

Abstract
Background
Loss-of-function mutations in the myotubularin (MTM1) gene cause X-linked myotubular myopathy (XLMTM), a fatal, inherited pediatric disease that affects the entire skeletal musculature. Labrador retriever dogs carrying an MTM1 missense mutation exhibit strongly reduced synthesis of myotubularin, the founder member of a lipid phosphatase required for normal skeletal muscle function. The resulting canine phenotype resembles that of human patients with comparably severe mutations, and survival does not normally exceed 4 months.
Methods
We studied MTM1 mutant dogs (n=7) and their age-matched control littermates (n=6) between the ages of 10 and 25 weeks. Investigators blinded to the animal identities sequentially measured limb muscle pathology, fore- and hind limb strength, walking gait, respiratory function and neurological impairment.
Results
MTM1-mutant puppies display centrally-nucleated myofibers of reduced size and disrupted sarcotubular architecture progressing until the end of life, an average of 17 weeks. In-life measures of fore- and hind limb strength establish the rate at which XLMTM muscles weaken, and their corresponding decrease in gait velocity and stride length. Pulmonary function tests in affected dogs reveal a right-shifted relationship between peak inspiratory flow (PIF) and inspiratory time (TI); neurological assessments indicate that affected puppies as young as 10 weeks show early signs of neurological impairment (neurological severity score, NSS =8.6±0.9) with progressive decline (NSS =5.6±1.7 at 17 weeks-of-age).
Conclusions
Our findings document the rate of disease progression in a large animal model of XLMTM and lay a foundation for preclinical studies.
Keywords: Animal models, muscle disease, myopathy, muscular dystrophy, myotubular myopathy, dog
Introduction
X-linked myotubular myopathy (XLMTM; OMIM 310400) is a fatal non-dystrophic disease of skeletal muscle that affects approximately one in 50,000 male births. Patients typically present with marked hypotonia, generalized muscle weakness and respiratory failure at birth (1). Survival beyond the postnatal period requires intensive support, often including gastrostomy feeding and mechanical ventilation. XLMTM results from loss-of-function mutations in the myotubularin 1 (MTM1) gene (2), which encodes the founder of a family of 3-phosphoinositide phosphatases acting on the second messengers phosphatidylinositol 3-monophosphate [PI(3)P] and phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] (3,4). Although myotubularin is expressed ubiquitously, loss of this enzyme primarily affects skeletal muscles. Myogenesis occurs, but muscle fibers throughout the body are hypotrophic and display structural abnormalities with associated weakness (5). No cure currently exists for the disease but putative therapies seek to replace either the defective gene or gene product (6,7).
Animal models of the disease currently exist in zebrafish, mouse and dog (5,8,9). Analogous to human XLMTM patients, mice engineered with a targeted deletion of MTM1 demonstrate marked hypotonia and shortened lifespan (5). In Labrador retriever dogs, a p.N155K missense mutation in canine MTM1 results in profound skeletal muscle weakness in affected puppies (10). After approximately 9 weeks-of-age, the canine phenotype parallels human patients with comparably severe mutations; survival does not normally exceed four months in affected dogs (9). Because dogs are similar in size and much closer physiologically to patients compared to mice, experiments in dogs are exceptionally informative for clinical trial design. We previously reported a gene therapy pilot where systemic administration of a single dose of a recombinant serotype-8 adeno-associated virus (AAV8) vector expressing canine MTM1 to MTM1-mutant puppies markedly improved severe muscle weakness and prolonged survival (6). In the present study, we set out to explore and further establish biomarkers of disease over the natural lifespan of the p.N155K dog model of XLMTM to provision additional dose-finding studies of gene therapy.
Materials and methods
Dogs were handled according to principles outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and as approved by the University of Washington Institutional Animal Care and Use Committee (IACUC). Investigators performing assessments were blinded to the identities of animals. Some offspring of the colony’s founding Labrador retriever were crossbred to beagle or mixed-breed dogs. To generate affected male and female dogs for the present study, two hemizygous males were previously infused with an adeno-associated viral (AAV) vector expressing the myotubularin (MTM1) gene as described (6). These AAV-infused affected males a year later produced both hemizygous males and homozygous female offspring expressing the disease. Criteria for humane euthanasia included inability to stand or walk, and continued weight loss with pronounced muscle wasting despite supplemental feeding.
Genotyping
Puppies were genotyped from DNA isolated from oral swabs. Affected hemizygous males, and homozygous females carrying the MTM1 c.465C>A variant were identified using a TaqMan assay as described (9). All genotypes were confirmed on two independent specimens for each dog.
Histology
Tissue collection
Muscle samples were collected and processed as described (11). For biopsies, tissue from the vastus lateralis, gastrocnemius, biceps brachialis, and sartorius muscles were collected from normal and MTM1-mutant dogs at 10, 18, and 25 weeks-of-age (unless the animal succumbed earlier to disease) and autopsy tissue collection was performed when animals required euthanasia (age range 17 to 27 weeks).
Histological evaluation
Muscle histology was performed based on staining with hematoxylin and eosin (H&E), reduced nicotinamide adenine dinucleotide (NADH) and an ATPase stain performed at pH 9.4. Slides were evaluated by a board-certified anatomic pathologist and neuropathologist (MWL) with respect to the full range of possible pathologies. Muscle tissue (vastus lateralis) was fixed in 2.5% glutaraldehyde, processed at the Medical College of Wisconsin’s electron microscopy (EM) Core Facility and evaluated at each time point using a standard approach previously described (6,7). Comprehensive reports of pathological findings at the light and EM level were prepared using an adaptation of the NINDS Common Data Elements muscle biopsy reporting form (12).
In vivo measures of limb strength
Contractile properties in canine hindlimb muscles were assessed in vivo as described (13,14). Briefly, hind limb torque of anesthetized dogs was measured by wrapping the foot to a pedal mounted on the shaft of a servomotor that also functioned as a force transducer. Percutaneous nerve stimulation activated hind limb muscles to either pull the paw up toward the body or push the paw down toward the ground. Isometric contractions were performed over a range of stimulation frequencies to determine torque-frequency relationships. Forelimb torque was measured in an analogous manner as described (15).
Gait
An instrumented carpet (GAITRite Electronic Walkway, CIR Systems Inc.) was used to measure gait as described (16). Generally 5-10 passes across the carpet were taken but testing ended immediately if the dog showed signs of fatigue. Fewer passes were collected for young puppies or for very weak animals. Data were collected using software (GAITFour version 4.1, CIR Systems Inc.) with simultaneous video recording by a synchronized camera (Logitech) for quality control. Walks were selected based on consistency of pattern and speed, as well as body position and behavior along the carpet. Spatiotemporal measures of gait including stride velocity and length were calculated by the software as described (16).
Pulmonary function
Respiratory assessment was carried out in anesthetized dogs before and after stimulation by a centrally acting stimulant, doxapram hydrochloride (1 mg/kg), as described (17-19). Briefly, data were collected in intubated dogs using a calibrated pneumotachometer, where changes in pressure across the device determined airflow. Flow-volume loops were created by replaying experimental data through the appropriate analyzer (iox 2.8.0.13, EMKA Technologies) and capturing 10 consecutive breaths during doxapram stimulation. Software (EMKA Technologies) calculated peak inspiratory flow (PIF), peak expiratory flow (PEF), inspired volume (IV), expired volume (EV), inspiratory time (TI), and expiratory time (TE).
Neurological assessment
Neurological severity score (NSS) was measured in dogs by a board-certified veterinary neurologist (JMS) as previously described (20). Parameters assessed included: cranial nerve function, postural reactions, segmental spinal reflexes, gait stride length, ability to run and jump, and muscle atrophy. Dogs were observed for exercise intolerance or increased respiratory rate and effort following activity. The presence or absence of a dropped jaw (ability to hold the jaw in a closed position) was also noted. Following each examination, a NSS was assigned on a scale of 10 to 1, with 10 indicating a normal examination and 1 indicating an inability to maintain sternal recumbency. However, predetermined humane euthanasia criteria were usually met by a NSS score of 3. Results of neurological scoring for wild type and some of the XLMTM dogs here were previously reported (20).
Segmental spinal reflexes assessed included the forelimb and hindlimb withdrawal, the extensor carpi radialis, the patellar, and the cranial tibial reflexes. Reflexes were graded from 0-4 with “0” indicating an absent response, “1” indicating a decreased response, “2” indicating a normal response, “3” indicating an increased response, or “4” indicating a clonic response. Reflex scores were assigned to each reflex and then averaged for the five reflexes assessed for each dog. Average reflex scores for each dog were then used to create a mean reflex score (sum of average reflex score for all dogs divided by the number of dogs in the group) for the wild type and affected groups at each time point.
Statistics analysis
Variability in some of the measures reduced statistical power. As such, descriptive statistics were primarily used to report findings at 10 and 17 weeks-of-age as well as the maximal survival age for XLMTM dogs. Group mean differences over time were determined by repeated measure analysis of variance, with Bonferoni confidence interval adjustment (SPSS Statistics v22, IBM). Values were significant (P<0.05) where indicated.
Results
Mutation in MTM1 (c.465C>A) leads to markedly shortened lifespan in the p.N155K canine model of XLMTM
About half of MTM1-mutant dogs were stillborn or died within the first week after birth and were excluded from study. Surviving affected dogs (n=7) lived to an average of 17 weeks with only a single animal alive beyond 25 weeks-of-age (Figure 1). Affected dogs generally weighed less than their littermate controls: 4.28±1.2 vs. 5.25±1.1 kg; and 7.11±1.7 vs. 8.19±1.9 kg, for XLMTM and control dogs 10 and 17 weeks-of-age, respectively. Hind limb length measurements of affected dogs were comparable to control dog lengths at 10- and 17-weeks-of-age (Table 1). These data indicate that during their average lifespan, MTM1-mutant dogs weigh slightly less than their WT or carrier littermates while growing to a comparable size.
Figure 1.
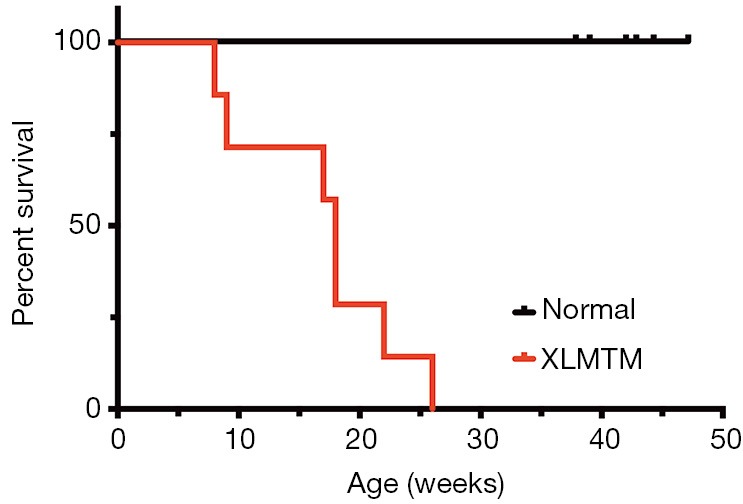
MTM1 mutation shortens the normal lifespan in the p.N155K canine model. Kaplan-Meier survival curve for WT and carriers (N=6) (black) and XLMTM (N=7) (red) dogs. XLMTM, X-linked myotubular myopathy.
Table 1. Body mass and hindlimb length of dogs harboring an MTM1 mutation (c.465C>A), and their age-matched littermate controls at 10 and 17 weeks-of-age.
| Dog | Sex | Genotype | Mass at 10 weeks (kg) | Mass at 17 weeks (kg) | Limb length at 10 weeks (cm) | Limb length at 17 weeks (cm) |
|---|---|---|---|---|---|---|
| XLMTM dogs | ||||||
| SSAN 7 | F | Affected | 5.01 | 6.71 | 13.2 | 15.3 |
| SSAN 9 | F | Affected | 4.91 | 5.20 | 13.3 | 14.6 |
| SSAN 13 | F | Affected | 3.93 | 6.67 | 12.6 | 15.3 |
| SSAN 16 | M | Affected | 5.94 | 9.95 | 14.6 | 17.5 |
| SSAN 101 | F | Affected | 4.06 | 7.03 | 10.6 | 14.5 |
| SSAN 103 | F | Affected | 3.81 | – | 11.3 | – |
| SSAN 114 | M | Affected | 2.27 | – | 9.9 | – |
| Mean ± SD | 4.28±1.16 | 7.11±1.74 | 4.28±1.16 | 7.11±1.74 | ||
| Normal dogs | ||||||
| SSAN 1 | F | Wildtype | 5.44 | 8.94 | 12.9 | 15.4 |
| SSAN 3 | F | Wildtype | 4.65 | 6.91 | 12.8 | 15.4 |
| SSAN 5 | F | Carrier | 5.77 | 7.93 | 12.9 | 14.2 |
| SSAN 2 | M | Wildtype | 3.82 | 5.93 | 11.1 | 13.2 |
| SSAN 6 | M | Wildtype | 4.77 | 8.12 | 12.5 | 15 |
| SSAN 12 | M | Wildtype | 7.03 | 11.31 | 14.6 | 17.5 |
| Mean ± SD | 5.25±1.10 | 8.19±1.85 | 5.25±1.10 | 8.19±1.85 | ||
XLMTM, X-linked myotubular myopathy.
Skeletal muscles harboring MTM1 (c.465C>A) mutation reveal early pathological abnormalities and tend to parallel XLMTM clinical disease severity
Muscle histopathology was assessed at 10, 18, and 25 weeks-of-age in MTM1-mutant dogs (when possible, n=6) and normal littermates (n=6) and additionally at end-stage of disease. Only one dog (SSAN16) survived long enough for tissue collection at 25 weeks, and this dog reached end-stage 2 weeks later. Histological findings in normal dogs were essentially unremarkable with the exception of scattered atrophic fibers corresponding to either fiber type, which constituted the minimal degree of pathology historically observed in dogs from this colony (Figure 2A). There was minimal to absent central or internal nucleation, fibrosis, and fatty infiltration in samples from normal dogs, except in one biopsy (CTRL2) at 17 weeks-of-age where a site of prior focal injury was included in the biopsy site (not shown). Mitochondrial localization was even and appropriate by NADH staining and EM in normal dogs. EM evaluation also identified normal sarcotubular organization with easily identifiable triad structures in normal dogs (Figure 2B, arrows). In contrast, MTM1-mutant dogs showed significant pathological abnormalities at all time points, including variable degrees of myofiber hypotrophy (fiber smallness), central nucleation, and mitochondrial mislocalization (Figure 2A). Sarcotubular disorganization was additionally evident at the ultrastructural level, where triad structures were extremely rare (Figure 2B). Differences in the rate of disease progression among affected dogs caused considerable variability at 17-18 weeks-of-age, as some animals were end-stage at this point while others showed a mixture of disease pathology and compensatory hypertrophy (Figure 2A). MTM1-mutant dogs that survived past 19 weeks had similar pathological findings to one another, including marked myofiber hypotrophy in a subset of fibers and marked compensatory hypertrophy in a different subset of fibers. Organelle mislocalization (as indicated by either central aggregates of mitochondria and/or peripheral aggregation of mitochondria and sarcotubular material on NADH stain) was widespread in both the hypotrophic and hypertrophic fiber populations. In contrast, MTM1-mutant dogs that reached end-stage at 18 weeks-of-age showed histological findings similar to those seen at end stage in the longer-lived MTM1-mutant dogs. These findings included fairly uniform fiber smallness (a combination of hypotrophy and atrophy, given the presence of larger fibers at earlier time points) and organelle mislocalization in essentially all myofibers. Ultrastructural examination at 18 weeks and at end stage showed findings similar to those seen at 10 weeks, including marked organelle disorganization and only very rare triad structures (Figure 2B). The main distinction between time points on ultrastructural examination was reflective of the fiber populations present in the sample (i.e., the presence of hypertrophic or atrophic fibers), but the degree of disorganization within individual fibers did not appear different between time points.
Figure 2.
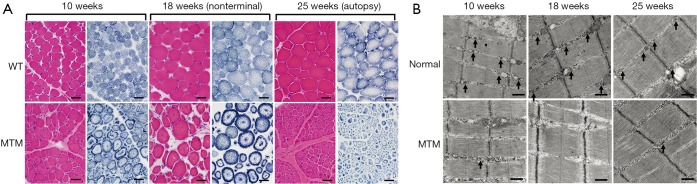
Progression of skeletal muscle pathology in the p.N155K canine model of XLMTM. (A) Representative micrographs of vastus lateralis biopsies stained with H&E (red) or NADH (blue) illustrate myofiber hypotrophy, central nucleation and mislocalization of organelles (necklace fibers, arrows) in the MTM1 mutant muscles. Bar =40 µm. (B) Representative electron micrographs of vastus lateralis biopsies show that the sarcotubular apparatus is progressively disrupted in MTM1-mutant dogs. The architecture of T-tubules and triads (arrows) observed in normal littermates appear altered in MTM1-mutant dogs. Abnormal longitudinal tubules (L-tubules, arrows) L-tubules were observed along with disruption of the normal triad structures in MTM1-mutant dogs. Bar =500 nm. XLMTM, X-linked myotubular myopathy; H&E, hematoxylin and eosin; NADH, nicotinamide adenine dinucleotide.
MTM1 mutation leads to preferential limb muscle weakness
To evaluate the effects of MTM1 c.465C>A mutation on muscle function over time in dogs, we used in vivo isometric torque (force) transduction assays to assess flexion and extension strength of the hind- and forelimbs. Muscle strength was recorded as the average maximum isometric torque normalized to total body mass. Muscles of forelimb extension and hindlimb flexion exhibited the most rapid decline among the limb muscles tested.
In the forelimbs, MTM1-mutant extensor muscles (mainly the extensor carpi radialis that pulls the front paw up from the ground) rapidly weakened. In young animals 10 weeks-of-age, isometric strength measured 0.022±0.01 and 0.018±0.01 N·m/kg in normal and XLMTM dogs, respectively. By 17 weeks-of-age, isometric strength measured 0.037±0.01 and 0.008±0.01 N·m/kg in normal and XLMTM dogs, respectively. At the final assessment time at 25 weeks-of-age, isometric forelimb extension measured 0.043±0.01 N·m/kg in normal controls and 0.010 N·m/kg in the last surviving XLMTM (Figure 3A).
Figure 3.
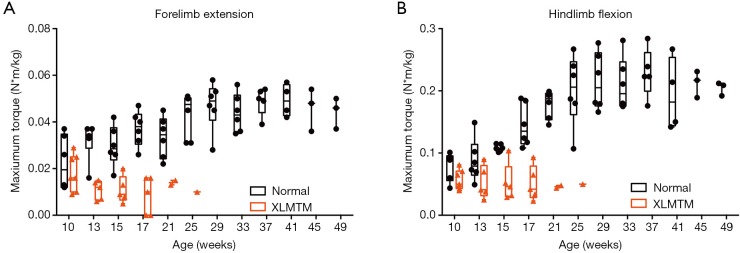
MTM1 mutation in dogs results in progressive loss of limb muscle strength in the first 4 months of life. (A) In vivo maximal isometric torque measured in forelimb extensor muscles (mainly the extensor carpi radialis) and (B) in vivo maximal isometric torque measured in hindlimb flexor muscles (mainly the cranial tibialis). Control dogs (WT and carriers) are shown in black; XLMTM dogs in red. XLMTM, X-linked myotubular myopathy.
In contrast to the rapid weakening observed in MTM1-mutant forelimb extensors, the forelimb flexors (mainly the flexor carpi radialis that moves the front paw down against the ground) weakened at a slower rate. At the initial assessment time (10 weeks-of-age) isometric strength measured 0.098±0.03 and 0.119±0.04 N·m/kg, for normal and XLMTM littermates, respectively. At 17 weeks-of-age, isometric strength measured 0.130±0.03 and 0.107±0.02 N·m/kg for normal and XLMTM dogs, respectively. At the end of the assessment period (25 weeks-of-age) isometric flexion strength measured 0.157±0.03 N·m/kg in normal controls and 0.126 N·m/kg in the last surviving affected dog.
In MTM1-mutant hind limbs, the flexor muscles weakened more rapidly than the extensor muscles (Figure 3B). At 10 weeks-of-age, flexors (mainly the cranial tibialis, a muscle that pulls the hind paw up away from the ground) measured 0.080±0.02 and 0.057±0.02 N·m/kg in normal and XLMTM dogs, respectively. At 17 weeks-of-age, isometric hindlimb flexion measured 0.145±0.03 and 0.051±0.03 N·m/kg in normal and XLMTM dogs, respectively. At the last timepoint, 25 weeks-of-age, flexion strength measured 0.201±0.06 N·m/kg in normal controls compared to 0.050 N·m/kg in the last surviving XLMTM dog (Figure 3B).
In contrast to the rapid decline in strength observed in MTM1-mutant flexors, at 10 weeks-of-age, extensors (mainly the gastrocnemius and soleus, muscles that move the hind paw down toward the ground) measured 0.239±0.11 and 0.211±0.06 N·m/kg in normal and XLMTM dogs, respectively. At 17 weeks-of-age, extension strength measured 0.311±0.10 and 0.238±0.09 N·m/kg in normal and XLMTM dogs, respectively. At the final 25 weeks-of-age assessment, extension strength measured 0.441±0.15 N·m/kg in the normal controls and 0.050 N·m/kg in the last surviving XLMTM dog.
Taken together, in vivo fore- and hind limb assays indicate that MTM1 c.465C>A mutation leads to limb weakness as early as 10 weeks-of-age and thereafter strength rapidly declines over the remaining short lifespan, typically by 17 weeks-of-age. The forelimb extensors and hind limb flexors appear to weaken more rapidly than the forelimb flexors or hind limb extensors.
MTM1 mutation results in abnormally slow gait with shortened strides and steps
To evaluate effects of MTM1 mutation on functional mobility an instrumented carpet was used to measure spatiotemporal gait variables, including velocity, stride and step length, stride, stance and swing times. As affected dogs progressively declined later in life, about half of XLMTM dogs were too weak for successful gait assessment by 17 weeks-of-age and were excluded from further analysis. In general, results indicate that MTM1 mutation in dogs’ results in reduced gait velocity, shortened stride and step lengths, and prolonged stance time.
Gait velocity
XLMTM dogs walked more slowly than their normal littermates (P=0.001), with velocities that were 26% of normal at 10 weeks-of-age (N=6), 35% of normal at 17 weeks (N=3) and 24% of normal at 21 weeks-of-age (N=2; Table 2, Figure 4A).
Table 2. Spatiotemporal measures of gait in normal and XLMTM dogs measured at 10, 17 and 25 weeks-of-age.
| Dog | Velocity (cm/s) | Stride length (cm) | Stride time (s) | Step length (cm) | Step time (s) | Stance time (s) | Stance % of cycle | Swing time (s) | Swing % of cycle |
|---|---|---|---|---|---|---|---|---|---|
| Normal | |||||||||
| 10 weeks (n=6) | 175.8±26.0 | 61.6±11.0 | 0.35±0.05 | 30.5±5.4 | 0.18±0.02 | 0.13±0.02 | 36±4.0 | 0.22±0.04 | 64.0±4.0 |
| 17 weeks (n=6) | 175.6±16.7 | 68.5±5.2 | 0.39±0.05 | 68.5±5.2 | 0.20±0.02 | 0.17±0.03 | 42.1±4.0 | 0.22±0.02 | 57.9±3.9 |
| 21 weeks (n=6) | 164.6±16.9 | 70.3±9.2 | 0.43±0.04 | 34.9±4.6 | 0.22±0.02 | 0.19±0.02 | 44.4±2.5 | 0.24±0.02 | 55.6±2.5 |
| XLMTM | |||||||||
| 10 weeks (n=6) | 129.4±45.7 | 48.1±11.0 | 0.39±0.06 | 23.9±5.5 | 0.19±0.03 | 0.16±0.04 | 42.0±4.8 | 0.22±0.02 | 55.7±8.1 |
| 17 weeks (n=3) | 114.2±1.5 | 51.5±6.6 | 0.45±0.05 | 25.6±3.3 | 0.23±0.03 | 0.24±0.04 | 51.8±2.6 | 0.22±0.01 | 48.2±2.6 |
| 21 weeks (n=2) | 125.8±3.5 | 55.0±4.9 | 0.44±0.02 | 27.4±2.4 | 0.22±0.01 | 0.22±0.02 | 50.6±1.4 | 0.22±0.00 | 49.4±1.4 |
Data are presented as mean ± SD. XLMTM, X-linked myotubular myopathy.
Figure 4.

Effects of MTM1 mutation on walking gait in dogs. XLMTM (red) and their control littermates (black) walked repeatedly over an instrumented carpet to capture spatiotemporal measures of gait including (A) gait speed, (B) stride length, (C) stance time, (D) stride time, (E) stance percentage, (F) swing time. XLMTM, X-linked myotubular myopathy.
Stride and step length
XLMTM dogs took shorter strides and steps than normal dogs. Both stride and step lengths were 22% shorter than normal by 10 weeks-of-age, 25% shorter by 17 weeks-of-age and 22% shorter at 21 weeks-of-age (Table 2, Figure 4B).
Stride, stance, and swing times
Compared to normal dogs, XLMTM dogs demonstrated slightly longer stride times at all gait assessments, with marked increases in stance time to 30% above normal at 10 weeks-of-age, 42% above normal at 17 weeks and 16% above normal at 21 weeks-of-age (P=0.018, Figure 4C-E). However, swing times remained at or near normal in XLMTM dogs. As a result, XLMTM dogs spent a larger proportion of the gait cycle in stance phase than control dogs (P=0.008, Figure 4F). At 10 weeks-of-age, XLMTM dogs spent 42.0%±4.8% of the gait cycle in stance phase, compared to 36.0%±4.0% in control dogs. At 21 weeks-of-age, stance times remained longer in XLMTM dogs, with XLMTM dogs spending 50.6%±1.4% of the gait cycle in stance compared to 44.4%±2.5% for control dogs.
Altogether, gait assessments indicate that myotubularin deficiency impacts functional mobility in dogs by a gait pattern characterized by slowed velocity, shortened stride and step length, and increased stance time compared to normal dogs.
MTM1-mutant dogs display qualitative abnormalities in respiratory flow-volume loops
XLMTM is associated with neonatal hypotonia and respiratory failure. To test for the presence and possible progression of respiratory muscle weakness in the p.N155K canine model of XLMTM, we evaluated tidal breathing in anesthetized dogs using a pneumotachometer. To test whether central respiratory stimulation would uncover mechanisms by which affected dogs maintain ventilatory homeostasis, we measured pulmonary function in anesthetized XLMTM dogs and their littermate controls before and immediately following bolus infusion of doxapram hydrochloride. In keeping with previous findings (21), normal dogs generate large, ‘D’ shaped flow-volume loop patterns at 17 weeks-of-age, where PIF plateaued in mid-late inspiration and dropped rapidly, while peak expiratory occurred early and then gradually reduced (Figure 5A). In contrast, the flow-volume loops of age-matched MTM1-mutant dogs appeared smaller or irregularly-shaped (Figure 5B). We frequently observed a reduced PIF (Figure 5B) in comparison to control dogs, suggesting impairment of diaphragm function. Tables 3,4 show standard respiratory parameters of the two genotypes over time, during resting breathing and doxapram stimulation.
Figure 5.

Effects of MTM1 mutation on respiration during direct stimulation. Dogs given intravenous doxapram to place the respiratory muscles at work. Representative flow volume loops captured after respiratory stimulation for individual (A) controls (N=2), dark blue loops and (B) XLMTM (N=4) dogs, red loops, at 17 weeks-of-age. (C) The flow-volume loop, with highlighted areas of measurement. XLMTM, X-linked myotubular myopathy.
Table 3. Baseline ventilatory parameters measured in normal and XLMTM dogs at 10, 17 and 25 weeks-of-age.
| Dog | Inspiratory time (ms) | Expiratory time (ms) | Peak inspiratory flow (mL/s) | Peak expiratory flow (mL/s) | Tidal volume (mL/kg) | Expiratory volume (mL/kg) | Minute ventilation (mL/s/kg) | Respiratory rate (bpm) |
|---|---|---|---|---|---|---|---|---|
| Normal | ||||||||
| 10 weeks (n=6) | 542.4±155 | 1,093.6±595 | 95.1±22 | 231.1±37 | 5.9±1 | 6.1±1 | 249.7±105 | 41.0±13 |
| 17 weeks (n=6) | 774.8±134 | 1,612.8±681 | 102.6±11 | 302.2±46 | 6.6±2 | 7.1±2 | 181.6±73 | 27.5±9 |
| 25 weeks (n=6) | 856.7±191 | 1,827.5±368 | 137.4±41 | 375.8±75 | 8.8±5 | 8.4±4 | 207.7±108 | 24.3±5 |
| XLMTM | ||||||||
| 10 weeks (n=6) | 529.8±116 | 1,093±472 | 115.6±28 | 152.6±53 | 7.0±2 | 6.7±1 | 283.0±132 | 39.0±12 |
| 17 weeks (n=3) | 755.6±216 | 1,560±583 | 121.0±35 | 239.1±61 | 6.3±1 | 6.3±1 | 174.9±48 | 27.8±8 |
| 21 weeks (n=2) | 1,552 | 1,270 | 256.4 | 307.9 | 14.4 | 11.4 | 281.0 | 20.0 |
Data are presented as mean ± SD. XLMTM, X-linked myotubular myopathy.
Table 4. Ventilatory parameters under doxapram (1 mg/kg) stimulation measured in normal and XLMTM dogs at 10, 17 and 25 weeks-of-age.
| Dog | Inspiratory time (ms) | Expiratory time (ms) | Peak inspiratory flow (mL/s) | Peak expiratory flow (mL/s) | Tidal volume (mL/kg) | Expiratory volume (mL/kg) | Minute ventilation (mL/s/kg) | Respiratory rate (bpm) |
|---|---|---|---|---|---|---|---|---|
| Normal | ||||||||
| 10 weeks (n=6) | 399.6±97 | 394.2±209 | 247.3±63 | 364.8±46 | 12.1±4 | 12.4±2 | 1,001.8±477 | 78.5±22 |
| 17 weeks (n=6) | 538.7±86 | 572.5±160 | 343.8±76 | 571.8±93 | 16.4±3 | 16.2±3 | 954.8±245 | 58.7±12 |
| 25 weeks (n=6) | 791.0±217 | 731.8±192 | 407.4±65 | 668.0±106 | 19.6±2 | 20.0±2 | 823.6±166 | 42.3±10 |
| XLMTM | ||||||||
| 10 weeks (n=6) | 439.1±101 | 589.0±252 | 224.1±79 | 286.9±115 | 13.4±2 | 12.5±3 | 865.6±253 | 65.4±22 |
| 17 weeks (n=3) | 772.6±319 | 760.9±260 | 268.9±78.2 | 418.7±133 | 16.8±8 | 16.4±8 | 675.0±262 | 40.0±16 |
| 21 weeks (n=2) | 1,542 | 907.5 | 458.0 | 602.6 | 24.6 | 21.8 | 654.5 | 27.0 |
Data are presented as mean ± SD. XLMTM, X-linked myotubular myopathy.
Peak inspiratory and expiratory flow
Between 10 and 25 weeks, PIF gradually increased in healthy dogs (N=6) with physical growth (Table 3). The PIF of affected dogs also increased over time, and resting PIF rates remained significantly higher than control animals (P=0.012, Table 3). Doxapram infusion acutely increased the PIF of controls. PIF of affected dogs also increased with doxapram, but the degree of change was modest, resulting in a generally reduced doxapram PIF response in affected dogs (10 weeks: −9%, 17 weeks: −22% compared to normal values, Table 4, Figure 6). However, the last surviving XLMTM dog maintained a PIF 12% above normal at 25 weeks-of-age. PEF and expiratory volume were consistently lower for affected animals, both during rest and in response to doxapram (Tables 3,4).
Figure 6.

MTM1 mutation alters standard measures of pulmonary function in dogs during direct stimulation. (A) Peak inspiratory flow, (B) minute ventilation, (C) inspiratory time, (D) respiratory rate, (E) tidal volume and (F) expired volume as recorded by pneumotach after stimulation with 1.0 mg/kg doxapram.
Volumetric response
Volume responses were variable for affected animals and appeared to change with growth/maturation. Initially, 10-week-old XLMTM dogs had higher resting tidal and EVs, compared to their littermate controls (Tidal: +19%, Expiratory: +10%, NS, Table 3), and near-normal volumes with doxapram stimulation (Tidal: +11%, Expiratory: +1%, NS). Volumes trended downward at 17 weeks-of-age, both at rest (Tidal: –5%, Expiratory: –11%, NS) and after stimulation (Tidal: +2%, Expiratory: +1%, NS). By 25 weeks, the only surviving animal had higher resting volume (Tidal: +63%, Expiratory: +36%, Table 3) and a variable response to doxapram stimulation (Tidal: +26%, Expiratory –10%, Table 4).
Ventilatory response
Prior to doxapram stimulation, the respiratory rate was similar between the XLMTM dogs and their unaffected littermates (10 weeks: –5%, 17 weeks: +1%, 25 weeks: –18% compared to normal, NS, Table 3). XLMTM dogs inspired at near normal times (–2% at 10 and 17 weeks and +81% at 25 weeks, compared to normal, NS, Table 3). However, after doxapram administration, XLMTM dogs breathed more slowly (10 weeks: –17%, 17 weeks: –32%, 25 weeks: –36%, Table 4, Figure 7), and this lower rate was attributed to a significantly prolonged TI (10%, 43% and 95% longer at 10, 17 and 25 weeks-of-age respectively) (P=0.027) (Table 4) (Figure 6). Minute ventilation, which reflects both the volume of breaths and the respiratory rate, was generally lower in quietly breathing XLMTM dogs (10 weeks: –13%, 17 weeks: +4%, 25 weeks: –35% compared to normal, NS, Table 3). Minute ventilation remained low in affected dogs after stimulation (10 weeks: –14%, 17 weeks: –29%, 25 weeks: –21% compared to normal, Table 4, Figure 6). Altogether, these data indicate that MTM1 mutation leads to an attenuated ventilatory response due to a prolonged TI and a lower relative change in volume during direct central stimulation.
Figure 7.
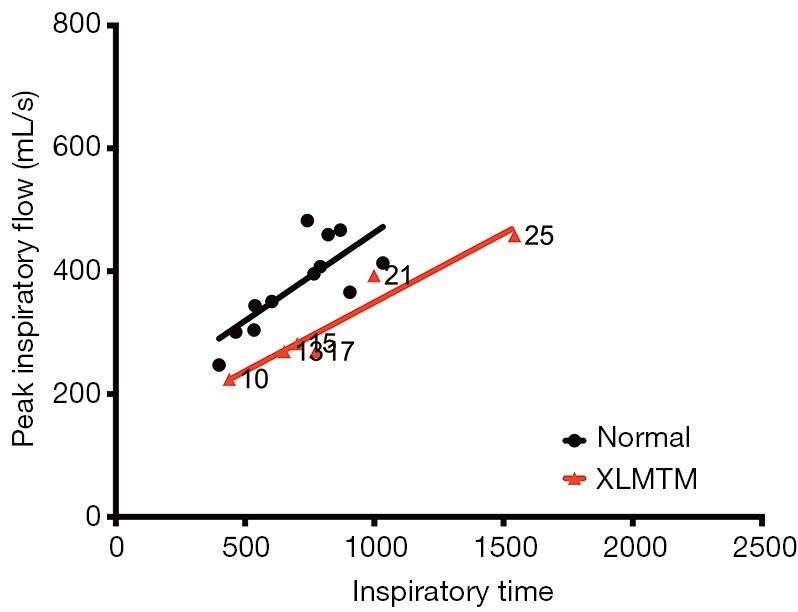
XLMTM dogs increase inspiratory time with a concomitant decrease in volume of inspiratory flow to maintain respiratory homeostasis. Peak inspiratory flow against inspiratory time, with associated linear regression, for controls (black) and XLMTM dogs (red) measured over time. Weeks-of-age at the time of measurement are indicated on the graph for affected animals. XLMTM, X-linked myotubular myopathy.
PIF as a function of time
Given the observed changes in respiratory flow and timing in XLMTM dogs, we examined the relationship between PIF and TI under doxapram stimulation in the two cohorts (Figure 7). The slope of the linear regression plots represents the rate of acceleration of airflow during an inspiratory effort. In general, linear regression plots for affected animals fell below those of normal animals, with a rightward shift. These data suggest that affected dogs required a longer inspiratory effort to generate airflow during each breath, compared to their littermate controls.
Neurological function progressively declines in MTM1-mutant dogs
To measure changes in neurological function over time, dogs were regularly tested using the NSS, a validated clinical scoring instrument developed for dogs (20). Results indicate that as early as 10 weeks-of-age, when normal dogs have an essentially normal score (mean NSS =9.8±0.4), affected dogs begin to show signs of neurological impairment (NSS =8.6±0.9) and progressively lose neurological function over time (NSS =5.6±1.7 at 17 weeks-of-age). A marked neurological decline was evident at the end of the assessment period at 25 weeks-of-age, where a NSS score of 3 was recorded for the last surviving affected dog (Figure 8). At 10 weeks-of-age, normal dogs had a mean reflex score of 1.8±0.2; whereas, affected dogs had reduced reflexes (mean score of 1.4±0.2). The mean reflex score in affected dogs decreased over time such that at 17 weeks-of-age, when normal dogs had a mean reflex score of 2.0±0.04, affected dogs had a mean reflex score of 1.0±0.3.
Figure 8.
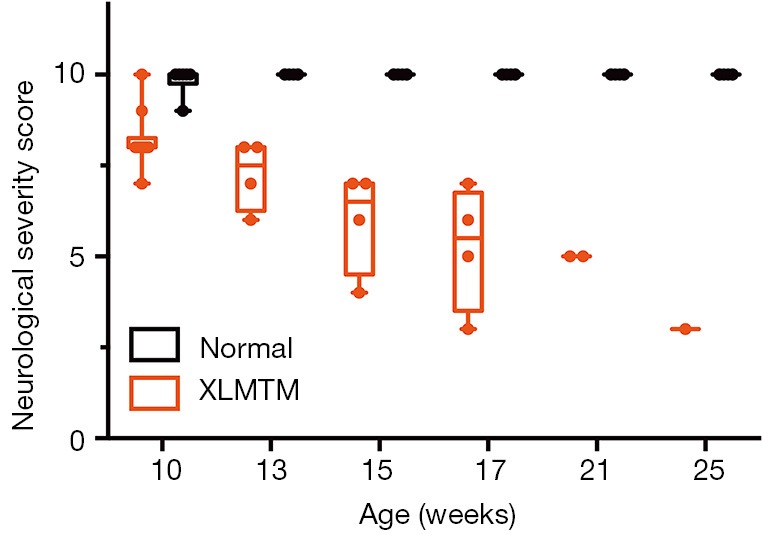
MTM1 mutation results in early and progressive decline in neurological function in dogs. (B) Box plots of neurological severity scores measured between 10 and 17 weeks-of-age.
Discussion
Establishing pathophysiological endpoints in myotubularin-deficient animals is essential for translation of preclinical treatment trials into the human clinical trial stage. Findings reported here establish a number of these endpoints and define the rate at which the p.N155K dog model recapitulates clinical features of XLMTM including shortened survival, progressive limb weakness, gait abnormalities, and neurological impairment. Because the disease is X-linked, only male dogs were previously reported (9,10,22). However, a single infusion of AAV8-MTM1 at 10 weeks-of-age ameliorated severe muscle pathology and increased the survival of two affected male dogs in our colony. Subsequent breeding of these AAV8-infused affected males a year later produced both hemizygous males and homozygous female offspring expressing the disease. This allowed us to study disease in both male and female XLMTM animals in the present study. As in human patients, carrier females remain phenotypically normal.
Pathological findings reported here demonstrate that MTM1-mutant canine muscle displays analogous histological changes observed in patients, including internal nucleation, organelle mislocalization, reduced fiber size and disrupted sarcotubular structures (23). Interestingly, the XLMTM dogs that lived longest (SSAN13, SSAN16, and SSAN101) showed some degree of myofiber growth at the 17 weeks-of-age, corresponding to the presence of a subpopulation of large hypertrophic myofibers amid small myofibers with classic XLMTM pathology. These hypertrophic fibers often displayed organelle mislocalization characteristic of XLMTM, and almost always corresponded to a subpopulation of the type 2 (glycolytic) myofiber population (not shown). At the time of necropsy, all MTM1-mutant specimens showed similar characteristics, with near-uniform small myofibers (due to a combination of hypotrophy and atrophy) and organelle mislocalization.
MTM1 mutation leads to weakness by interrupting normal excitation-contraction coupling (ECC) as indicated by studies of contractile function in myotubularin-deficient mice (7,24). The gene product of MTM1 is myotubularin, a small (~66 kD) cytoplasmic protein with pleotropic activity including endosomal trafficking, cytoskeletal organization, neuromuscular junction structure, autophagy, satellite cell proliferation and survival, and ECC (8,24-27). In skeletal muscle, myotubularin is involved in the formation and maintenance of the “triad,” the set of juxtaposed sarcoplasmic reticulum and t-tubule structures responsible for mediating ECC (8,24,28,29). Electron micrographs from the present study indicate that MTM1-mutant canine muscles display disrupted sarcotubular architecture, based on the scarcity of triad structures (Figure 2B). Disruption of the sarcoplasmic reticulum and t-tubule triad relationship in myotubularin-deficient skeletal muscle leads to abnormal Ca2+ exchange and impaired ECC (8,24). Findings reported here support this idea and point to evaluation of triadic structures as an important potential translational endpoint.
In vivo muscular contraction torque elicited by peripheral nerve stimulation in the hind limb is an established surrogate of muscle strength and used as a primary readout in XLMTM and other canine models of muscle disease (6,10,13). We previously reported hind limb contraction torque data in fourteen normal and affected XLMTM dogs (10), but forelimb torque assays have not previously been reported in this model. For translational applications, forelimb contraction torque measures may have more translational value than hindlimb measures since upper limb impairment negatively impacts quality of life in non-ambulatory patients (30). Here we adapted an established forelimb torque assay (15) to evaluate the extent and rate of weakening of the flexors and extensors of the distal upper limb. Our data suggest that forelimb extensor torque (Figure 3A) provides more reliable data compared to forearm flexor torque due to technical issues with peripheral nerve stimulation. Together, the present data point to the use of forelimb extension and hindlimb flexion torque assays as co-primary endpoints of muscular strength in future interventional trials in the XLMTM dog.
Gait assessment captures domains of limb function in awake animals principally by measuring variables such as joint angles, velocity and other spatiotemporal parameters of movement (31-33). We previously used two dimensional kinematic gait analyses in a pilot study of MTM1-mutant dogs (16). In that study, we found that gait velocity, step and stride length were reduced in the XLMTM dog compared to WT animals, but we did not detect joint angle abnormalities. Unlike dystrophin-deficient dogs who develop joint contractures over several years of life (34), myotubularin-deficient dogs do not develop contractures, perhaps due to shortened lifespan of less than 6 months. Thus in the present study, we used an instrumented carpet to track gait velocity, step and stride length and other spatiotemporal characteristics. Slower, shorter strides previously observed as an indicator of muscle weakness due to disease were serially measured in the present study of XLMTM dogs and point to the consequences of limb weakness in this model (16,35).
Our findings indicate that the MTM1 p.N155K dog model of XLMTM differs from human patients in the extent of respiratory impairment; affected dogs generally maintained ventilatory homeostasis even in the presence of profound limb muscle weakness and neurological impairment. Differences in respiratory impairment observed between XLMTM dogs and patients may reflect the deep cardiopulmonary reserves harbored by dogs through evolutionary processes (36). For example, sled dogs are famous for their extraordinary oxygen extraction capacity (VO2max) that more than doubles that of an elite cross-country skier. Yet, despite the lack of profound respiratory muscle weakness observed in XLMTM dogs in the present investigation, respiratory data in one of the affected animals provides a clue for translational application. The single affected dog to survive to 25 weeks-of-age maintained near normal PIF and myofiber size, but ultimately became too weak to ambulate. In patients, mild, moderate and severe forms of the disease can stratify severity in XLMTM, where the mild/moderate form associates with increased lifespan and reduced requirement for ventilatory support (31,32,37). Perinatal deaths and shortened survival of XLMTM dogs in our colony appear to correlate with the poor respiratory function (unpublished observations) suggesting that similar classifications could be applied to the canine model. We speculate that PIF and inspiratory duration may prove useful predictors of increased survival in the canine model, and by translational application inform future clinical trials.
Neurological severity scoring and reflex scoring were consistently lower in XLMTM versus wild type dogs at all ages. Similar clinical scoring systems have been reported for large animal models of the GM1 and GM2 gangliosidosis (38,39) and stroke models (40-42) and are useful because they represent noninvasive assessment methods that can be performed as serial tests in awake animals. The finding that the NSSs and reflex scores differed between affected and normal dogs at both early and late time points suggests that these assessments may be useful in judging response to therapy in future interventional trials.
Acknowledgements
Thanks to Emily Troiano for assistance genotyping the dogs. Some microscopic images were obtained using the Children’s Hospital of Wisconsin Research Institute’s Imaging Core Facility. Electron microscopy was performed using the Medical College of Wisconsin’s EM Core Facility.
This work was supported in part by United States National Institute of Health (NIH) grants R21 AR064503 and R01 HL115001 to MKC, K08 AR059750 to MWL, R01 AR044345 to AHB; Senator Paul D Wellstone Muscular Dystrophy Cooperative Research Center, Seattle (NIH U54AR065139); Association Française contre les Myopathies and Muscular Dystrophy Association (United States) to MKC; Where There’s a Will There’s a Cure, and Joshua Frase Foundation to AHB and MKC, Peter Khuri Myopathy Research Foundation to M.K.C.; and Audentes Therapeutics to MKC and MWL.
Footnotes
Conflicts of Interest: Two of the authors (AH Beggs and MK Childers) are inventors of a patent on gene therapy for myotubular myopathy and are paid members of the scientific advisory board for Audentes Therapeutics.
References
- 1.Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis 2008;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 1996;13:175-82. [DOI] [PubMed] [Google Scholar]
- 3.Laporte J, Blondeau F, Buj-Bello A, et al. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet 1998;7:1703-12. [DOI] [PubMed] [Google Scholar]
- 4.Robinson FL, Dixon JE. Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol 2006;16:403-12. [DOI] [PubMed] [Google Scholar]
- 5.Buj-Bello A, Laugel V, Messaddeq N, et al. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A 2002;99:15060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Childers MK, Joubert R, Poulard K, et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med 2014;6:220ra10. [DOI] [PMC free article] [PubMed]
- 7.Lawlor MW, Armstrong D, Viola MG, et al. Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet 2013;22:1525-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dowling JJ, Vreede AP, Low SE, et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet 2009;5:e1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beggs AH, Böhm J, Snead E, et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A 2010;107:14697-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grange RW, Doering J, Mitchell E, et al. Muscle function in a canine model of X-linked myotubular myopathy. Muscle Nerve 2012;46:588-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng H, Janssen PM, Grange RW, et al. Tissue triage and freezing for models of skeletal muscle disease. J Vis Exp 2014;(89). [DOI] [PMC free article] [PubMed]
- 12.Dastgir J, Rutkowski A, Alvarez R, et al. Common Data Elements for Muscle Biopsy Reporting. Arch Pathol Lab Med 2015. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornegay JN, Bogan DJ, Bogan JR, et al. Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci 1999;166:115-21. [DOI] [PubMed] [Google Scholar]
- 14.Childers MK, Grange RW, Kornegay JN. In vivo canine muscle function assay. J Vis Exp 2011;(50). [DOI] [PMC free article] [PubMed]
- 15.Le Guiner C, Montus M, Servais L, et al. Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol Ther 2014;22:1923-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goddard MA, Burlingame E, Beggs AH, et al. Gait characteristics in a canine model of X-linked myotubular myopathy. J Neurol Sci 2014;346:221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CJ, McKiernan BC, Pace J, et al. The effects of doxapram hydrochloride (dopram-V) on laryngeal function in healthy dogs. J Vet Intern Med 2002;16:524-8. [DOI] [PubMed] [Google Scholar]
- 18.Yost CS. A new look at the respiratory stimulant doxapram. CNS Drug Rev 2006;12:236-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coglianese CJ, Peiss CN, Wurster RD. Rhythmic phrenic nerve activity and respiratory activity in spinal dogs. Respir Physiol 1977;29:247-54. [DOI] [PubMed] [Google Scholar]
- 20.Snyder JM, Meisner A, Mack D, et al. Validity of a Neurological Scoring System for Canine X-Linked Myotubular Myopathy. Hum Gene Ther Clin Dev 2015;26:131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamama-Moraitou KK, Pardali D, Menexes G, et al. Tidal breathing flow volume loop analysis of 21 healthy, unsedated, young adult male Beagle dogs. Aust Vet J 2013;91:226-32. [DOI] [PubMed] [Google Scholar]
- 22.Snead EC, Taylor SM, van der Kooij M, et al. Clinical phenotype of X-linked myotubular myopathy in Labrador Retriever puppies. J Vet Intern Med 2015;29:254-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biancalana V, Beggs AH, Das S, et al. Clinical utility gene card for: Centronuclear and myotubular myopathies. Eur J Hum Genet 2012;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Qusairi L, Weiss N, Toussaint A, et al. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A 2009;106:18763-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowling JJ, Joubert R, Low SE, et al. Myotubular myopathy and the neuromuscular junction: a novel therapeutic approach from mouse models. Dis Model Mech 2012;5:852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hnia K, Tronchère H, Tomczak KK, et al. Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J Clin Invest 2011;121:70-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsujita K, Itoh T, Ijuin T, et al. Myotubularin regulates the function of the late endosome through the gram domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem 2004;279:13817-24. [DOI] [PubMed] [Google Scholar]
- 28.Al-Qusairi L, Laporte J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle 2011;1:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowling BS, Chevremont T, Prokic I, et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest 2014;124:1350-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzone ES, Vasco G, Palermo C, et al. A critical review of functional assessment tools for upper limbs in Duchenne muscular dystrophy. Dev Med Child Neurol 2012;54:879-85. [DOI] [PubMed] [Google Scholar]
- 31.Biancalana V, Caron O, Gallati S, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet 2003;112:135-42. [DOI] [PubMed] [Google Scholar]
- 32.McEntagart M, Parsons G, Buj-Bello A, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord 2002;12:939-46. [DOI] [PubMed] [Google Scholar]
- 33.Chanzy S, Routon MC, Moretti S, et al. Unusual good prognosis for X-linked myotubular myopathy. Arch Pediatr 2003;10:707-9. [DOI] [PubMed] [Google Scholar]
- 34.Kornegay JN, Sharp NJ, Schueler RO, et al. Tarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab Anim Sci 1994;44:331-3. [PubMed] [Google Scholar]
- 35.Marsh AP, Eggebeen JD, Kornegay JN, et al. Kinematics of gait in golden retriever muscular dystrophy. Neuromuscul Disord 2010;20:16-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnurr TM, Reynolds AJ, Komac AM, et al. The effect of acute exercise on GLUT4 levels in peripheral blood mononuclear cells of sled dogs. Biochem Biophys Rep 2015;2:45-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laporte J, Biancalana V, Tanner SM, et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat 2000;15:393-409. [DOI] [PubMed] [Google Scholar]
- 38.McCurdy VJ, Rockwell HE, Arthur JR, et al. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther 2015;22:181-9. [DOI] [PubMed] [Google Scholar]
- 39.McCurdy VJ, Johnson AK, Gray-Edwards HL, et al. Sustained normalization of neurological disease after intracranial gene therapy in a feline model. Sci Transl Med 2014;6:231ra48. [DOI] [PMC free article] [PubMed]
- 40.Brown A, Woods S, Skinner R, et al. Neurological assessment scores in rabbit embolic stroke models. Open Neurol J 2013;7:38-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zu QQ, Liu S, Xu XQ, et al. An endovascular canine stroke model: middle cerebral artery occlusion with autologous clots followed by ipsilateral internal carotid artery blockade. Lab Invest 2013;93:760-7. [DOI] [PubMed] [Google Scholar]
- 42.Purdy PD, Devous MD, Sr, Batjer HH, et al. Microfibrillar collagen model of canine cerebral infarction. Stroke 1989;20:1361-7. [DOI] [PubMed] [Google Scholar]