTD potentiation.
In conclusion, we demonstrated that naltrexone-induced plasticity in excitatory synapses, via the transitory increase of GluA1 insertion at the PSD and GluA1-S845 phosphorylation, facilitates learning. The presence of enhanced mnemonic cognition following treatment with naltrexone through the stimulation of AMPAR trafficking found in normal animals could be explored in a mouse model of memory disorder in an attempt to delay the onset of memory deficits.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health: DA011806 and DA031442.
Footnotes
Financial Disclosures
All authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Introini-Collison IB, Ford L, McGaugh JL. Memory impairment induced by intraamygdala beta-endorphin is mediated by noradrenergic influences. Neurobiol Learn Mem. 1995;63:200–205. doi: 10.1006/nlme.1995.1021. [DOI] [PubMed] [Google Scholar]
- 2.Ukai M, Watanabe Y, Kameyama T. Endomorphins 1 and 2, endogenous mu-opioid receptor agonists, impair passive avoidance learning in mice. Eur J Pharmacol. 2001;421:115–119. doi: 10.1016/s0014-2999(01)01009-3. [DOI] [PubMed] [Google Scholar]
- 3.Spain JW, Newsom GC. Chronic opioids impair acquisition of both radial maze and Y-maze choice escape. Psychopharmacology. 1991;105:101–106. doi: 10.1007/BF02316870. [DOI] [PubMed] [Google Scholar]
- 4.Kamboj SK, Tookman A, Jones L, Curran HV. The effects of immediate-release morphine on cognitive functioning in patients receiving chronic opioid therapy in palliative care. Pain. 2005;117:388–395. doi: 10.1016/j.pain.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Izquierdo I. Effect of naloxone and morphine on various forms of memory in the rat: possible role of engogenous opiate mechanisms in memory consolidation. Psychopharmacology (Berl) 1979;66:199–203. doi: 10.1007/BF00427631. [DOI] [PubMed] [Google Scholar]
- 6.Messing RB, Jensen RA, Martinez JL, Jr, Spiehler VR, Vasquez BJ, Soumireu-Mourat B, et al. Naloxone enhancement of memory. Behav Neural Biol. 1979;27:266–275. doi: 10.1016/s0163-1047(79)92328-8. [DOI] [PubMed] [Google Scholar]
- 7.Gallagher M. Naloxone enhancement of memory processes: effects of other opiate antagonists. Behav Neural Biol. 1982;35:375–382. doi: 10.1016/s0163-1047(82)91020-2. [DOI] [PubMed] [Google Scholar]
- 8.Gallagher M, Bostock E, King R. Effects of opiate antagonists on spatial memory in young and aged rats. Behav Neural Biol. 1985;44:374–385. doi: 10.1016/s0163-1047(85)90688-0. [DOI] [PubMed] [Google Scholar]
- 9.Aigner TG, Mishkin M. Improved recognition memory in monkeys following naloxone administration. Psychopharmacology. 1988;94:21–23. doi: 10.1007/BF00735874. [DOI] [PubMed] [Google Scholar]
- 10.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 11.Keifer J, Zheng Z. AMPA receptor trafficking and learning. Europ J Neurosci. 2010;32:269–277. doi: 10.1111/j.1460-9568.2010.07339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu W, Lu B. Synapses and dendritic spines as pathogenic targets in Alzheimer’s disease. Neural Plast. 2012;2012:247150. doi: 10.1155/2012/247150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 14.Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X, Choe ES, Mao L. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–249. doi: 10.1385/MN:32:3:237. [DOI] [PubMed] [Google Scholar]
- 15.Liao D, Lin H, Law PY, Loh HH. Mu-opioid receptors modulate the stability of dendritic spines. Proc Natl Acad Sci U S A. 2005;102:1725–1730. doi: 10.1073/pnas.0406797102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao D, Grigoriants OO, Loh HH, Law PY. Agonist-dependent postsynaptic effects of opioids on miniature excitatory postsynaptic currents in cultured hippocampal neurons. J Neurophysiol. 2007;97:1485–1494. doi: 10.1152/jn.00790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao D, Grigoriants OO, Wang W, Wiens K, Loh HH, Law PY. Distinct effects of individual opioids on the morphology of spines depend upon the internalization of mu opioid receptors. Mol and Cell Neurosci. 2007;35:456–469. doi: 10.1016/j.mcn.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson TE, Gorny G, Savage VR, Kolb B. Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse. 2002;46:271–279. doi: 10.1002/syn.10146. [DOI] [PubMed] [Google Scholar]
- 19.Kam AY, Liao D, Loh HH, Law PY. Morphine induces AMPA receptor internalization in primary hippocampal neurons via calcineurin-dependent dephosphorylation of GluR1 subunits. J Neurosci. 2010;30:15304–15316. doi: 10.1523/JNEUROSCI.4255-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia Y, Portugal GS, Fakira AK, Melyan Z, Neve R, Lee HT, Russo SJ, Liu J, Moron JA. Hippocampal GluA1-containing AMPA receptors mediate context-dependent sensitization to morphine. J Neurosci. 2011;31:16279–16291. doi: 10.1523/JNEUROSCI.3835-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Neurosci. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- 23.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 25.Liao D, Zhang X, O’Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- 26.Lin H, Huganir R, Liao D. Temporal dynamics of NMDA receptor-induced changes in spine morphology and AMPA receptor recruitment to spines. Biochem Biophys Res Commun. 2004;316:501–511. doi: 10.1016/j.bbrc.2004.02.086. [DOI] [PubMed] [Google Scholar]
- 27.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 29.Rossato JI, Bevilaqua LR, Medina JH, Izquierdo I, Cammarota M. Retrieval induces hippocampal-dependent reconsolidation of spatial memory. Learn Mem. 2006;13:431–440. doi: 10.1101/lm.315206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- 31.Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;1:180–187. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- 32.Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- 33.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 34.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Chang PK, Verbich D, McKinney RA. AMPA receptors as drug targets in neurological disease--advantages, caveats, and future outlook. Europ J Neurosci. 2012;35:1908–1916. doi: 10.1111/j.1460-9568.2012.08165.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang D, Raehal KM, Bilsky EJ, Sadée W. Inverse agonists and neutral antagonists at mu opioid receptor (MOR): possible role of basal receptor signaling in narcotic dependence. J Neurochem. 2001;77:1590–1600. doi: 10.1046/j.1471-4159.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Raehal KM, Lin ET, Lowery JJ, Kieffer BL, Bilsky EJ, et al. Basal signaling activity of mu opioid receptor in mouse brain: role in narcotic dependence. J Pharmacol Exp Ther. 2004;308:512–520. doi: 10.1124/jpet.103.054049. [DOI] [PubMed] [Google Scholar]
- 42.Lubbers ME, van den Bos R, Spruijt BM. Mu opioid receptor knockout mice in the Morris Water Maze: a learning or motivation deficit? Behav Brain Res. 2007;180:107–111. doi: 10.1016/j.bbr.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 43.Cominski TP, Ansonoff MA, Turchin CE, Pintar JE. Loss of the mu opioid receptor induces strain-specific alterations in hippocampal neurogenesis and spatial learning. Neuroscience. 2014;278:11–19. doi: 10.1016/j.neuroscience.2014.07.039. [DOI] [PubMed] [Google Scholar]
- 44.Olmstead MC, Ouagazzal AM, Kieffer BL. Mu and delta opioid receptors oppositely regulate motor impulsivity in the signaled nose poke task. PLoS One. 2009;4:e4410. doi: 10.1371/journal.pone.0004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magnan J, Paterson SJ, Tavani A, Kosterlitz HW. The binding spectrum of narcotic analgesic drugs with different agonist and antagonist properties. Naunyn-Schmiedeberg Arch Pharmacol. 1982;319:197–205. doi: 10.1007/BF00495865. [DOI] [PubMed] [Google Scholar]
- 47.Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, et al. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci. 1995;15:3328–3341. doi: 10.1523/JNEUROSCI.15-05-03328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller EC, Zhang L, Dummer BW, Cariveau DR, Loh H, Law PY, et al. Differential modulation of drug-induced structural and functional plasticity of dendritic spines. Mol Pharmacol. 2012;82:333–343. doi: 10.1124/mol.112.078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharp JW, Ross CM, Koehnle TJ, Gietzen DW. Phosphorylation of Ca2+/calmodulin-dependent protein kinase type ii and the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (ampa) receptor in response to a threonine-devoid diet. Neuroscience. 2004;126:1053–1062. doi: 10.1016/j.neuroscience.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 50.Oh JH, Yang JH, Ahn SM, Youn B, Choi BT, Wang JQ, et al. Activation of protein kinase C is required for AMPA receptor GluR1 phosphorylation at serine 845 in the dorsal striatum following repeated cocaine administration. Psychopharmacology (Berl) 2013;227:437–445. doi: 10.1007/s00213-013-2968-1. [DOI] [PubMed] [Google Scholar]
- 51.Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 53.Crombag HS, Sutton JM, Takamiya K, Lee HK, Holland PC, Gallagher M, et al. A necessary role for GluR1 serine 831 phosphorylation in appetitive incentive learning. Behav Brain Res. 2008;191:178–183. doi: 10.1016/j.bbr.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 55.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 56.Wong TP, Howland JG, Robillard JM, Ge Y, Yu W, Titterness AK, et al. Hippocampal long-term depression mediates acute stress-induced spatial memory retrieval impairment. Proc Natl Acad Sci U S A. 2007;104:11471–11476. doi: 10.1073/pnas.0702308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dalton GL, Wang YT, Floresco SB, Phillips AG. Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology. 2008;33:2416–2426. doi: 10.1038/sj.npp.1301642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.