

Abstract
Background
Distal Renal Tubular Acidosis is a disorder of acid-base regulation caused by functional failure of α-intercalated cells in the distal nephron. The recessive form of the disease (which is usually associated with sensorineural deafness) is attributable to mutations in ATP6V1B1 or ATP6V0A4, which encode the tissue-restricted B1 and a4 subunits of the renal apical H+-ATPase. ATP6V1B1 lies adjacent to the gene encoding the homeobox domain protein VAX2, at 2p13.3. To date, no human phenotype has been associated with VAX2 mutations.
Case presentation
The male Caucasian proband, born of a first cousin marriage, presented at 2 months with failure to thrive, vomiting and poor urine output. No anatomical problems were identified, but investigation revealed hyperchloremic metabolic acidosis with inappropriately alkaline urine and bilateral nephrocalcinosis. Distal Renal Tubular Acidosis was diagnosed and audiometry confirmed hearing loss at 2 years. ATP6V0A4 was excluded from genetic causation by intragenic SNP linkage analysis, but ATP6V1B1 completely failed to PCR-amplify in the patient, suggesting a genomic deletion. Successful amplification of DNA flanking ATP6V1B1 facilitated systematic chromosome walking to ascertain that the proband harbored a homozygous deletion at 2p13.3 encompassing all of ATP6V1B1 and part of VAX2; gene dosage was halved in the parents. This results in the complete deletion of ATP6V1B1 and disruption of the VAX2 open reading frame. Later ocular examinations revealed bilateral rod / cone photoreceptor dystrophy and mild optic atrophy. Similar changes were not detected in an adult harbouring a disruptive mutation in ATP6V1B1.
Conclusions
The genomic deletion reported here is firstly, the only reported example of a whole gene deletion to underlie Distal Renal Tubular Acidosis, where the clinical phenotype is indistinguishable from that of other patients with ATP6V1B1 mutations; secondly, this is the first reported example of a human VAX2 mutation and associated ocular phenotype, supporting speculation in the literature that VAX2 is important for correct retinal functioning.
Keywords: dRTA, H+-ATPase, ATP6V1B1, VAX2, Retina
Background
Most physiological functions and the ultimate survival of all multicellular organisms depend on the maintenance of constant extracellular pH (close to 7.4 in man) and appropriate adaptations to change. The normal human omnivorous diet creates a net acid load, which requires excretion. The kidney plays a vital part in pH regulation by extruding acid into nascent urine, with concomitant reabsorption of bicarbonate. The two main sites for acid-base regulation along the nephron (the functional unit of the kidney) are the proximal tubule, where major bicarbonate reabsorption takes place, and the distal nephron where fine regulation is achieved by specialized acid-handling epithelial cells, α-intercalated cells (α-ICs). At this site, the net effect in humans is acid secretion [1].
Alpha-ICs are polarized such that H+ secretion occurs across their apical plasma membrane and into the urine by specialized multi-subunit proton pumps (H+-ATPases). H+-ATPases are also vital for acidification of intracellular compartments such as endosomes and lysosomes. They consist of two domains, a catalytic V1 domain responsible for ATP hydrolysis, and a membrane-anchored V0 domain that uses the energy thus generated to drive H+ transport. Each domain comprises a number of separate components such that together, the pump contains a total of at least 13 different subunits, all separately encoded [2].
On the basolateral side of α-ICs, the anion exchanger AE1 (encoded by SLC4A1) acts as a 1:1 chloride/bicarbonate exchanger, where it reabsorbs bicarbonate. How apical H+-ATPases and AE1 are functionally coupled is not understood [1].
Failure of acid-base regulation in α-ICs results in distal Renal Tubular Acidosis (dRTA; MIM#602722 and 267300). This disease is characterized by sometimes-severe hyperchloremic metabolic acidosis with an inappropriately alkaline urine (pH >5.5), usually accompanied by hypokalemia, hypercalciuria, renal tract calcification and in untreated cases, rickets/osteomalacia. It can occur secondarily, particularly in autoimmune diseases such as Sjogren’s syndrome, or can be inherited as either an autosomal dominant or recessive disorder. Dominant dRTA is caused by mutations in SLC4A1 [3, 4]. We and others have previously shown that mutations in ATP6V1B1 and ATP6V0A4, which encode subunits B1 and a4 of the H+-ATPase respectively, are causative of recessive dRTA (rdRTA). These are associated with progressive sensorineural hearing loss (SNHL) [5–7], which is presumed due to endolymphatic pH abnormalities [5–7]. a4 and B1 are tissue-restricted forms of the a- and B subunits respectively, who replace the ubiquitously expressed a1 and B2 forms found on intracellular membranes. Their expression is largely restricted to the apical surface of the α-IC, male reproductive tract and inner ear.
ATP6V1B1 maps to 2p13.3, adjacent to VAX2, which encodes a homeobox protein transcription factor that is almost exclusively expressed in the ventral portion of the retina during development. To date, no human mutations have been found in VAX2, but GWAS meta-analysis has suggested it as a candidate to underlie astigmatism [8]. Vax2-null mice show disruptions to the establishment of a dorsoventral axis of the neural retina, a key step in eye development, and abnormal outgrowth of ventral retinal ganglion cells [9, 10]. Transcriptome analysis in these mice suggests that lack of Vax2 expression adversely affects the distribution of retinoic acid which itself plays a major part in correct patterning of the eye during development. Importantly, retinoic acid is also shown to be important in maintenance of appropriate gene expression in the photoreceptor cells of the mature retina [11]. Some of the Vax2-/- mice also display incomplete closure of the optic fissure and coloboma (with variable penetrance) and there is speculation in the literature that VAX2 might be responsible for a similar human phenotype [9, 12].
VAX2 shares 100 % identity in the homeobox domain with VAX1, which is strongly expressed both during early development in the neural ridge, and later in the rostro-medial olfactory placode, optic nerve and disc and anterior ventral forebrain. VAX1 is known to be essential both for corpus callosum development and correct axon guidance in the optic nerve and forebrain [13]. A single case has been reported wherein a mutation in VAX1 (R152S) was found in a patient with microphthalmia, optic nerve hypoplasia, cleft lip/palate and corpus callosum agenesis, a phenotype similar to that found in the Vax1 null mouse [13, 14]. VAX2 was also screened in an additional 70 patients with anophthalmia/microphthalmia but no mutations were found [14]. To date therefore, the single VAX1 case represents the only reported mutation in either human VAX gene.
Here, we describe a genomic deletion that causes both dRTA (due to the complete absence of ATP6V1B1) and a novel deleterious eye phenotype (due to disruption of VAX2). This deletion represents a novel mechanism of disease for dRTA and underpins the importance of VAX2 in maintenance of retinal integrity in man.
Case presentation
Clinical presentation
The consanguineous Caucasian kindred under study was referred via pediatric nephrology services in Tehran, Iran. The male patient, born to first cousin parents, presented at 2 months of age with failure to thrive and vomiting, and was described as having ‘difficulty in urination’. On examination there were no anatomical problems, but hyperchloremic metabolic acidosis with inappropriately alkaline urine were discovered on biochemical assessment (blood pH 7.01, HCO3 4mmol/l, urine pH > 6). Ultrasonography revealed bilateral nephrocalcinosis. Hearing impairment was suspected at two years old and confirmed by audiometry. Visual difficulties were not reported, and both parents and a sibling were clinically normal.
Genetic investigation of dRTA
Linkage analysis using previously described intragenic SNPs within both ATP6V0A4 and ATP6V1B1 excluded linkage to ATP6V0A4 in this family [7] (Fig. 1a). In contrast, none of the intragenic SNPs in ATP6V1B1 would PCR-amplify in the patient. Multiplex PCR amplification of both genes confirmed the integrity of the DNA template, suggesting a genomic deletion (Fig. 1b). To investigate the extent of the deletion, genes flanking ATP6V1B1 were also subjected to PCR in the proband and an unrelated unaffected individual. VAX2 lies immediately 5′ of ATP6V1B1, and ANKRD53 is 3′ (Fig. 1c). In the patient, successful PCR amplification was achieved for exon 1 but not exons 2 or 3 of VAX2, indicating that the deletion breakpoint lay in intron 1. All exons of ANKRD53 were successfully amplified. Further PCR was performed in the patient and an unrelated unaffected individual to ‘walk’ systematically inwards until a segment was amplified from control but not patient DNA. Reactions were performed using QIAGEN’s multiplex PCR kit in a total reaction volume of 15 μl containing 50 ng DNA. Finally, the outermost primers of these pairs (VAX2, intron 1 and between ATP6V1B1 and ANKRD53) were used in combination to amplify across the presumed deleted region in the proband (Fig. 2a). Primer sequences are available on request. Sequencing of this product was performed and analyzed by BLAT [15]. The first 225 bp exactly matched sequence in intron 1 of VAX2. This was followed by GG, and the remainder of the product exactly matched sequence from the region between ATP6V1B1 and ANKRD53 (Fig. 2b). This confirmed that the deleted region was 56.8 Kb and contained 2 of the 3 coding exons of VAX2 and the known dRTA gene.
Fig. 1.
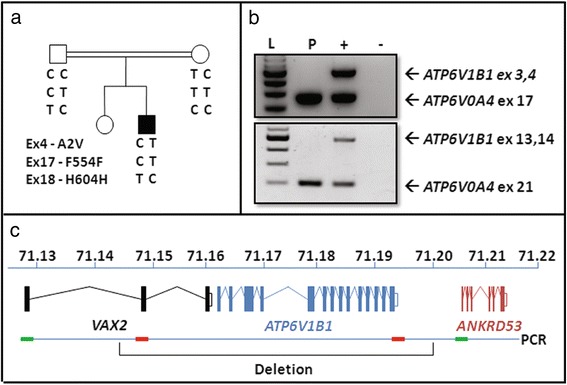
Evidence that the deletion of ATP6V1B1 underlies dRTA in this family (a) ATP6V0A4 was excluded by heterozygosity of SNPs in the patient in exons 4, 17 and 18. b Failure of PCR to amplify exons 3–4 and 13–14 of ATP6V1B1 in the patient (P) suggested a large deletion compared to positive control (+). c Successful PCR amplification in the patient for exon 1 of VAX2 ( ) and exon 1 of ANKRD53 (
) and exon 1 of ANKRD53 ( ), but not exon 2 of VAX2 (
), but not exon 2 of VAX2 ( ) and exon 21 of ATP6V1B1 (
) and exon 21 of ATP6V1B1 ( ) define regions on either side of ATP6V1B1 for the deletion breakpoints on chromosome 2. L = 100bp ladder
) define regions on either side of ATP6V1B1 for the deletion breakpoints on chromosome 2. L = 100bp ladder
Fig. 2.
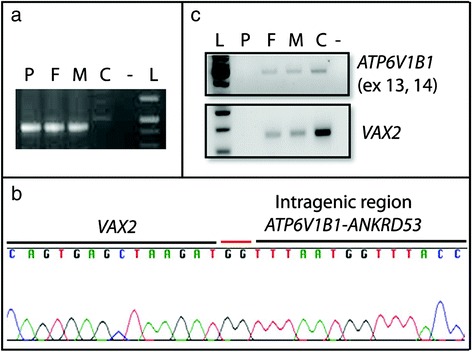
Characterization and gene dosage of the genomic deletion at 2p13.3 (a) Successful PCR amplification across the junction breakpoint in the patient (P) and his parents (F, M) but not in a control individual with normal genomic DNA (C). b Sequencing revealed the insertion of 2 nucleotides (GG) at the site of the deletion. c Semi-quantitative PCR confirmed that gene dosage of ATP6V1B1 and exons 2 and 3 of VAX2 is reduced in the parents (F, M) compared to the control (C)
Gene dosage in family members was investigated by semi-quantitative PCR, using primers to amplify exons 13–14 of ATP6V1B1 and exon 3 of VAX2 from genomic DNA of the patient, both parents and an unrelated unaffected individual. This confirmed that both parents were heterozygous for the deletion (Fig. 2c).
Investigation of a possible ocular phenotype due to loss of VAX2
No visual problems were initially reported in this patient, now aged 13. However, recent retrospective ophthalmological examination revealed subtle abnormalities. Corrected visual acuity was 18/20 in each eye. Refraction showed [OD +1.50 DS -0.75 DC at 120] and [OS +1.0 DS -0.75 DC at 1600]. Morphology of the external eye was normal bilaterally, with normal ocular motility and anterior segments. Fundal examination revealed bilateral pigmentary changes at the macula and retinal periphery consistent with a retinal dystrophy, and bilateral optic atrophy (Fig. 3). Electroretinography demonstrated reduced amplitude a-waves in keeping with a photoreceptor dystrophy. Visual evoked potential amplitude was mildly reduced, likely secondary to ganglion cell loss from the retinal dystrophy.
Fig. 3.
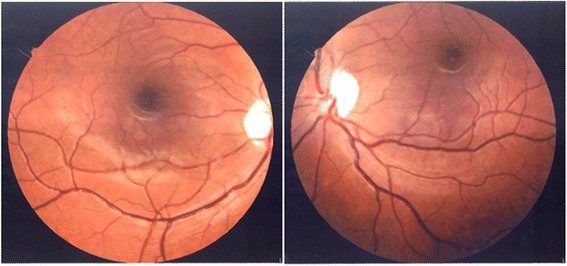
Fundus photographs of the right eye (left image) and left eye (right image). Abnormal pigmentation around the macula and mild bilateral optic atrophy can be seen.
To confirm that the phenotype seen in this patient was not attributable to B1 deficiency, we arranged for detailed retinal examination of an unrelated adult (32 year old) dRTA patient with an underlying homozygous B1 mutation (c.687 + 1G > T; a previously reported [5] essential splice site mutation at the exon 7/intron 7 boundary). This revealed normal retinal function with no cellular changes (data not shown), which supports the conclusion that VAX2 deficiency in the patient described here is responsible for the eye phenotype.
Conclusions
This is the first time that a whole gene deletion has been found to underlie dRTA. The clinical phenotype in this patient was indistinguishable from cases caused by nonsense or missense mutations in ATP6B1V1. However, on finding the deletion of two of the three coding exons of VAX2, a post-hoc ocular examination revealed an undiagnosed eye phenotype. The patient did not manifest coloboma but this is not particularly surprising as in Vax2-/- mice this abnormality showed variable penetrance [9, 10]. However, the patient described here was found to have electrophysiological evidence of photoreceptor degeneration. Additionally, there was optic atrophy similar to Vax2-/- mice which may be primary or secondary to the retinal dystrophy.
A possible mechanism for the abnormality is disruption to retinoic acid disposition in the retina, for which VAX2 is necessary, and which is important for photoreceptor function [11]. Both in this patient and in the Vax2-null mouse, the phenotype is mild compared to that associated both with a previously reported human VAX1 mutation and a targeted murine Vax1 deletion, where coloboma was fully penetrant and moderately severe [13, 14]. The mild VAX2-null phenotype is likely due to compensation by VAX1 with which VAX2 has been shown to work in concert to ventralize the eye. Vax1/Vax2 double mutant mice have a fully penetrant, severe phenotype where the optic nerve is transformed into differentiated retina. This is caused by loss of repression of the Pax6 transcription factor, which promotes a dorsal phenotype/retinal differentiation [16].
The normal retinal function we report in an unrelated, adult dRTA patient with a homozygous B1 splice site mutation excludes the possibility that the eye phenotype seen in the case described here is attributable to a failure of the H+-ATPase. We and others have previously demonstrated expression of the tissue-restricted ATP6V0A4 and ATP6V1B1 in the eye [17–19] where it is suggested that the proton pump contributes to intraocular fluid homeostasis. Additionally, the role of the a3 subunit of the H+-ATPase in correct ocular function is well documented; mutations cause severe autosomal recessive osteopetrosis often associated with impaired vision [20] but here, loss of sight is attributed to compression of the optic nerve caused by failure of bone resorption by osteoclasts [18].
This study is important in terms of clinical and research findings as it has confirmed that VAX2 plays an important role in maintenance of the human neural retina. In order to further understand the pathogenesis, more patients with VAX2 mutations will need to be investigated. Although presentation of these patients has not to date been forthcoming, they may now be easier to identify as we have some a priori knowledge of how the phenotype might present.
Consent
Written informed consent was obtained from the parents of the index case (Cambridge Local Research Ethics Committee 99/078). A copy of the written consent is available for review by the Editor of this journal.
Acknowledgements
This study was supported by the Wellcome Trust (awards 088489 to FEK and 079895 to Cambridge Institute for Medical Research).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EEN drafted the manuscript; EEN, AY and KGB identified and characterized the deletion; MS and AK have clinical responsibility for the patient; AN and LEA performed and analyzed the ophthalmological examination respectively; FEK coordinated the study and critically revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Elizabeth E. Norgett, Email: een22@cam.ac.uk
Anthony Yii, Email: anthony.yii@cantab.net.
Katherine G. Blake-Palmer, Email: kgb24@cantab.net
Mostafa Sharifian, Email: sharifian.dorche@gmail.com.
Louise E. Allen, Email: louise.allen@addenbrookes.nhs.uk
Abdolhamid Najafi, Email: hamidnajafi20@gmail.com.
Ariana Kariminejad, Email: arianakariminejad@yahoo.com.
Fiona E. Karet Frankl, Email: fek1000@cam.ac.uk.
References
- 1.Fry AC, Karet FE. Inherited renal acidoses. Physiology. 2007;22:202–11. doi: 10.1152/physiol.00044.2006. [DOI] [PubMed] [Google Scholar]
- 2.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H + -ATPase. Physiol Rev. 2004;84:1263–314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 3.Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest. 1997;100:1693–707. doi: 10.1172/JCI119694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karet FE, Gainza FJ, Gyory AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci U S A. 1998;95:6337–42. doi: 10.1073/pnas.95.11.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H + -ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 6.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–5. doi: 10.1038/82492. [DOI] [PubMed] [Google Scholar]
- 7.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopes MC, Hysi PG, Verhoeven VJ, Macgregor S, Hewitt AW, Montgomery GW, Cumberland P, Vingerling JR, Young TL, van Duijn CM, Oostra B, Uitterlinden AG, Rahi JS, Mackey DA, Klaver CC, Andrew T, Hammond CJ. Identification of a candidate gene for astigmatism. Invest Ophthalmol Vis Sci. 2013;54:1260–7. doi: 10.1167/iovs.12-10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbieri AM, Broccoli V, Bovolenta P, Alfano G, Marchitiello A, Mocchetti C, Crippa L, Bulfone A, Marigo V, Ballabio A, Banfi S. Vax2 inactivation in mouse determines alteration of the eye dorsal-ventral axis, misrouting of the optic fibres and eye coloboma. Development. 2002;129:805–13. doi: 10.1242/dev.129.3.805. [DOI] [PubMed] [Google Scholar]
- 10.Mui SH, Hindges R, O’Leary DD, Lemke G, Bertuzzi S. The homeodomain protein Vax2 patterns the dorsoventral and nasotemporal axes of the eye. Development. 2002;129:797–804. doi: 10.1242/dev.129.3.797. [DOI] [PubMed] [Google Scholar]
- 11.Alfano G, Conte I, Caramico T, Avellino R, Arno B, Pizzo MT, Tanimoto N, Beck SC, Huber G, Dolle P, Seeliger MW, Banfi S. Vax2 regulates retinoic acid distribution and cone opsin expression in the vertebrate eye. Development. 2011;138:261–71. doi: 10.1242/dev.051037. [DOI] [PubMed] [Google Scholar]
- 12.Barbieri AM, Lupo G, Bulfone A, Andreazzoli M, Mariani M, Fougerousse F, Consalez GG, Borsani G, Beckmann JS, Barsacchi G, Ballabio A, Banfi S. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc Natl Acad Sci U S A. 1999;96:10729–34. doi: 10.1073/pnas.96.19.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertuzzi S, Hindges R, Mui SH, O’Leary DD, Lemke G. The homeodomain protein vax1 is required for axon guidance and major tract formation in the developing forebrain. Genes Dev. 1999;13:3092–105. doi: 10.1101/gad.13.23.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slavotinek AM, Chao R, Vacik T, Yahyavi M, Abouzeid H, Bardakjian T, Schneider A, Shaw G, Sherr EH, Lemke G, Youssef M, Schorderet DF. VAX1 mutation associated with microphthalmia, corpus callosum agenesis, and orofacial clefting: the first description of a VAX1 phenotype in humans. Hum Mutat. 2012;33:364–8. doi: 10.1002/humu.21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Human BLAT Search [http://genome.ucsc.edu/cgi-bin/hgBlat?command=start]
- 16.Mui SH, Kim JW, Lemke G, Bertuzzi S. Vax genes ventralize the embryonic eye. Genes Dev. 2005;19:1249–59. doi: 10.1101/gad.1276605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norgett EE, Golder ZJ, Lorente-Canovas B, Ingham N, Steel KP, Karet Frankl FE. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci U S A. 2012;109:13775–80. doi: 10.1073/pnas.1204257109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawamura N, Tabata H, Sun-Wada GH, Wada Y. Optic nerve compression and retinal degeneration in Tcirg1 mutant mice lacking the vacuolar-type H-ATPase a3 subunit. PLoS One. 2010;5:e12086. doi: 10.1371/journal.pone.0012086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wax MB, Saito I, Tenkova T, Krupin T, Becker B, Nelson N, Brown D, Gluck SL. Vacuolar H + -ATPase in ocular ciliary epithelium. Proc Natl Acad Sci U S A. 1997;94:6752–7. doi: 10.1073/pnas.94.13.6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–63. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]