

Abstract
Chordoma is a rare low-grade primary malignant skeletal tumor, which is presumed to derive from notochord remnants. The pathogenesis of chordoma has not been fully elucidated. However, recent advances in the molecular biology studies have identified brachyury underlying the initiation and progression of chordoma cells. More efforts have been made on accumulating evidence of the notochordal origin of chordoma, discovering signaling pathways and identifying crucial targets in chordomagenesis. In this review, we summarize the most recent research findings and focus on the pathophysiology and molecular mechanisms of chordoma.
Keywords: Chordoma, Pathophysiology, Chromosomal alterations, microRNA, DNA methylation
Introduction
Chordoma is a rare, low-grade, primary malignant skeletal tumor [1]. It accounts for 1–4 % of all primary skeletal tumors, and its incidence rate is inferior to 1.0 individual per million a year with a dominance in males (a male-to-female ratio of 2:1) [1, 2, 3•]. It is a tumor with lobules and vacuolated, moderately atypical, neoplastic cells across a myxoid stroma separated by fibrous bands. It predominantly arises at the cranial and caudal ends of the axial skeleton and has a slow aggressive and locally invasive character [4–7]. Histologically, chordomas are categorized as classical (or conventional), chondroid, and dedifferentiated. Surgical resection is the most effective treatment modality for chordoma, as chordoma is known to be relatively resistant to conventional chemotherapy and radiotherapy. Due to some puzzling features, there has been a longstanding discussion on the origin of chordoma related to notochord or not. For better understanding of the pathophysiology of this disease, studies have identified some genomic and epigenetic alterations underlying the development of chordoma, using robust techniques, such as comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), single nucleotide polymorphisms (SNP), and micro-ribonucleic acid (microRNA, miRNA, miR)-expression arrays. We would like to present an overview of recent significant advances on the pathophysiology and molecular mechanisms of chordoma.
The embryological formation of chordoma
Physaliferous cells are typical of chordoma, appearing as clusters of gray-white large cells separated by fibrous septa into lobules and surrounded by a basophilic extracellular matrix rich in mucin and glycogen. This distinctive histological appearance led to the first hypothesis in 1858 that chordoma was of notochordal origin [8]. Later, ecchordosis physaliphora was introduced to designate hamartomatous lesions of notochordal origin, which are considered the benign counterparts of chordoma and hypothesized as a precursor of chordoma up to date [9]. It has been generally accepted that chordoma cells originate from remnants of the embryonic notochord [10]. In humans, the notochord is a transient embryonic structure with critical developmental role. After inducing vertebral column formation, the notochord disappears, leaving cellular remnants in the nucleus pulposus. It has been proposed that notochordal remnants are derived from the embryonic notochord since notochordal remnants are similar in size to notochord cells and reside in the region of the embryo in which the embryonic notochord was present [3•, 11]. To clarify the pathophysiology, we constructed a figure to describe that chordomas arose via transformation of notochordal cells (Fig. 1). Since the occurrence of notochordal remnants in humans is much higher than the incidence of chordoma, it is presumed that notochordal remnants stay dormant in most cases but may transform into malignancies when stimulated by a mutation, environmental insult, or other events [11]. Yamaguchi et al. documented the link between persistent notochordal remnants and chordoma, i.e., the site of the vestiges corresponds closely with the distribution of chordoma, the morphological similarities by both transmitted and electron light microscopy, and the overlapped immunophenotype (cytokeratin and S100 protein expression) [12]. Several authors reported cases indicating an association between benign notochordal cell tumor (BNCT) and chordoma [13]. BNCTs are notochordal remnants that have the potential for malignant transformation. Moreover, the anatomical distribution of BNCT and chordoma completely overlaps. This distribution pattern indirectly supports the evidence that BNCT is a precursor of chordoma [13].
Fig. 1.
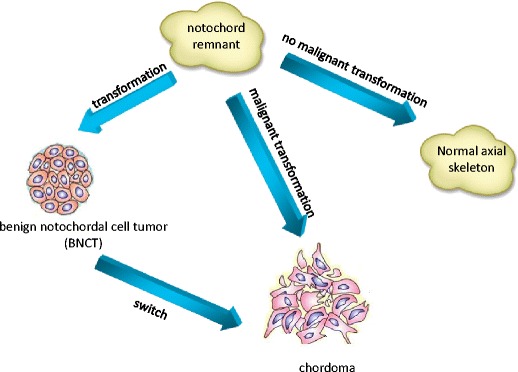
A hypothesized model of chordomagenesis. Chordomas might also be a result of direct malignant transformation of the notochordal remnant, without a benign notochordal tumor intermediary stage. Or chordoma might be derived from benign notochordal cell tumor due to a malignant transformation in phenotype (switch) in the chordomagenesis
The recent cancer stem cell (CSC) theory has shed more light on the embryonic transformations, which drives the notochordal cells to differentiate into the benign or malignant variant. This hypothesis suggests that a small subpopulation of tumor cells which exhibit characteristics of CSC are the driving force behind tumor growth and the presence of different tumor cells. Stem-like cells in chordoma display stemness gene expression, differentiation, sphere formation, and soft agar anchorage-independent growth [14]. Further evidences for the existence of a cell population with stem cell properties in chordoma have been reported by Hsu et al. recently [15]. They illustrated the formation of sarcospheres in the human chordoma cell line CHJ-7. Chordoma sarcospheres were found to be self-perpetuating and exhibited higher expression of the functional stem cell marker ALDH1 compared to classic chordoma cells. Moreover, sarcospheres enabled to differentiate into neuroepithelial and mesodermal cell types, suggesting a hierarchical model for transformation and differentiation of chordoma cells. Further investigations concerning the initiating factors for differentiation are crucial for our understanding of the embryological formation of chordoma.
Molecular mechanisms in chordoma
The molecular events in chordomagenesis have not been fully delineated, particularly with respect to differentially expressed genes involved in the origin of chordoma. Recent studies are looking for novel mutations for better understanding of the molecular mechanisms and potentially identifying therapeutic targets [16, 17].
Chromosomal instability in chordoma
Chordoma is a cytogenetically heterogeneous tumor which displays complex karyotypes. It has some chromosomal aberrations and is characterized by chromosomal gains and losses at various regions throughout the wide genome. Furthermore, quantitative changes in the tumor cell genome are frequently detected using CGH [18, 19].
Molecular cytogenetic studies have demonstrated that a variety of chromosomal markers exist in primary and recurrent chordomas, as well as frequent quantitative changes in the tumor cell genome. Molecular cytogenetics showed that gains of chromosomal material in chordoma were most prevalent at 7q (42 %), 12q (21 %), 17q (21 %), 20q (27 %), and 22q (21 %), and DNA sequence losses occurred most frequently at 1p (21 %), 3p (36 %), 4q (27 %), 10q (21 %), and 13q (24 %) [18]. It seemed that losses were more frequent than gains in chordoma. Bayrakli et al. found that chromosomes 1p36, 1q25, 2p13, and 7q33 were affected in seven primary chordomas, and these aberrations persisted in 11 recurrent tumors [20]. Walter et al. investigated changes in copy number of chromosome 7 by chromogenic in situ hybridization (CISH) [21]. Molecular cytogenetic techniques have mapped the most frequent gains at the following loci: 7p15, 7p21–p22, and portions of arms 7q; 7q22, 7q33, 7q34, and 7q36. The study also confirmed polysomy of chromosome 7 resulted in c-MET expression alterations, presenting its genetic role in chordoma progression. Loss of heterozygosity (LOH) and genome-wide linkage studies have already been used to narrow down and define candidate regions for chordoma development. Lononi et al. observed 1p36 LOH in 75–85 % of chordoma samples and determined the expression pattern of eight apoptotic genes mapped at 1p36, including CASP9, DFFA, DFFB, TP73, TNFRSF1B, TNFRSF8, TNFRSF9, and TNFRSF14 [22]. Among them, the deregulation of TNFRSF8, TNFRSF9, and TNFRSF14 were detected more common. They had unique structural attributes that coupled directly to signaling pathways for cell proliferation, survival, and differentiation, as members of the tumor necrosis factor receptor superfamily.
Rinner et al. demonstrated common losses including chromosome 1, 4, 9, 10, 13, 14, 18, 20, and 22 as well as common gains in 7, 12, and 19 using CGH in ten chordoma specimens [23]. They found significant genomic instability a loss of 3q26.32 (PIK 3CA) and 3q27.3 (BCL6) thus underlining the potential importance of the PI3K pathway in chordoma development. Phi Le et al. reported the copy change analysis of 21 sporadic chordomas [17]. The study demonstrated large copy number losses, involving chromosomes 1p, 3, 4, 9, 10, 13, 14, and 18, were more common than copy number gains. They elucidated that majority of chordoma might rely on mechanisms of large copy number losses, like loss of CDKN2A on 9p21.3, and loss of PTEN on 10q23.31, other than copy number gain [24]. In another study, CDKN2A gene on chromosome 9p21 was found to be homozygously or heterozygously lost in 70 % of chordomas [25]. Choy et al. tested 865 hotspot mutations within 45 human chordoma tumor samples and reported the largest comprehensive mutational analysis of chordoma [24]. The study showed that 18 of 21 chordoma samples displayed copy loss at the locus for CDKN2A, 17 of 21 chordoma samples displayed copy loss at PTEN, and 3 of 4 chordoma samples displayed deletion at the SMARCB1 locus (at chromosome 22q). Thus, it was inferred that a loss of heterozygosity at CDKN2A, PTEN, as well as SMARCB1 might play a significant role in chordomagenesis.
Embryological pathways in chordoma
To verify the hypothesis that chordoma differentiates down a notochordal lineage and derives from notochord, many studies have investigated the proteins implicated in preservation of this embryonic structure in chordoma.
Brachyury is a transcription factor encoded by T gene in humans, a member of the T-box gene family [26]. It helps with the promotion of cell movement and adhesion, which are fundamental for both morphogenesis and tumorigenesis. Long before it has been proposed as a unique specific diagnostic marker of chordoma, brachyury has been identified as a major regulator of notochord formation and a specific marker for the notochord and notochord-derived tumors [26, 27]. Vujovic et al. confirmed brachyury was localized and restricted to chordoma cells at a high protein expression in all 53 primary chordoma tissue samples, but not in a wide variety of other neoplasms or normal tissues [27]. Its expression is required for the specification of posterior mesodermal identity, representing one of the key genes regulating notochord formation during embryogenesis. They also found that brachyury was detected in both the chondroid and chordoid components of chordomas, which argued that the chondroid component of chordoma was supposed to arise from the notochord [27, 28]. It was the first time to specifically link chordoma and the notochord together by a specific molecule. As the evolution of multicellular organisms proceeded, brachyury involved in the specification of an area of the blastopore with distinctive properties in axis formation and later on turned into a driver of mesoderm specification. Eventually, through the synergistic interaction with other transcription factors, such as members of the Fox family, brachyury acquired its crucial role in notochord formation.
In the early embryo, the notochord produces and secretes some signaling factors (Sonic hedgehog, Wnt/β-catenin, BMP/Nodal, and FGF) to direct organogenesis by providing fate and positional guidance [29–31]. Further researches should be investigated in order to gain a better understanding of that whether aberrant activation of these signaling pathways contributes to chordomagenesis.
Prognostic significance of other pathways in chordoma
Recent studies have shed lights on the genomic analysis in predicting chordoma growth and the ability to genetically modify or inhibit certain chromosomal loci or their protein products. These efforts may contribute to identify diagnostic and prognostic biomarkers, which can be applied to adjuvant therapies and improve oncologic outcomes in chordoma.
The cell cycle aberrations have been proved to contribute to tumorigenesis and progression in many human malignancies, including chordoma [25, 32]. Some percentage of chordomas present alterations of p53, MDM2, cyclin D1, and pRb proteins [32]. Among them, p53 overexpression was the only independent factor for higher MIB-1 LI. In the same study, MDM2 gene amplification was found in 15.4 % of chordoma samples and associated with higher MIB-1 LI [32]. Yakkioui et al. noted that MDM2 expression within chordomas negatively regulates tumor suppressor functions of p53 by binding at the N-terminal and modifying it for degradation [33]. Combined with the interesting finding that the 5-year survival rate was 38.9 % for patients with p53 overexpression and 79.4 % for patients without p53 overexpression, there are reasons to believe that p53 overexpression in particular indicates an unfavorable prognosis in patients with chordoma. All these studied were based on immunohistochemical staining, so p53 overexpression should be interpreted carefully. Not all the overexpression was of mutant p53, as wild-type p53 could also be detected at a small concentration [34]. CDK4 is well known as a regulator in the growth 1-synthesis phases of the cell cycle, and its expression is highly correlated to MIB1-LI [33]. It exerts a negative effect on the tumor suppressor retinoblastoma gene, increasing cell proliferation and growth [25]. CDKN2A gene which is located on 9p21 encodes the p14ARF protein. This protein has been reported to interfere with the function of MDM2 and, by blocking the MDM2-mediated ubiquitination, facilitates the action of p53 with overexpression of CDK4 [33]. As the results of the study, expression of CDK4 and p53 were both significantly correlated with poor overall survival of chordoma patients. It could be identified CDK4 and p53 as possible potential targets for future studies aimed at inhibiting tumor progression.
Fibroblast growth factor (FGF) has been found to be associated with chordoma pathophysiology. Hu et al. explored that fibroblast growth factor receptor 2 (FGFR2), FGFR3, mitogen-activated protein kinase (MEK), and extracellular signal-regulated kinase (ERK) were expressed in both UCH1 and UCH2 chordoma cell lines [35]. Neutralization of FGF2 inhibited MEK/ERK phosphorylation, decreased brachyury expression, and induced apoptosis while reducing cell growth. The selective inhibition of FGFR, MEK, and ERK phosphorylation decreased brachyury expression, induced apoptosis, and inhibited cell growth and epithelial-mesenchymal transition (EMT). Moreover, knockdown of brachyury by small hairpin RNA reduced FGF2 secretion, inhibited FGFR/MEK/ERK phosphorylation, and blocked the effects of FGF2 on cell growth, apoptosis, and EMT. The FGFR/MEK/ERK/brachyury pathway has been proposed to coordinately regulate chordoma cell growth and survival and represent a novel chemotherapeutic target for chordoma.
DNA methylation in chordoma
DNA methylation is now a well-documented aspect of cancer development, and hyper-/hypo-methylation of specific gene loci has been shown to be strongly associated with malignancy [36]. DNA methylation has already provided useful biomarkers for diagnosing cancer, monitoring treatment, and predicting the prognosis. Aberrant DNA hypermethylation of CpG islands in the promoter region of genes is well established as a common mechanism for the silencing of tumor suppressor genes and serve as an alternative mechanism of functional inactivation in malignances, including chordoma [37]. Rinner et al. demonstrated DNA methylation of tumor suppressor genes exited in chordoma and served as a marker for early tumor detection [23]. They found 20 significantly differentially methylated genes, 15 hypermethylated in chordoma (RASSF1, KL, HIC1, and others), and 5 hypomethylated. Among them, RASSF1, KL, and HIC1 are known to be tumor suppressor genes. Inactivation of RASSF1 was found to be correlated with CpG island promoter region hypermethylation. KL potently inhibits ligand-dependent activation of the insulin and IGF-1 pathways and binds to FGFR. Expression of KL gene was inversely correlated to chordomagenesis. HIC1 is a transcriptional target of p53. HIC1 methylation could be used as a target for pharmacologic DNA-methyl transferase and could suit as a potential new target to treat chordoma patients.
Alholle et al. identified a preliminary set of nine probes (eight gene loci, FAM181B, KANK2, NPR3, PON3, RAB32, RAI1, SLC16A5, and ZNF397OS) that were differentially methylated in 26 chordomas and normal nucleus pulposus samples plus UCH1 chordoma cell line [36]. Seven probes were located in gene promoter region CpG islands and the rest two were located in CGI shores, likely to be associated with gene expression. The study demonstrated that these genes were re-expressed and/or regulated in methylated chordoma cell lines. Among them, KANK1 is a candidate tumor suppressor gene and was shown to be involved in gene fusion in a myeloproliferative neoplasm.
Promoter methylation of the MGMT gene represents a well-established molecular feature with prognostic and predictive significance in some tumors. Marucci et al. found methylation of MGMT promoter was present in a significant portion of recurring clival chordomas. On the contrary, MGMT promoter was unmethylated in clival chordomas without recurrence [38]. Furthermore, promoter methylation of the MGMT gene contributes to predict the efficaciousness of temozolomide. These results indicated that methylation of MGMT promoter was related to recurrent chordomas and prompted further investigation into the potential role of temozolomide as an adjuvant treatment of chordoma.
MicroRNA expression in chordoma
Aberrant miRNA expression enables to affect some pathways important for tumor initiation and maintenance in the diagnosis and prognosis of many malignancies, including chordoma [39, 40] (Table 1).
Table 1.
Dysregulated microRNAs in chordoma
| miRNA | Expression | Target gene(s) | Function and/or pathway involvement in chordoma | Reference |
|---|---|---|---|---|
| miR1 | Downregulated | MET, HDAC4 | Inhibit chordoma cell growth and proliferation | Duan Z et al., 2014 [40] |
| miR-149-3p | Downregulated | MAP, PKA | Tumor suppressor, MAPK pathway-related gene | Long C et al. 2013 [39] |
| miR-663a, miRNA-1908, miRNA-2861 | Downregulated | TGFB,JUND | Tumor suppressor, MAPK pathway-related gene | Long C et al. 2013 [39] |
| miR-3185 | Downregulated | FGF, CNrasGEF | Tumor suppressor, MAPK pathway-related gene | Long C et al. 2013 [39] |
| miR-762 | Downregulated | NCX1, PMCA1, NCKX4 | loss of calcification | Long C et al. 2013 [39] |
| miR-1228 | Downregulated | MOAP1, BMP2K | loss of osteoblast differentiation | Long C et al. 2013 [39] |
| miR-2861 | Downregulated | BMP | BMP-induced osteoblastogenesis | Long C et al. 2013 [39] |
| miR-222 | Downregulated | NA | Decrease epithelial-to-mesenchymal transition, cell migration and invasion | Bayrak, O.F. et al. 2013 [42] |
| miR-31 | Downregulated | c-MET | Apoptotic effect on chordoma cells | Bayrak, O.F. et al. 2013 [42] |
| miR-608 | Downregulated | EGFR/Bcl-xL | Tumor suppressor | Zhang, Y.et al. 2014 [44] |
| miR-34a | Downregulated | MET | Tumor suppressor, deregulated RTKs | Zhang, Y. et al. 2014 [44] |
| miR-155 | Upregulated | NA | Regulating cell proliferation, apoptosis, differentiation, angiogenesis, epithelial-to-mesenchymal transition;c correlating with advanced disease stage and the present metastasis | Zhang, Y.et al. 2014 [44] |
| miR-1237-3p | Downregulated | MMP-2 | Chordoma invasion and prognosis | Zou, M.X. et al. 2015 [46] |
Duan et al. first established a direct connection between a cell signaling pathway implicated in the molecular pathogenesis of chordoma and the miRNA machinery. Twenty-one miRNAs were profiled that were differentially expressed in chordoma cell lines (UCH1, CH22), and similar expression patterns were also found in primary chordoma tissues. The expression of miRNA-1 and miRNA-206 were markedly decreased in both chordoma tissues and cell lines. The proto-oncogene MET, which harbors two conserved miR-1 cognate sites, is expressed in most chordomas (94.4 %). With stable transfect miR-1 expression vectors in chordoma cells, re-introduction of miR-1 mimics by transient transfection suppresses Met expression and inhibits growth and proliferation of chordoma cells [40, 41]. Thirty-three miRNAs and 2791 mRNAs were indicted to be significantly dysregulated in chordoma relative to fetal notochord tissue using microarrays with hierarchical clustering analysis [39]. MAPK signaling pathway is the most highly overrepresented genetic pathway, governing cellular processes such as proliferation, differentiation, and survival. A pathway analysis showed that the five significantly downregulated miRNAs (miR-149-3p, miR-663a, miR-2861, miR-1908, and miR-3185) were predicted to target MAPK signaling pathway-related genes, including FGF2, JUND, DUSP4, MAP3K3, TGFB1, PRKACA, and RAPGEF2 through an inverse correlation. In the meantime, another three downregulated miRNAs (miR-762, miR-1228, and miR-2861) were reported to be associated to the calcification or osteoblast differentiation of notochord tissues. Loss of the capability of calcification or osteoblast differentiation might result in the formation of undifferentiated notochordal remnants and chordomagenesis. Another study has found 53 dysregulated miRNAs were in chordoma with the comparison between the miRNA profile of chordoma samples and the profile of normal nucleus pulposus samples, including 30 upregulated miRNAs and 23 downregulated miRNAs [42]. For example, miR-140-3p and miR-148a were upregulated, while miR-31 and miR-222 were downregulated in most chordomas [43]. These findings delineated the differential regulation of cancer-related genes in chordoma tumor cells and defined the complex molecular biology of the initiation and progression mechanisms. Zhang et al. uncovered miR-608 and miR-34a as novel tumor suppressors in chordoma. They also identified the commonly deregulated oncogenes EGFR/Bcl-xL as direct targets of miR-608 and MET as direct target of miR-34a. Both the inhibition of EGFR and MET inhibits chordoma cell proliferation and survival. In the study, both of miRNAs contributed to induce apoptosis and inhibit proliferation, growth and invasion in chordoma cells. As miRNAs enable to target multiple genes, overexpression of miR-608 or miR-34a is supposed to exert greater anti-tumor effects than EGFR or MET inhibitions [44]. Global miRNA expression and activity analysis suggested miR-155 was biologically active in chordoma [45]. Its upregulated expression was correlated with tumor progression in chordoma. Osaka et al. demonstrated that controlled downregulation of miR-155 expression inhibited multiple in vitro correlates of tumor aggression, including suppressed proliferation, and the migratory and invasive activities of chordoma cells. miR-155 expression was also analyzed to be significantly correlated with poor outcomes for chordoma patients, which suggested that modulating miR-155 expression could inhibit local recurrence and metastasis. Based on these differential expression of miRNAs and overlapping target genes selected in GO and pathway analysis, Zou et al. established a miRNA-gene regulatory network and revealed the interactions of these differentially expressed miRNAs and the potential target genes [46]. Five upregulated (miR-107, miR-20a-5p, miR-665, miR-202-3p, and miR-181a-5p) and one downregulated miRNAs (miR1237-3p) formed a gene regulation network, which included major target genes, such as CNOT4, KIF1B, NR4A3, CTNND1, KALRN, PCDHA9, CNOT6, ZAK, CIT, NTRK2, TSC22D2, HMGA1, COL19A1, EPHA7, SENP1, EFNB3, FNDC3B, TET3, PLAGL2, and CSNK1G1. The study showed that miR-1237-3p expression was an independent predictor with recurrence-free survival of the chordoma patients in the multivariate analysis. As the potential target gene, MMP-2 was associated with local invasion and worse prognosis of chordoma. Thus, downregulation of miR-1237-3p expression is supposed to promote MMP-2 expression, exerting a negative effect on recurrence and invasion of chordoma.
As is known, miRNAs can act as either oncogenes or tumor suppressors in chordoma. While some issues remain unresolved regarding the monitoring of circulating miRNA as biomarkers or the efficacy of miRNA delivery, miRNA reexpression therapy constitutes a novel approach and represents a therapeutic challenge in chordoma.
Receptor tyrosine kinase expression in chordomas
Previous studies have focused on the expression of receptor tyrosine kinases (RTKs) and their related secondary messengers in chordoma [47, 48]. Some RTKs have demonstrated dysregulated expression in chordoma and played a considerable role in chordoma’s makeup, including platelet-derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), human epidermal growth factor receptor (HER2/neu), and c-Met (hepatocyte growth factor receptor), as well as their downstream effectors phosphatidylinositide 3-kinase/protein kinase B (PI3K/AKT) and mammalian target of rapamycin (mTOR) [3•, 49, 50]. Concerning the downstream signaling following RTK’s phosphorylation, a considerable number of second messengers are reported to be overexpressed, and these are cross-linked in a very complex manner. Once activated, GTP-Ras instigates an intricate network of intracellular phosphorylation cascades, of which activation of the PI3K/RAS/Akt/mTOR pathway is most frequently described.
PDGFR-β expression and evidence of receptor phosphorylation were detected in all of the 18 samples analyzed in the study of Tamborini [48]. As an inhibitor of tyrosine kinases (especially PDGFR-β), imatinib acts as a competitive antagonist at the ATP binding site of the kinase domain and disrupts the ability of the kinase to phosphorylate its substrates. The absence of the pivotal role of KIT activating mutations and the constitutive pathologic activation of wild-type imatinib-related receptors in chordoma could explain why chordoma responds more slowly than gastrointestinal stromal tumor (GIST) to imatinib. The co-expression and co-immunoprecipation of PDGFR-β and EGFR have been described at the preclinical level [49, 51]. It is known that PDGF-β stimulates the activation of PDGFR-β and subsequently stimulates EGFR unidirectionally. In vitro studies demonstrated that two small molecule inhibitors of EGFR, erlotinib and gefitinib, inhibited proliferation of the chordoma cell line UCH1 [52]. Both EGFR and HER2/neu are members of the ErbB/HER family of RTKs. Variable amounts of HER2 immunoreactivity has been reported in chordoma [49]. In addition to the PDGFR family, which is inhibited by imatinib, the presence of activated receptors of the EGFR family may provide a rationale for chordoma treatment with small molecules that are effective on both EGFR and HER2/neu [50]. Chromosome 7 copy number was evaluated by chromogenic in situ hybridization (CISH) and protein expression of EGFR and c-Met in 22 chordoma samples [21]. Only c-Met protein expression was significantly correlated with chromosome 7 aneusomy. Its overexpression might represent an early chromosome 7 alteration that could play an important role during chordoma pathogenesis. Presneau et al. hypothesized that 75 % of chordoma samples would be responsive to mTOR inhibitors based on the expression of either the activated phosphorylated mTOR or the downstream activated protein phosphorylated S6K, which they considered as a surrogate marker for phosphorylated mTOR activity. Key downstream proteins within the PI3K/AKT/mTOR pathway were also shown to be phosphorylated in human chordomas tested including PDK, AKT, mTOR, and S6 [53]. The activation of important effectors of signaling downstream of RTK was analyzed in the study of Dewaele et al.. Using kinase antibody arrays, AKT was the most frequent (9 out of 10 cases analyzed) and highest phosphorylated in chordomas [51]. The consistent activation of AKT, the recurrent activation of upstream EGFR and of downstream effectors like p70S6K and mTOR, together with frequent loss of TSC1 and PTEN gene loci, all indicated that the PI3K/AKT pathway was an important mediator of transformation in chordoma. Since the downstream molecules have been observed to be activated, an explanation has yet to be offered for the failed regulation by inhibitory phosphatases such as PTEN, which is responsible for the regulation of mTOR. Another study found a significant correlation between negative expression (10/40) of PTEN and positive expression (25/40) of mTOR in sacral chordomas, indicating that mTOR and PTEN might co-regulate the progression of the tumor and participate in proliferation, invasion, and metastasis of sacral chordoma [54].
These results contribute to the understanding of the molecular alterations related to chordomas and support the development of targeted therapies with RTK inhibitors, which may have a synergistic effect for chemotherapy in patients with chordoma.
In vivo models of chordoma
Several in vivo models have recently been developed for further understanding the molecular and cellular biology of chordomagenesis [55–57].
Zebrafish models have a rapid onset of the tumor phenotype, and they contribute to readily accomplish the stable expression of transgenic constructs. Burger et al. described a zebrafish model of chordoma, driven by notochord-specific GFP-tagged HRASV12 expression during notochordal development. HRASV12 is well known as oncogenic mutation. The tumors of zebrafish models shared histological characteristics of human chordoma. The mTORC1 inhibitor rapamycin was demonstrated to delay the onset of tumor formation in this model and improve survival of tumor-bearing fish [55].
Chordoma cell lines have been shown to generate tumors in established mouse xenograft models and retain molecular characteristics of the parent tumor and expanded cells [57]. Subsequently, recent studies have established chordoma xenografts from patient-derived primary human chordoma cells, which also inherit and recapitulate the molecular features of parent tumors [56, 58, 59]. SF8894 was a chordoma xenograft derived from a recurrent clival chordoma [57]. The PI3K/Akt/mTOR pathway was activated in both the parent tumor and the xenograft. Treatment with ATP-competitive mTOR inhibitor, MLN0128, resulted in decreased activity of the PI3K/Akt/mTOR signaling pathway as indicated by decreased phospho-mTOR levels in the mice harboring chordoma xenografts. A primary sacral dedifferentiated chordoma was xenografted into NOD/SCID/IL-2R γ-null mice in another study [59]. Sunitinib and bortezomib, as proteasome inhibitor by inhibiting the activity of nuclear factor κB (NF-κB), were identified by a high-throughput in vitro screen. Both of the agents significantly slowed the growth of the established xenograft tumors. The results strongly implicated NF-κB signaling as a critical mediator of chordoma growth.
Conclusion
Chordoma is a malignant tumor with some puzzling features, such as its origin, and its local invasive and slow aggressive character. Here, we presented some investigations concerning the embryologic formation of chordoma for better understanding of the complex pathophysiology. We also summarized molecular researches yielding significant findings. Brachyury is discovered as both a crucial regulator of notochordal development and a novel biomarker for chordoma. Losses of CDKN2A and PTEN, as representative karyotype changes, have functional significance to the underlying biology and potential therapeutics for chordoma. We reviewed other discoveries of molecular pathways including cell cycle regulatory pathway and activated receptor tyrosine kinase pathway, both of which were well known to play considerable roles in chordoma process. DNA methylation of tumor suppressor genes has been demonstrated to modify both during notochord cell differentiation and malignant transformation. We also defined a set of miRNA candidates, which were dysregulated in chordomas, and proposed to utilize this miRNA profiling in the diagnosis and survival prediction of chordoma. We hope these integrative approaches could provide useful novel biomarkers advancing diagnostic workup and prognostic predictors.
Compliance with ethical standards
Conflict of interest
Dr. Schwab has been a consultant for Stryker, Biom’up, and Synthes, plus a speaker for Synthes. Dr. Sun and Dr. Hornicek have nothing to disclose.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on Orthopedic Oncology: New Concepts and Techniques
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–76. doi: 10.1016/S1470-2045(11)70337-0. [DOI] [PubMed] [Google Scholar]
- 2.Bjornsson J, Wold LE, Ebersold MJ, et al. Chordoma of the mobile spine. A clinicopathologic analysis of 40 patients. Cancer. 1993;71(3):735–40. doi: 10.1002/1097-0142(19930201)71:3<735::AID-CNCR2820710314>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 3.•.Yakkioui Y, van Overbeeke JJ, Santegoeds R, et al. Chordoma: the entity. Biochim Biophys Acta. 2014;1846(2):655–69. doi: 10.1016/j.bbcan.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Ferraresi V, Nuzzo C, Zoccali C, et al. Chordoma: clinical characteristics, management and prognosis of a case series of 25 patients. BMC Cancer. 2010;10:22. doi: 10.1186/1471-2407-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casali PG, Stacchiotti S, Sangalli C, et al. Chordoma. Curr Opin Oncol. 2007;19(4):367–70. doi: 10.1097/CCO.0b013e3281214448. [DOI] [PubMed] [Google Scholar]
- 6.Wiacek MP, Kaczmarek K, Sulewski A, et al. Unusual location of chordoma metastasis. Pol Orthop Traumatol. 2014;79:47–9. [PubMed] [Google Scholar]
- 7.Lee SH, Ahn BK. Sacral chordoma: challenging for resection margin. Ann Coloproctol. 2014;30(3):104–5. doi: 10.3393/ac.2014.30.3.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.M¨uller H, Ueber das Vorkommen von Resten der Chorda dorsalisbeiMenschennachderGeburtund ¨uber ihrVerh¨altniss zu den Gallertgeschw¨ulsten am Clivus. Zeitung F¨ur Rationelle Medizin. 1858; 2: 202.
- 9.Ribbert H, Uber die Ecchondrosis Physalifora Sphenooccipitalis. Zentralblatt F¨ur Allgemeine Pathologie Und Pathologische Anatomie. 1894; 5: 457–61.
- 10.Chauvel A, Taillat F, Gille O, et al. Giant vertebral notochordal rest: a new entity distinct from chordoma. Histopathology. 2005;47(6):646–9. doi: 10.1111/j.1365-2559.2005.02168.x. [DOI] [PubMed] [Google Scholar]
- 11.Choi KS, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn. 2008;237(12):3953–8. doi: 10.1002/dvdy.21805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi T, Suzuki S, Ishiiwa H, et al. Benign notochordal cell tumors: a comparative histological study of benign notochordal cell tumors, classic chordomas, and notochordal vestiges of fetal intervertebral discs. Am J Surg Pathol. 2004;28(6):756–61. doi: 10.1097/01.pas.0000126058.18669.5d. [DOI] [PubMed] [Google Scholar]
- 13.Kreshak J, Larousserie F, Picci P, et al. Difficulty distinguishing benign notochordal cell tumor from chordoma further suggests a link between them. Cancer Imaging. 2014;14:4. doi: 10.1186/1470-7330-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aydemir E, Bayrak OF, Sahin F, et al. Characterization of cancer stem-like cells in chordoma. J Neurosurg. 2012;116(4):810–20. doi: 10.3171/2011.12.JNS11430. [DOI] [PubMed] [Google Scholar]
- 15.Hsu ea W. Identification of cancer stem cells in human chordoma, 27th annual meeting of th AANS/CNS section on disorders of the spine and peripheral nerves. J Neurosurg. 2011;30(3):A1–25. doi: 10.1227/NEU.0b013e31820ccf90. [DOI] [Google Scholar]
- 16.Feng Y, Shen JK, Hornicek FJ, et al. Genomic and epigenetic instability in chordoma: current insights. Clin Cosmet Investig Dent. 2014;6:45–56. [Google Scholar]
- 17.Le LP, Nielsen GP, Rosenberg AE, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS One. 2011;6(5):e18846. doi: 10.1371/journal.pone.0018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheil-Bertram S, Kappler R, von Baer A, et al. Molecular profiling of chordoma. Int J Oncol. 2014;44(4):1041–55. doi: 10.3892/ijo.2014.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura Y, Sasaki H, Kimura T, et al. Molecular and clinical risk factors for recurrence of skull base chordomas: gain on chromosome 2p, expression of brachyury, and lack of irradiation negatively correlate with patient prognosis. J Neuropathol Exp Neurol. 2013;72(9):816–23. doi: 10.1097/NEN.0b013e3182a065d0. [DOI] [PubMed] [Google Scholar]
- 20.Bayrakli F, Guney I, Kilic T, et al. New candidate chromosomal regions for chordoma development. Surg Neurol. 2007;68(4):425–30. doi: 10.1016/j.surneu.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Walter BA, Begnami M, Valera VA, et al. Gain of chromosome 7 by chromogenic in situ hybridization (CISH) in chordomas is correlated to c-MET expression. J Neurooncol. 2011;101(2):199–206. doi: 10.1007/s11060-010-0250-5. [DOI] [PubMed] [Google Scholar]
- 22.Longoni M, Orzan F, Stroppi M, et al. Evaluation of 1p36 markers and clinical outcome in a skull base chordoma study. Neuro Oncol. 2008;10(1):52–60. doi: 10.1215/15228517-2007-048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rinner B, Weinhaeusel A, Lohberger B, et al. Genomic instability and characteristic DNA methylation pattern in chordoma. In VIRCHOWS ARCHIV. 2013. Springer, New York.
- 24.Choy E, MacConaill LE, Cote GM, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One. 2014;9(7):e101283. doi: 10.1371/journal.pone.0101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaloostian PE, Gokaslan ZL. Understanding the cell cycle in the pathophysiology of chordomas: a molecular look. World Neurosurg. 2014;82(1–2):e135–7. doi: 10.1016/j.wneu.2013.01.126. [DOI] [PubMed] [Google Scholar]
- 26.Barresi V, Ieni A, Branca G, et al. Brachyury: a diagnostic marker for the differential diagnosis of chordoma and hemangioblastoma versus neoplastic histological mimickers. Dis Markers. 2014;2014:514753. doi: 10.1155/2014/514753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vujovic S, Henderson S, Presneau N, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209(2):157–65. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 28.Nibu Y, Jose-Edwards DS, Di Gregorio A. From notochord formation to hereditary chordoma: the many roles of brachyury. Biomed Res Int. 2013;2013:826435. doi: 10.1155/2013/826435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cates JM, Itani DM, Coffin CM, et al. The sonic hedgehog pathway in chordoid tumours. Histopathology. 2010;56(7):978–9. doi: 10.1111/j.1365-2559.2010.03572.x. [DOI] [PubMed] [Google Scholar]
- 30.Kozmikova I, Candiani S, Fabian P, et al. Essential role of Bmp signaling and its positive feedback loop in the early cell fate evolution of chordates. Dev Biol. 2013;382(2):538–54. doi: 10.1016/j.ydbio.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Satoh N, Tagawa K, Takahashi H. How was the notochord born? Evol Dev. 2012;14(1):56–75. doi: 10.1111/j.1525-142X.2011.00522.x. [DOI] [PubMed] [Google Scholar]
- 32.Naka T, Boltze C, Kuester D, et al. Alterations of G1-S checkpoint in chordoma: the prognostic impact of p53 overexpression. Cancer. 2005;104(6):1255–63. doi: 10.1002/cncr.21296. [DOI] [PubMed] [Google Scholar]
- 33.Yakkioui Y, Temel Y, Creytens D, et al. A comparison of cell-cycle markers in skull base and sacral chordomas. World Neurosurg. 2014;82(1–2):e311–8. doi: 10.1016/j.wneu.2013.01.131. [DOI] [PubMed] [Google Scholar]
- 34.Wynford-Thomas D. P53 in tumour pathology: can we trust immunocytochemistry? J Pathol. 1992;166(4):329–30. doi: 10.1002/path.1711660402. [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Mintz A, Shah SR, et al. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis. 2014;35(7):1491–9. doi: 10.1093/carcin/bgu014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alholle A, Brini AT, Bauer J, et al. Genome-wide DNA methylation profiling of recurrent and non-recurrent chordomas. Epigenetics. 2015;10(3):213–20. doi: 10.1080/15592294.2015.1006497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulis M, Queiros AC, Beekman R, et al. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013;1829(11):1161–74. doi: 10.1016/j.bbagrm.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Marucci G, Morandi L, Mazzatenta D, et al. MGMT promoter methylation status in clival chordoma. J Neurooncol. 2014;118(2):271–6. doi: 10.1007/s11060-014-1445-y. [DOI] [PubMed] [Google Scholar]
- 39.Long C, Jiang L, Wei F, et al. Integrated miRNA-mRNA analysis revealing the potential roles of miRNAs in chordomas. PLoS One. 2013;8(6):e66676. doi: 10.1371/journal.pone.0066676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan Z, Shen J, Yang X, et al. Prognostic significance of miRNA-1 (miR-1) expression in patients with chordoma. J Orthop Res. 2014;32(5):695–701. doi: 10.1002/jor.22589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan Z, Choy E, Nielsen GP, et al. Differential expression of microRNA (miRNA) in chordoma reveals a role for miRNA-1 in Met expression. J Orthop Res. 2010;28(6):746–52. doi: 10.1002/jor.21055. [DOI] [PubMed] [Google Scholar]
- 42.Bayrak OF, Gulluoglu S, Aydemir E, et al. MicroRNA expression profiling reveals the potential function of microRNA-31 in chordomas. J Neurooncol. 2013;115(2):143–51. doi: 10.1007/s11060-013-1211-6. [DOI] [PubMed] [Google Scholar]
- 43.Zou MX, Huang W, Wang XB, et al. Identification of miR-140-3p as a marker associated with poor prognosis in spinal chordoma. Int J Clin Exp Pathol. 2014;7(8):4877–85. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Schiff D, Park D, et al. MicroRNA-608 and microRNA-34a regulate chordoma malignancy by targeting EGFR, Bcl-xL and MET. PLoS One. 2014;9(3):e91546. doi: 10.1371/journal.pone.0091546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osaka E, Kelly AD, Spentzos D, et al. MicroRNA-155 expression is independently predictive of outcome in chordoma. Oncotarget. 2015;6(11):9125–39. doi: 10.18632/oncotarget.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou MX, Huang W, Wang XB, et al., Reduced expression of miRNA-1237-3p associated with poor survival of spinal chordoma patients. Eur Spine J. 2015. [DOI] [PubMed]
- 47.Heymann D, Redini F. Targeted therapies for bone sarcomas. Bonekey Rep. 2013;2:378. doi: 10.1038/bonekey.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamborini E, Miselli F, Negri T, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12(23):6920–8. doi: 10.1158/1078-0432.CCR-06-1584. [DOI] [PubMed] [Google Scholar]
- 49.Tamborini E, Virdis E, Negri T, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro-Oncol. 2010;12(8):776–89. doi: 10.1093/neuonc/noq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Castro CV, Guimaraes G, Aguiar S, Jr, et al. Tyrosine kinase receptor expression in chordomas: phosphorylated AKT correlates inversely with outcome. Hum Pathol. 2013;44(9):1747–55. doi: 10.1016/j.humpath.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 51.Dewaele B, Maggiani F, Floris G, et al. Frequent activation of EGFR in advanced chordomas. Clin Sarcoma Res. 2011;1(1):4. doi: 10.1186/2045-3329-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siu IM, Ruzevick J, Zhao Q, et al. Erlotinib inhibits growth of a patient-derived chordoma xenograft. PLoS One. 2013;8(11):e78895. doi: 10.1371/journal.pone.0078895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwab J, Antonescu C, Boland P, et al. Combination of PI3K/mTOR inhibition demonstrates efficacy in human chordoma. Anticancer Res. 2009;29(6):1867–71. [PubMed] [Google Scholar]
- 54.Chen K, Mo J, Zhou M, et al. Expression of PTEN and mTOR in sacral chordoma and association with poor prognosis. Med Oncol. 2014;31(4):886. doi: 10.1007/s12032-014-0886-7. [DOI] [PubMed] [Google Scholar]
- 55.Burger A, Vasilyev A, Tomar R, et al. A zebrafish model of chordoma initiated by notochord-driven expression of HRASV12. Dis Model Mech. 2014;7(7):907–13. doi: 10.1242/dmm.013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davies JM, Robinson AE, Cowdrey C, et al. Generation of a patient-derived chordoma xenograft and characterization of the phosphoproteome in a recurrent chordoma. J Neurosurg. 2014;120(2):331–6. doi: 10.3171/2013.10.JNS13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karikari IO, Gilchrist CL, Jing L, et al. Molecular characterization of chordoma xenografts generated from a novel primary chordoma cell source and two chordoma cell lines. J Neurosurg Spine. 2014;21(3):386–93. doi: 10.3171/2014.4.SPINE13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siu IM, Salmasi V, Orr BA, et al. Establishment and characterization of a primary human chordoma xenograft model. J Neurosurg. 2012;116(4):801–9. doi: 10.3171/2011.12.JNS111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trucco MM, Awad O, Wilky BA, et al. A novel chordoma xenograft allows in vivo drug testing and reveals the importance of NF-kappaB signaling in chordoma biology. PLoS One. 2013;8(11):e79950. doi: 10.1371/journal.pone.0079950. [DOI] [PMC free article] [PubMed] [Google Scholar]