

Abstract
Altered tryptophan–kynurenine (KYN) metabolism has been implicated in major depressive disorder (MDD). The l-[1-13C]tryptophan breath test (13C-TBT) is a noninvasive, stable-isotope tracer method in which exhaled 13CO2 is attributable to tryptophan catabolism via the KYN pathway. We included 18 patients with MDD (DSM-IV) and 24 age- and sex-matched controls. 13C-tryptophan (150 mg) was orally administered and the 13CO2/12CO2 ratio in the breath was monitored for 180 min. The cumulative recovery rate during the 180-min test (CRR0–180; %), area under the Δ13CO2-time curve (AUC; %*min), and the maximal Δ13CO2 (Cmax; %) were significantly higher in patients with MDD than in the controls (p = 0.004, p = 0.008, and p = 0.002, respectively). Plasma tryptophan concentrations correlated negatively with Cmax in both the patients and controls (p = 0.020 and p = 0.034, respectively). Our results suggest that the 13C-TBT could be a novel biomarker for detecting a subgroup of MDD with increased tryptophan–KYN metabolism.
The kynurenine (KYN) pathway of l-tryptophan metabolism has gained increased attention in research into depression1,2,3,4,5,6,7. Indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO, tryptophan pyrrolase) are the two initial and rate-limiting enzymes that convert tryptophan to KYN1,3,8,9. IDO is activated by proinflammatory cytokines, such as interferon (IFN)-α, IFN-γ, and tumor necrosis factor (TNF)-α6,9,10,11,12, which in addition to other inflammatory molecules such as interleukins, chemokines, and prostaglandins have been associated with major depressive disorder (MDD)2,6,11,13,14,15. The neuroactive KYN metabolites kynurenic acid (KA) and quinolinic acid (QUIN) are synthesized in the KYN–KA and KYN–nicotinamide adenine dinucleotide (NAD) pathways, respectively (Fig. 1). KA acts as an excitatory amino acid receptor antagonist, whereas QUIN has an excitotoxic effect. The activation of IDO promotes tryptophan catabolism in the KYN pathway and may cause reduced tryptophan availability for serotonin synthesis in the brain, resulting in imbalance or misregulation of the neuroactive metabolites. This mechanism may explain both MDD and the depressive symptoms observed in patients with inflammatory diseases and in those undergoing cytokine therapies3,16,17.
Figure 1. Schematic view of 13C-tryptophan metabolism.

After utilization for protein synthesis, over 90% of dietary tryptophan is converted to KYN by one of two rate-limiting enzymes: by TDO in the liver under healthy conditions and by IDO in various human tissues (e.g., intestines, lungs, placenta, kidneys, spleen, blood, and brain) under inflammatory conditions. The 13CO2 derived from 13C-tryptophan is produced by the conversion of 3-OH-KYN into 3HAA via the KYN–NAD pathway (box with solid line) as the post-KYN metabolism. In addition, although 13CO2 is produced to a minor degree in the tryptophan–serotonin and anthranilic acid pathways, it is not produced by the KA (box with dotted line) or xanthurenic acid pathways. Bold arrows indicate quantitatively major pathways. Minor pathways and pathways without release of 13CO2 are omitted. Red circles indicate the 13C-labeled carbon. Abbreviations: IDO, indoleamine 2,3-dioxygenase; KA, kynurenic acid; KYN, kynurenine; NAD, nicotinamide adenine dinucleotide; TDO, tryptophan 2,3-dioxygenase; 3HAA, 3-hydroxyanthranilic acid; 3-OH-KYN, 3-hydroxy-l-kynurenine.
To clarify this mechanism, a number of clinical studies have used blood and cerebrospinal fluid (CSF) samples from patients undergoing cytokine therapy. These have evidenced decreased tryptophan levels, increased KYN levels, increased KYN/tryptophan ratios (used to estimate peripheral IDO activity), and correlations between depressive symptoms, and levels of tryptophan, and its metabolites15,16,17,18,19. However, contradictory results have also been reported20,21.
Several lines of evidence, including our recent meta-analysis22, have demonstrated decreased tryptophan levels in MDD23,24,25,26,27. However, it remains controversial whether the KYN pathway is activated in MDD. Indeed, although several studies have reported increased KYN/tryptophan ratios23,28,29, or its association with depression severity in MDD30, some negative results have also been reported24,31. These inconsistent findings, therefore, require further investigation. Furthermore, few studies have investigated the role of post-KYN metabolism: specifically, distinguishing between the KYN–KA and the KYN–NAD pathways in MDD28.
Cross-sectional quantitative data, obtained from peripheral blood or CSF samples, may provide a reasonable “snapshot”, reflecting total accumulation at the sampling time and site, but cannot measure changes in the synthesis or turnover rates of the metabolites. Indeed, these rates can alter without change in the total amount of metabolites in a dynamic steady-state equilibrium32. Additionally, metabolite turnover rates could be altered after any compensatory changes for the total amount of metabolites. Furthermore, because tryptophan, KYN and 3-hydroxykynurenine (3-OH-KYN) readily cross the blood–brain barrier (BBB)33,34,35, fluctuations in peripheral blood levels can directly affect metabolism of KYN pathway metabolites, including the synthesis of KA, 3-hydroxyanthranilic acid (3HAA), and QUIN in the brain, although these three metabolites do not readily cross the BBB36,37. IDO is distributed in various human tissues, including the intestine, lungs, placenta, kidneys, spleen, brain, and blood8, and when peripherally activated, IDO can affect the concentration of QUIN in the brain and CSF38. Raison et al. reported that there were significant positive correlations between peripheral and central measures of KYN and QUIN, but not tryptophan, and that peripheral tryptophan decreased while CSF tryptophan remained unchanged in patients receiving IFN-α therapy16. These factors point to the merit of monitoring KYN pathway activation in the whole body.
Tracer tests are advantageous for assessing in vivo metabolism of biological compounds as a more direct approach. Using a 13C-phenylalanine breath test, our group reported that altered phenylalanine metabolism could be identified in patients with schizophrenia39. Hankes et al. administered a 14C-tryptophan breath test to patients with scleroderma and reported that expiratory 14CO2 was decreased and labeled QUIN in the urine was increased40,41. Therefore, they suggested that metabolism after 3HAA may be altered in these patients. They also reported that the production of 14CO2 from 14C-labeled d-tryptophan was different from that of 14C-labeled l-tryptophan, and suggested that labeled l-tryptophan should be used for studies of tryptophan metabolism. In the present study, we used a stable isotope (i.e., non-radioactive tracer) in an l-[1-13C]tryptophan breath test (13C-TBT) to examine the whole-body kinetics and metabolism of orally administered tryptophan. In this method, the 13C label of l-[1-13C]tryptophan is released as 13CO2 when 3HAA, an immediate precursor for QUIN, is produced from tryptophan via the KYN–NAD pathway both peripherally and in the brain. This method also selectively observes the KYN–NAD pathway, but not the KYN–KA pathway, because 13CO2 is not produced during the synthesis of KA or xanthurenic acid. Therefore, 13CO2 formation measured by the 13C-TBT is an index of whole-body tryptophan metabolism via the KYN pathway and excludes the pathways for KA and xanthurenic acid production.
The aim of this study was to assess KYN pathway activity in patients with MDD. We hypothesized that the 13C-TBT would detect altered tryptophan metabolism via the KYN pathway in patients with MDD, and that the 13C-TBT indices would correlate negatively with plasma tryptophan concentrations.
Methods
Participants
We enrolled 18 patients with MDD (age range: 25–62 years; 10 women) and 24 healthy volunteers (age range: 21–61 years; 12 women), matched for age and sex, and who were biologically unrelated. Participants were recruited through advertisements in free local information magazines, by word of mouth, and via a website announcement; all were Japanese residents from the western part of Tokyo. Patients were also recruited from the National Center of Neurology and Psychiatry (NCNP) Hospital, Tokyo, Japan.
Consensus diagnoses were made for MDD by at least two experienced psychiatrists according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV)42, based on the Japanese version of the Mini-International Neuropsychiatric Interview (MINI)43, careful examination of medical records, observations, and additional unstructured interviews.
Healthy volunteers were screened by psychiatrists using unstructured interviews and the MINI. We confirmed that they had no history of psychiatric illness or contact with psychiatric services. Participants were excluded from both the patient and control groups if they met the DSM-IV criteria for intellectual disability, had a history of substance dependence or abuse, or had a history of severe head injury. We also excluded participants if they had a current serious physical disorder, particularly an inflammatory or liver disease (where tryptophan is primarily metabolized) or if they received any immunotherapy. All participants underwent blood tests for aspartate aminotransferase (AST), alanine aminotransferase (ALT), total protein, albumin, total bilirubin, blood urea nitrogen (BUN), and creatinine levels to exclude liver and kidney dysfunction. Individuals with abnormal blood test results were excluded.
The present study was conducted in accordance with the principles of the Declaration of Helsinki. The capacity of patients to understand the nature and purpose of the study and to provide consent was confirmed by an experienced psychiatrist. After explaining the study aim and protocol, signed informed consent was obtained from each participant. The study was approved by the ethics committee of the NCNP, Japan.
Principle of the 13C-TBT
The kinetic values for l-[1-13C]tryptophan were assumed to be the same as for unlabeled tryptophan44. The following theory underpinned the 13C-TBT. After protein synthesis, dietary tryptophan, an essential amino acid for humans, is primarily metabolized along the KYN pathway in the liver, which accounts for over 90% of tryptophan catabolism35; in contrast, only 1%–3% of dietary tryptophan is metabolized via the serotonin pathway34. Under healthy conditions, the conversion of tryptophan to KYN via N-formyl-l-kynurenine is initiated by TDO, which is activated by stress and high glucocorticoid levels1,2,45. Thus, this is the product of elevated hypothalamic pituitary-adrenal (HPA) axis activity, which is often implicated in the development of depressive disorders46,47. In conditions where the immune system is stimulated, IDO is induced by proinflammatory cytokines2,3,7,48. Crucial to the test is that the 13CO2 derived from orally administered 13C-tryptophan can be produced by the conversion of 3-OH-KYN into 3HAA via the KYN–NAD pathway, but not by the KA or xanthurenic acid pathways (Fig. 1). The 13CO2 is also produced to much lesser degrees by minor pathways, such as the tryptophan–serotonin and anthranilic acid pathways. The 13CO2 measured by the 13C-TBT was, therefore, expected to reflect whole-body tryptophan–KYN–3HAA metabolism initiated by IDO or TDO.
The maximal Δ13CO2 (Cmax; %), time to reach the Cmax (Tmax; min), area under the Δ13CO2-time curve (AUC; %*min), and the ratio of the total amount of exhaled 13CO2 to the administered dose of 13C-tryptophan (i.e., the cumulative recovery rate, CRR; %) were calculated from the Δ13CO2 (%) at each sampling point. Observations were made by infrared (IR) spectrometry in which IR absorption intensities of 12CO2 (2380 ± 10 cm−1) and 13CO2 (2280 ± 10 cm−1) were measured, as described previously39,49.
Protocol for the 13C-TBT
The 13C-TBT was performed as previously described for the 13C-phenylalanine breath test39,50. Blood samples were collected after an overnight fast and stored at −20 °C until biochemical analysis (AST, ALT, total protein, albumin, total bilirubin, BUN, creatinine, and tryptophan levels) by the SRL Corporation (Tokyo, Japan). The plasma tryptophan level was measured by liquid chromatography–mass spectrometry. A dose of 150 mg/body of l-[1-13C]tryptophan (99 atom% 13C; Cambridge Isotope Laboratories, Cambridge, UK) was orally administered in 100 ml of water at 1000 h. Breath samples were collected in 250-ml breath-sampling bags (Otsuka Electronics Co., Ltd., Osaka, Japan) at 10, 15, 20, 30, 45, 60, 90, 120, 150, and 180 min after ingestion of 13C-tryptophan. The 13CO2 /12CO2 ratio in exhaled air was analyzed by IR spectrometry (UBiT-IR300 and UBiT-AS10, Otsuka Pharmaceutical, Tokyo, Japan).
Clinical status and antidepressant medication
Current depression severity in patients was assessed using a 21-item version of the Hamilton Depression Rating Scale (HAMD-21) administered by a single experienced psychiatrist. Antidepressant doses were converted to imipramine-equivalent (IMIeq) doses according to published guidelines51,52.
Statistical analysis
The Statistical Package for the Social Sciences (SPSS) version 11.0 (SPSS Japan, Tokyo, Japan) was used to conduct all statistical tests. Averages were expressed as mean ± standard deviation (SD). Statistical differences in the demographic data between groups were compared using the χ2 test and t-test for categorical variables and continuous variables, respectively. We then performed stepwise multiple regression analyses with two models, using the 13C-TBT indices (Cmax, AUC, and CRR0–180) as the dependent variable: in model 1, diagnosis, sex, age, and body weight were entered as possible predictors; and in model 2, diagnosis, sex, age, body weight, and plasma tryptophan concentration were entered as possible predictors.
A 2 × 10 (group × sampling point) repeated measures analysis of covariance (ANCOVA) was conducted to examine group differences in Δ13CO2 or CRR over time, controlling for sex, age, and body weight. Greenhouse–Geisser corrections were used for lack of sphericity. We used ANCOVA (controlling for sex, age, and body weight) and t-tests to compare 13C-TBT indices between the two groups. The Tmax, a discrete variable, was compared between the two groups using the Mann–Whitney U test. The correlations of each 13C-TBT index (AUC, CRR0–180, and Cmax) with the liver parameters and the plasma tryptophan concentrations were examined by Spearman’s and Pearson’s correlation analyses, respectively. Cramer’s V, Cohen’s d, η2, and partial η2 were calculated as measures of effect size for χ2 test, t-tests, ANOVAs, and ANCOVAs, respectively. In the patients, the correlations of each 13C-TBT index (AUC, CRR0–180, and Cmax) with IMIeq and HAMD-21 total score were examined using Spearman’s correlation analysis. A stepwise multiple regression was employed to assess independent predictors of Cmax, AUC, and CRR0–180, using sex, IMIeq, HAMD-21 total score, and plasma tryptophan concentration as candidate predictors. Statistical significance was two-tailed and set at p < 0.05.
Results
Demographic and clinical characteristics
The demographic and clinical characteristics of the patients and controls are presented in Table 1. There was no significant difference in sex, age, body weight, body mass index, education length, liver or kidney blood test results, or plasma tryptophan concentrations between the two groups.
Table 1. Demographic and clinical characteristics of patients with major depressive disorder, and healthy controls.
| Patients (n = 18) | Controls (n = 24) | statistic analysis | |
|---|---|---|---|
| Sex, male: n (%) | 8 (44.4) | 12 (50.0) | χ2(1) = 0.13, p = 0.76, Cramer’s V = 0.06 |
| Age (year) | 39.9 ± 10.1 | 45.1 ± 11.9 | t(40) = 1.49, p = 0.14, Cohen’s d = 0.46 |
| Body weight (kg) | 60.3 ± 9.4 | 63.0 ± 15.9 | t(38.1) = 0.68, p = 0.50, Cohen’s d = 0.20 |
| Body mass index (kg/m2) | 22.5 ± 2.6 | 22.8 ± 4.3 | t(38.5) = 0.34, p = 0.73, Cohen’s d = 0.10 |
| Education (year) | 15.1 ± 2.2 | 14.2 ± 2.1 | t(40) = 1.41, p = 0.17, Cohen’s d = 0.44 |
| Comorbid dysthymia/anxiety disorders (n) | 6 | ||
| HAMD-21 total score | 12.6 ± 7.6 | ||
| IMIeq (mg per day) | 69.4 ± 114.9 | ||
| Tryptophan (μmol l−1) | 49.6 ± 7.4 | 49.7 ± 10.1 | t(40) = 0.05, p = 0.96, Cohen’s d = 0.02 |
| AST (U l−1) | 19.4 ± 6.5 | 19.0 ± 5.2 | t(40) = 0.22, p = 0.83, Cohen’s d = 0.07 |
| ALT (U l−1) | 12.9 ± 7.6 | 14.4 ± 8.1 | t(40) = 0.62, p = 0.54, Cohen’s d = 0.19 |
| Total protein (g dl−1) | 7.3 ± 0.3 | 7.4 ± 0.4 | t(40) = 0.51, p = 0.61, Cohen’s d = 0.16 |
| Albumin (g dl−1) | 4.7 ± 0.3 | 4.5 ± 0.3 | t(40) = 1.74, p = 0.09, Cohen’s d = 0.54 |
| Total bilirubin (mg dl−1) | 0.6 ± 0.2 | 0.5 ± 0.2 | t(40) = 0.74, p = 0.47, Cohen’s d = 0.23 |
| BUN (mg dl−1) | 13.7 ± 2.7 | 12.2 ± 3.3 | t(40) = 1.59, p = 0.12, Cohen’s d = 0.49 |
| Cr (mg dl−1) | 0.73 ± 0.16 | 0.71 ± 0.15 | t(40) = 0.38, p = 0.71, Cohen’s d = 0.12 |
Abbreviations: HAMD-21, 21-item version of the Hamilton Depression Rating Scalecreatinine; IMIeq, Imipramine-equivalent antidepressant dose; BUN, blood urea nitrogen; Cr, creatinine.
Predictors for 13C-TBT indices
Stepwise regression analyses were performed (Table 2). In model 1, diagnosis and sex were identified as significant predictors for Cmax, AUC, and CRR0–180. In model 2, diagnosis and sex remained significant predictors for Cmax, AUC, and CRR0–180, but the plasma tryptophan concentration was not a significant predictor in the model.
Table 2. Stepwise multiple regression for l-[1-13C]tryptophan breath test indices as dependent variables.
| Dependant variable | Significant predictor variablea | Adjusted R2 | Standardized Coefficient (β) | p value |
|---|---|---|---|---|
| Model 1 | ||||
| Cmax | sexb | 0.36 | 0.436 | 0.001 |
| diagnostic statusc | 0.426 | 0.002 | ||
| AUC | sexb | 0.48 | 0.617 | <0.001 |
| diagnostic statusc | 0.316 | 0.008 | ||
| CRR0–180 | diagnostic status | 0.25 | 0.378 | 0.008 |
| sex | 0.361 | 0.011 | ||
| Model 2 | ||||
| Cmax | sexb | 0.36 | 0.436 | 0.001 |
| diagnostic statusc | 0.426 | 0.002 | ||
| AUC | sexb | 0.48 | 0.617 | <0.001 |
| diagnostic statusc | 0.316 | 0.008 | ||
| CRR0–180 | diagnostic status | 0.25 | 0.378 | 0.008 |
| sex | 0.361 | 0.011 | ||
Abbreviations: Cmax, the maximal Δ13CO2 (%); AUC, area under the Δ13CO2-time curve (min %); CRR0-180, the cumulative recovery rate during the 180 min test (%).
aPossible predictor variables. Model 1: diagnostic status, sex, age, and body weight; model 2: diagnostic status, sex, age, body weight, and plasma tryptophan concentration.
bSex was measured on a nominal scale: 1 = male; 2 = female.
cDiagnostic status was measured on a nominal scale: 1 = healthy control; 2 = major depressive disorder.
Increased Cmax, AUC, and CRR in MDD
Figure 2 shows the time course of mean values of Δ13CO2 (Fig. 2a) and CRR (Fig. 2b) in patients and controls. The repeated measures ANCOVA, controlling for sex, age, and body weight, indicated significant differences between the two groups in both Δ13CO2 and CRR over time. The repeated measures ANCOVA with Greenhouse–Geisser adjustment revealed significant interactions between the diagnosis and sampling point for both the Δ13CO2 and the CRR, indicating different patterns of change over time between the two groups. The Δ13CO2 and CRR values were higher in the patients than in the controls at all sampling points during the 180-min test (Fig. 2 and Table S1).
Figure 2.
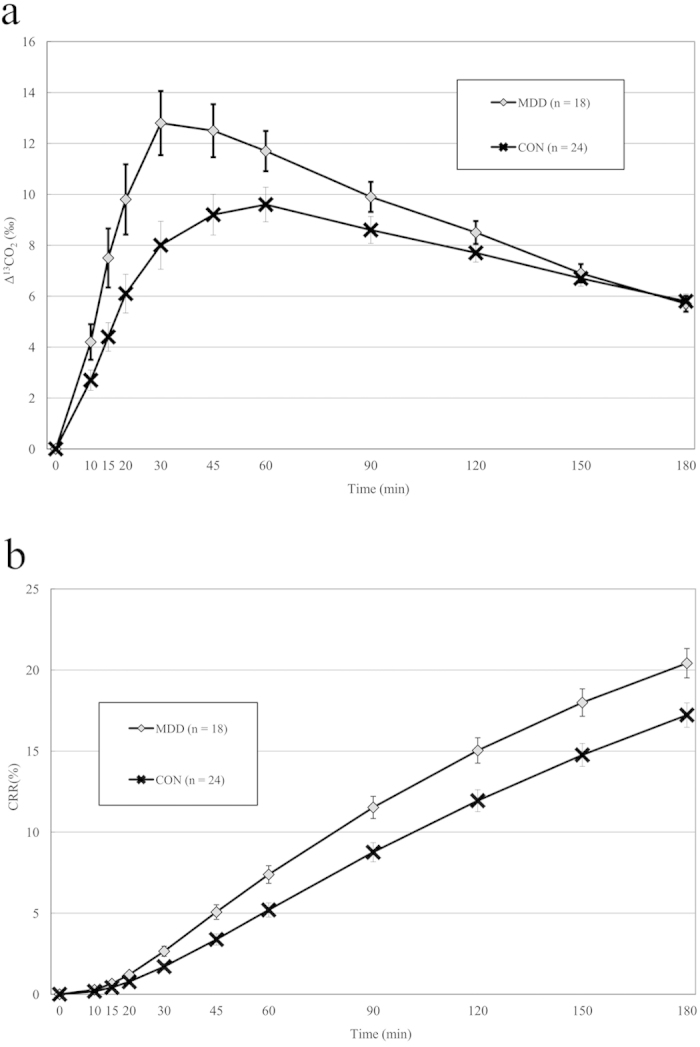
(a) Time course for the Δ13CO2 in expired air after the 13C-tryptophan breath test was applied to patients with major depressive disorder (MDD) and to healthy controls. (b) Time course for the cumulative recovery rate (CRR; %) in expired air for patients with MDD and for healthy controls. Abbreviations: MDD, major depressive disorder group; CON, control group; Time, time after ingesting the solution of 13C-tryptophan (150 mg).
Using t-tests, the Cmax, AUC, and CRR0–180 were all significantly higher in the patients than in the controls, and these differences remained significant after ANCOVA controlling for sex, age, and body weight (Table 3). The Tmax was reduced in the patients compared to the controls at the trend level.
Table 3. l-[1-13C]tryptophan breath test results in patients with major depressive disorder and healthy controls.
| Parameter | Patients (n = 18) Mean ± SD | Controls (n = 24) Mean ± SD | t–test/Mann–Whitney U test | ANCOVAa |
|---|---|---|---|---|
| Cmax | 14.5 ± 4.3 | 10.9 ± 3.1 | p = 0.003 Cohen’s d = 0.99 | p = 0.002 partial η2 = 0.23 |
| AUC | 1596.2 ± 405.3 | 1320.0 ± 350.2 | p = 0.023 Cohen’s d = 0.74 | p = 0.008 partial η2 = 0.18 |
| CRR0-180 | 20.4 ± 3.8 | 17.2 ± 3.7 | p = 0.009 Cohen’s d = 0.86 | p = 0.004 partial η2 = 0.21 |
| Tmaxb | 30 | 45 | p = 0.068c r = 0.28 |
Abbreviations: Cmax, the maximal Δ13CO2 (%); Tmax, time to reach the Cmax (min); AUC, area under the Δ13CO2-time curve (min %); CRR0-180, the cumulative recovery rate during the 180 min test (%); r, the effect size of the Mann–Whitney U test.
aHealthy controls versus patients with major depressive disorder in analysis of covariance controlled for sex, age, and body weight.
bMedian.
cHealthy controls versus patients with major depressive disorder by the Mann–Whitney U test.
Association of 13C-TBT indices with tryptophan concentrations and clinical variables in patients
Both Cmax (Pearson’s correlation coefficient [r] = −0.54, p = 0.020) and CRR0–180 (r = −0.48, p = 0.045) showed significantly negative correlations with plasma tryptophan concentrations in the patient group. In the controls, a significantly negative correlation was also observed for the plasma tryptophan concentrations with the Cmax of the 13C-TBT (r = −0.43, p = 0.034). Further, stepwise multiple regression analysis demonstrated that plasma tryptophan concentrations were significant predictors of Cmax, while sex predicted the AUC and CRR0–180 (Table S2). In addition, both the AUC [Spearman’s rank correlation coefficient (ρ) = −0.53, p = 0.023] and CRR0–180 (ρ = −0.50, p = 0.033) were significantly and negatively correlated with the IMIeq dose. There were no significant correlations between 13C-TBT indices and depression severity (HAMD-21 score; data not shown). There were also no significant correlations between the 13C-TBT indices and the liver tests in the participants (Table S3).
Discussion
We examined, for the first time, whole-body tryptophan metabolism using a 13C-TBT in patients with MDD and found that levels of exhaled 13CO2 were significantly increased in patients compared with healthy controls. Although plasma tryptophan concentrations were not significantly different between the two groups, tryptophan showed significant and negative correlations with the Cmax in both the patient and control groups. In our 13C-TBT, exhaled 13CO2 is mainly attributable to the conversion of tryptophan via KYN into 3HAA. Therefore, our results support the study hypotheses and corroborate previous findings of KYN-pathway activation in MDD (i.e., the serotonin–KYN hypothesis)48,53.
The 13CO2 measured by our 13C-TBT can be produced by either the KYN pathway or the serotonin pathway (Fig. 1). Although just 1%–3% of dietary tryptophan is metabolized via the serotonin pathway34, expiration of 14CO2 (produced by the conversion of 14C-labeled 5-hydroxytryptophan to serotonin) can be measurably decreased in depression54. Therefore, in our patients with MDD, the observed increase in expired 13CO2 could be attributed to increased metabolism in the KYN pathway rather than the serotonin pathway. Specifically, the observed increase in 13CO2 could relate to increased conversion of 3-OH-KYN to 3HAA (Fig. 1), suggesting an increased production of KYN and 3-OH-KYN (precursors of 3HAA), as well as QUIN (the immediate catabolite of 3HAA). Importantly, 3-OH-KYN and 3HAA have neurotoxic effects that are mediated by oxidative stress2,55. In addition, QUIN not only has neurotoxic effects mediated by the activation of NMDA-type glutamate receptors2,56 but also induces lipid peroxidation and oxidative stress57. Therefore, our results are consistent with the neurodegeneration hypothesis of MDD48. Although tryptophan, KYN, and 3-OH-KYN are able to cross the BBB33,34,35, KA, 3HAA, and QUIN cannot do so readily36,37. It has also been reported that there are significant positive correlations between peripheral and central measures of the KYN and QUIN, but not tryptophan16. Further studies are warranted to elucidate the relationship between concentrations of peripheral and central KYN pathway metabolites and indices of 13C-TBT. These neurotoxic metabolites in the KYN pathway are also implicated in several neurodegenerative disorders11,12,58. It may, therefore, be interesting to examine the possible clinical use of the 13C-TBT in these disorders.
We observed significant increases in 13C-TBT indices in the patient group, but found no significant difference in the plasma tryptophan concentrations between patients and controls. This result is inconsistent with previous data, including the results of our own meta-analysis22,23,24,25,26,27. The inconsistency may have occurred due to the small sample size and the modest severity of depression in our patients (mean HAMD-21 score of 12.6), as well as the modest effect size. Indeed, our meta-regression analysis found a negative correlation between plasma tryptophan concentrations and depression severity22. Moreover, some previous studies have reported no significant difference in plasma tryptophan levels between patients with MDD and healthy controls59,60,61. Our results may suggest that 13C-TBT is more sensitive than plasma tryptophan levels at detecting increased metabolism in the KYN pathway.
As shown in Table 3, differences existed between the 2 groups in most of the pharmacokinetic parameters, including the AUC, CRR0–180, and Cmax, but not the Tmax. All these parameters are affected by gastric emptying, digestive absorption, metabolism rates, and respiratory status62. The AUC reflects the global amount of exhaled 13CO2 during the test session. However, although the CRR0-180 also reflects the global process, it may be a better index because it takes into account the body surface area. The CRR may therefore be the best parameter for biomarker research in depression, particularly given the high statistical significance. Finally, while Tmax is particularly subject to the influence of digestion, Cmax could be more relevant because it more closely reflects the metabolic rate.
There are several limitations to this study. First, the sample size was small and the patients with MDD had only modestly severe disease. In addition, depression severity was not measured for the control sample. Despite conflicting results31, there is normal variability in mild depression symptoms in the healthy population together with evidence of a relationship between depressive symptoms and the KYN/tryptophan ratio30. Such methodological limitation might have minimized, rather than exaggerated the difference between the MDD and control groups in 13C-TBT indices. Further studies are needed to address these issues, and should include larger samples, patients with more severe forms of MDD, and assessment for depressive symptoms among healthy controls. A second limitation is that we included 6 patients who had co-morbid diagnoses of dysthymia or an anxiety disorder. There is some evidence suggesting that there is a different KYN metabolism pathway between anxiety and depression63. To examine the possible influence of the comorbidity on the current results, we re-analyzed the data, excluding the 6 patients with MDD comorbidities. However, the results were essentially unchanged and the significant differences in 13C-TBT indices remained between the 2 groups (data not shown). In addition, some studies have shown positive associations of suicidality with KYN and QUIN29,64,65,66. Almost all of the patients in our study (17 of 18) had no history of having attempted suicide. However, because the MDD group in our study might be the heterogeneous group with different biological mechanisms, further studies will be needed with a sample of MDD patients without comorbidity or suicidal behavior. Third, only 10 patients were drug-free, with a further 8 being antidepressant-treated. The AUC and CRR0–180 correlated negatively with the IMIeq dose. Therefore, because some antidepressants have anti-inflammatory effects67, inhibit TDO68, or can affect KYN/tryptophan31, it is possible that antidepressant treatment minimized the differences in the indices of the 13C-TBT and concentrations of plasma tryptophan between the patients and controls. Fourth, although we measured plasma tryptophan, we did not measure any KYN pathway metabolites. Unfortunately, no conclusive relationship can be determined between the measures in this study and the 13C-TBT indices, without knowing the data for other KYN pathway metabolites. In addition, we did not obtain data on inflammatory markers such as C-reactive protein, proinflammatory cytokines, or cortisol levels, which may affect depressive symptoms through the KYN pathway31. Thus, additional studies are required to examine the relationship between these data and the 13C-TBT indices. Lastly, we did not assess the effects of vitamin B6 deficiency, which can interfere with the breakdown of KYN69. Pyridoxal phosphate, the active form of vitamin B6, is needed for the conversions of KYN to anthranilic acid and KA, and for that of 3-OH-KYN to 3HAA and xanthurenic acid.
In conclusion, the 13C-TBT, a noninvasive tracer test, provided evidence of increased tryptophan metabolism along the KYN–NAD pathway in patients with MDD. Consistent with this, 13C-TBT indices correlated negatively with plasma tryptophan concentrations. Therefore, whole-body tryptophan metabolism detected by our 13C-TBT could be a novel and sensitive biomarker for the diagnosis and subtyping of MDD, with the potential to enhance the development of new drugs for the treatment of MDD. Moreover, when 13C-TBT is combined with the 13C-phenylalanine breath test39, discrimination between MDD and schizophrenia may be possible.
Additional Information
How to cite this article: Teraishi, T. et al.13C-tryptophan breath test detects increased catabolic turnover of tryptophan along the kynurenine pathway in patients with major depressive disorder. Sci. Rep. 5, 15994; doi: 10.1038/srep15994 (2015).
Supplementary Material
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (Exploratory Research) from the Japan Society for the Promotion of Science (JSPS) and the Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP (H.K.). We thank Yukiko Kinoshita, Ikki Ishida, Yuki Mitsuyama, Junko Iida, Manami Sakairi, Miku Fujita, Kentaro Yoda, Noriko Yamamoto, Anna Nagashima, Shinsuke Hidese and Fuyuko Yoshida for their assistance with the recruitment of participants and for their support, as well as all the volunteers for their participation. We would also like to thank Hideji Nonomura for his technical advice.
Footnotes
Author Contributions H.K. and T.T. designed the study. T.T., H.H. and D.S. managed the literature searches, collected the data, and undertook the statistical analyses. T.T. wrote the draft of the manuscript. J.M., S.O., M.O., K.H., M.K. and T.H. contributed to the data collection and gave critical comments on the manuscript. J.M. and S.O. contributed to the recruitment of clinical volunteers. H.K. supervised the entire project and gave critical comments on the manuscript. All authors contributed to and have approved the final manuscript.
References
- Maes M., Leonard B. E., Myint A. M., Kubera M. & Verkerk R. The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry 35, 702–721 (2011). [DOI] [PubMed] [Google Scholar]
- Oxenkrug G. F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci 47, 56–63 (2010). [PMC free article] [PubMed] [Google Scholar]
- Myint A. M. & Kim Y. K. Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuropsychopharmacol Biol Psychiatry 48, 304–313 (2014). [DOI] [PubMed] [Google Scholar]
- Lapin I. P. & Oxenkrug G. F. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet 1, 132–136 (1969). [DOI] [PubMed] [Google Scholar]
- Coppen A. The biochemistry of affective disorders. Br J Psychiatry 113, 1237–1264 (1967). [DOI] [PubMed] [Google Scholar]
- Dantzer R., O’Connor J. C., Freund G. G., Johnson R. W. & Kelley K. W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9, 46–56 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller N. Immunology of major depression. Neuroimmunomodulation 21, 123–130 (2014). [DOI] [PubMed] [Google Scholar]
- Yamazaki F., Kuroiwa T., Takikawa O. & Kido R. Human indolylamine 2,3-dioxygenase. Its tissue distribution, and characterization of the placental enzyme. Biochem J 230, 635–638 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. W. & Feng G. S. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. Faseb j 5, 2516–2522 (1991). [PubMed] [Google Scholar]
- Takikawa O., Kuroiwa T., Yamazaki F. & Kido R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J Biol Chem 263, 2041–2048 (1988). [PubMed] [Google Scholar]
- Oxenkrug G. Interferon-gamma - Inducible Inflammation: Contribution to Aging and Aging-Associated Psychiatric Disorders. Aging Dis 2, 474–486 (2011). [PMC free article] [PubMed] [Google Scholar]
- Guillemin G. J. & Brew B. J. Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep 7, 199–206 (2002). [DOI] [PubMed] [Google Scholar]
- Maes M. et al. Increased neopterin and interferon-gamma secretion and lower availability of l-tryptophan in major depression: further evidence for an immune response. Psychiatry Res 54, 143–160 (1994). [DOI] [PubMed] [Google Scholar]
- Tuglu C., Kara S. H., Caliyurt O., Vardar E. & Abay E. Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder. Psychopharmacology (Berl) 170, 429–433 (2003). [DOI] [PubMed] [Google Scholar]
- Capuron L. et al. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry 54, 906–914 (2003). [DOI] [PubMed] [Google Scholar]
- Raison C. L. et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15, 393–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L. et al. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry 7, 468–473 (2002). [DOI] [PubMed] [Google Scholar]
- Bonaccorso S. et al. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol 22, 86–90 (2002). [DOI] [PubMed] [Google Scholar]
- Wichers M. C. et al. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry 10, 538–544 (2005). [DOI] [PubMed] [Google Scholar]
- Bannink M. et al. Interferon-alpha influences tryptophan metabolism without inducing psychiatric side effects. Neuropsyc-hobiology 55, 225–231 (2007). [DOI] [PubMed] [Google Scholar]
- Van Gool A. R. et al. Neurotoxic and neuroprotective metabolites of kynurenine in patients with renal cell carcinoma treated with interferon-alpha: course and relationship with psychiatric status. Psychiatry Clin Neurosci 62, 597–602 (2008). [DOI] [PubMed] [Google Scholar]
- Ogawa S. et al. Plasma l-tryptophan concentration in major depressive disorder: new data and meta-analysis. J Clin Psychiatry 75, e906–915 (2014). [DOI] [PubMed] [Google Scholar]
- Gabbay V. et al. The possible role of the kynurenine pathway in adolescent depression with melancholic features. J Child Psychol Psychiatry 51, 935–943 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M. M. et al. Tryptophan depletion in depressed patients occurs independent of kynurenine pathway activation. Brain Behav Immun 26, 979–987 (2012). [DOI] [PubMed] [Google Scholar]
- Maes M. et al. Lower serum l-tryptophan availability in depression as a marker of a more generalized disorder in protein metabolism. Neuropsychopharmacology 15, 243–251 (1996). [DOI] [PubMed] [Google Scholar]
- Anderson I. M., Parry-Billings M., Newsholme E. A., Poortmans J. R. & Cowen P. J. Decreased plasma tryptophan concentration in major depression: relationship to melancholia and weight loss. J Affect Disord 20, 185–191 (1990). [DOI] [PubMed] [Google Scholar]
- Cowen P. J., Parry-Billings M. & Newsholme E. A. Decreased plasma tryptophan levels in major depression. J Affect Disord 16, 27–31 (1989). [DOI] [PubMed] [Google Scholar]
- Myint A. M. et al. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 98, 143–151 (2007). [DOI] [PubMed] [Google Scholar]
- Sublette M. E. et al. Plasma kynurenine levels are elevated in suicide attempters with major depressive disorder. Brain Behav Immun 25, 1272–1278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elovainio M. et al. Indoleamine 2,3-dioxygenase activation and depressive symptoms: results from the Young Finns Study. Psychosom Med 74, 675–681 (2012). [DOI] [PubMed] [Google Scholar]
- Quak J. et al. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology 45, 202–210 (2014). [DOI] [PubMed] [Google Scholar]
- Hasunuma T., Harada K., Miyake C. & Fukusaki E. Dynamic metabolic profiling using stable isotope labeling. Low Temperature Science 67, 169–174 (2009). [Google Scholar]
- Russo S. et al. Tryptophan as a link between psychopathology and somatic states. Psychosom Med 65, 665–671 (2003). [DOI] [PubMed] [Google Scholar]
- Sainio E. L., Pulkki K. & Young S. N. l-Tryptophan: Biochemical, nutritional and pharmacological aspects. Amino Acids 10, 21–47 (1996). [DOI] [PubMed] [Google Scholar]
- Richard D. M. et al. l-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int J Tryptophan Res 2, 45–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui S., Schwarcz R., Rapoport S. I., Takada Y. & Smith Q. R. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 56, 2007–2017 (1991). [DOI] [PubMed] [Google Scholar]
- Foster A. C., Miller L. P., Oldendorf W. H. & Schwarcz R. Studies on the disposition of quinolinic acid after intracerebral or systemic administration in the rat. Exp Neurol 84, 428–440 (1984). [DOI] [PubMed] [Google Scholar]
- Saito K., Crowley J. S., Markey S. P. & Heyes M. P. A mechanism for increased quinolinic acid formation following acute systemic immune stimulation. J Biol Chem 268, 15496–15503 (1993). [PubMed] [Google Scholar]
- Teraishi T. et al. 13C-phenylalanine breath test detects altered phenylalanine kinetics in schizophrenia patients. Transl Psychiatry 2, e119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankes L. V., Brown R. R., Leklem J., Schmaeler M. & Jesseph J. Metabolism of C14 labeled enantiomers of tryptophan, kynurenine and hydroxykynurenine in humans with scleroderma. J Invest Dermatol 58, 85–95 (1972). [DOI] [PubMed] [Google Scholar]
- Hankes L. V., Brown R. R., Lippincott S. & Schmaeler M. Effects of l-tryptophan load on the metabolism of tryptophan-2-C14 in man. J Lab Clin Med 69, 313–324 (1967). [PubMed] [Google Scholar]
- American Psychiatric Association. DSM-IV: Diagnostic and Statistical Manual of Mental Disorders, 4th edn. (American Psychiatric Association, 1994). [Google Scholar]
- Otsubo T. et al. Reliability and validity of Japanese version of the Mini-International Neuropsychiatric Interview. Psychiatry Clin Neurosci 59, 517–526 (2005). [DOI] [PubMed] [Google Scholar]
- Brown T. R. Stable isotopes in pharmaceutical research (ed. Brown T. R.) Ch. 2, 13–18 (Elsevier Science Publishers, 1997). [Google Scholar]
- Nomura J. Effect of Stress and Psychotropic Drugs on Rat Liver Tryptophan Pyrrolase. Endocrinology 76, 1190–1194 (1965). [DOI] [PubMed] [Google Scholar]
- Kunugi H., Hori H., Adachi N. & Numakawa T. Interface between hypothalamic-pituitary-adrenal axis and brain-derived neurotrophic factor in depression. Psychiatry Clin Neurosci 64, 447–459 (2010). [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23, 477–501 (2000). [DOI] [PubMed] [Google Scholar]
- Myint A. M. & Kim Y. K. Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses 61, 519–525 (2003). [DOI] [PubMed] [Google Scholar]
- Okano Y. et al. In vivo studies of phenylalanine hydroxylase by phenylalanine breath test: diagnosis of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Pediatr Res 56, 714–719 (2004). [DOI] [PubMed] [Google Scholar]
- Ishii T. et al. Optimum conditions for the 13C-phenylalanine breath test. Chem Pharm Bull (Tokyo) 46, 1330–1332 (1998). [DOI] [PubMed] [Google Scholar]
- Inagaki A., Inada T., Fujii Y. & Yagi G. Equivalent Dose of Psychotropics. (Seiwa Shoten, 1999). (in Japanese). [Google Scholar]
- Inagaki A. & Inada T. Dose equivalence of psychotropic drugs part X VIII dose equivalence of psychotropic drugs: 2006-version. J Clin Psychopharmacol, 1443–1447 (2006). [Google Scholar]
- Oxenkrug G. Serotonin-kynurenine hypothesis of depression: historical overview and recent developments. Curr Drug Targets 14, 514–521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppen A., Shaw D. M. & Malleson A. Changes in 5-hydroxytryptophan metabolism in depression. Br J Psychiatry 111, 105–107 (1965). [DOI] [PubMed] [Google Scholar]
- Okuda S., Nishiyama N., Saito H. & Katsuki H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J Neurochem 70, 299–307 (1998). [DOI] [PubMed] [Google Scholar]
- Stone T. W. & Perkins M. N. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol 72, 411–412 (1981). [DOI] [PubMed] [Google Scholar]
- Rios C. & Santamaria A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem Res 16, 1139–1143 (1991). [DOI] [PubMed] [Google Scholar]
- Guidetti P. & Schwarcz R. 3-Hydroxykynurenine and quinolinate: pathogenic synergism in early grade Huntington’s disease? Adv Exp Med Biol 527, 137–145 (2003). [DOI] [PubMed] [Google Scholar]
- Price L. H., Charney D. S., Delgado P. L. & Heninger G. R. Serotonin function and depression: neuroendocrine and mood responses to intravenous l-tryptophan in depressed patients and healthy comparison subjects. Am J Psychiatry 148, 1518–1525 (1991). [DOI] [PubMed] [Google Scholar]
- Møller S. E. Plasma amino acid profiles in relation to clinical response to moclobemide in patients with major depression. Danish University Antidepressant Group. J Affect Disord 27, 225–231 (1993). [DOI] [PubMed] [Google Scholar]
- Mauri M. C. et al. Plasma and platelet amino acid concentrations in patients affected by major depression and under fluvoxamine treatment. Neuropsychobiology 37, 124–129 (1998). [DOI] [PubMed] [Google Scholar]
- Treacy E. et al. Analysis of phenylalanine hydroxylase genotypes and hyperphenylalaninemia phenotypes using l-[1-13C]phenylalanine oxidation rates in vivo: a pilot study. Pediatr Res 42, 430–435 (1997). [DOI] [PubMed] [Google Scholar]
- Orlikov A. B., Prakhye I. B. & Ryzov I. V. Kynurenine in blood plasma and DST in patients with endogenous anxiety and endogenous depression. Biol Psychiatry 36, 97–102 (1994). [DOI] [PubMed] [Google Scholar]
- Bradley K. A. et al. The role of the kynurenine pathway in suicidality in adolescent major depressive disorder. Psychiatry Res 227, 206–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay-Richter C. et al. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav Immun 43, 110–117 (2015). [DOI] [PubMed] [Google Scholar]
- Erhardt S. et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 38, 743–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubera M. et al. Anti-Inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J Clin Psychopharmacol 21, 199–206 (2001). [DOI] [PubMed] [Google Scholar]
- Ara I. & Bano S. Citalopram decreases tryptophan 2,3-dioxygenase activity and brain 5-HT turnover in swim stressed rats. Pharmacol Rep 64, 558–566 (2012). [DOI] [PubMed] [Google Scholar]
- Linkswiler H. Biochemical and physiological changes in vitamin B6 deficiency. Am J Clin Nutr 20, 547–561 (1967). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.