

Abstract
Background
Visual system homeobox gene (VSX1) plays a major role in the early development of craniofacial and ocular tissues including cone opsin gene in the human retina. To date, few disease-causing mutations of VSX1 have been linked to familial and sporadic keratoconus (KC) in humans. In this study, we describe the clinical features and screening for VSX1 gene in families with KC from India.
Methods
Clinical data and genomic DNA were collected from patients with clinically diagnosed KC and their family members. The study was conducted on 20 subjects of eight families from India. The coding exons of VSX1 gene were amplified using PCR and amplicons were analyzed by direct sequencing. Predictive effect of the mutations was performed using Polyphen-2, SIFT and mutation assessor algorithms. Additionally, haplotypes of VSX1 gene were constructed for affected and unaffected individuals using SNPs.
Results
In the coding region of VSX1, one novel missense heterozygous change (p.Leu268His) was identified in five KC patients from two unrelated families. Another family of three members had a novel heterozygous change (p.Ser251Thr). These variants co-segregated with the disease phenotype in all affected individuals but not in the unaffected family members and 105 normal controls. In silico analysis suggested that p.Leu268His could have a deleterious effect on the protein coded by VSX1, while p.Ser251Thr has a neutral effect on the functional properties of VSX1. Haplotype examination revealed common SNPs around the missense change (p.Leu268His) in two unrelated KC families.
Conclusions
In this study, we add p.Leu268His, a novel missense variation in the coding region of VSX1 to the existing repertoire of VSX1 coding variations observed in Indian patients with the characteristic phenotype of KC. The variant p.Ser251Thr might be a benign polymorphism, but further biophysical studies are necessary to evaluate its molecular mechanism. The shared haplotype by two families with the same variant suggests the possibility of a founder effect, which requires further elucidation. We suggest that p.Leu268His might be involved in the pathogenesis of KC, which may help in the genetic counselling of patients and their family.
Keywords: Visual system homeobox gene (VSX1), Familial keratoconus, Mutation screening, CVC (Chx10/Vsx–1 and ceh–10) domain, In sillico analysis, Missense mutation, Haplotype analysis
Background
Keratoconus (KC: OMIM 14830) is a progressive ectatic disorder of the cornea characterized by thinning of the central cornea leading to protrusion and progressive, irregular astigmatism. Though there are several treatment modalities available, severe KC remains an indication for corneal transplantation [1, 2]. The mean age of onset is 15.39 years with a prevalence of 0.0003 %–2.3 % that affects both genders and all ethnicities across the globe [3]. The disease is a complex heterogeneous disorder with risk factors like chronic eye rubbing and atopy playing a significant role besides ultraviolet light induced oxidative stress [4–6]. The genetic basis for keratoconus has always been an accepted theory considering its familial occurrence and high concordance in monozygotic twins [4, 7, 8]. Though most KC cases are sporadic, it has been noted that 6–10 % of cases have a positive family history [9, 10]. Inheritance in KC can be dominant or recessive; with autosomal dominant inheritance, the disease exhibits variable phenotypes with incomplete penetrance [9].
Linkage analysis has identified several genomic loci in KC families thereby establishing genetic heterogeneity [11–15]. Genes with mutations (VSX1, DOCK9, TGFβI, SOD1, FLG, ZEB1) were found to be responsible for only a small fraction of KC cases in select populations around the world [16–21]. Nevertheless, VSX1 mutations have been identified in two different corneal phenotypes - posterior polymorphous corneal dystrophy (PPCD) and KC [16]. Genetic analysis of KC patients from different ethnic backgrounds has revealed several coding variations in the VSX1 gene [22–26]. So far, four pathogenic VSX1 mutations have been reported in the KC phenotype. Hence, the significance of a genetic basis for KC is still unclear [22–26]. VSX1 is a paired-like homeodomain transcriptional factor gene localized in 20p11.21. It is expressed in the adult cornea and adult retinal cDNA libraries [27], inner nuclear layer of the human retina and embryonic craniofacial tissue [28]. The human VSX1 gene has five exons that encodes for a 365–amino acid protein with homeobox DNA binding domain and a CVC (Chx10/Vsx–1 and ceh–10) domain, which is highly conserved among vertebrates. In this present study, we correlate the genetic, and clinical features of KC patients and their families of Indian origin with VSX1 gene variants.
Methods
Study subjects and clinical examination
Twenty affected individuals from eight unrelated KC families, 11 unaffected family members, and 105 ethnically matched normal controls were included in this study. All patients were examined at the Cornea and Refractive Surgery Department, Narayana Nethralaya Postgraduate Institute of Ophthalmology, Bangalore, India. The study followed the tenets of the Declaration of Helsinki and was approved by the Institutional Ethical Committee (IEC–C/2013/07/01). All patients underwent visual acuity assessment, a detailed slit lamp examination with topographic and pachymetric evaluation on the Pentacam HR (Oculus Inc.) and Orbscan (Orbtek, Baush, & Lomb). Keratoconus was graded according to the Amsler-Krumeich Classification [29]. If KC was detected in more than one member of the family, the entire family was counselled, detailed informed consent taken, and blood collected for genetic analysis.
Genetic study
A detailed family history was taken including history of ocular and non-ocular hereditary disorders and pedigree charts drawn accordingly. The total genomic DNA was isolated from peripheral blood leukocytes by the salt precipitation method [30] from all study subjects. For mutational analysis, the entire coding exons of VSX1 and their flanking intronic junctions were amplified by PCR in eight probands using the primer reported elsewhere [25]. The PCR products were sequenced on 3130xl Genetic Analyzer (Applied Biosystems) according to the manufacturer’s protocol. Sequencing results were analyzed in chromatogram viewer (FinchTV 1.40), pairwise BLAST (Basic Local Alignment Search Tool) [31] to examine if there were any changes from the normal VSX1 sequence available in the database (NM_014588, ENSG00000100987). The segregation of nucleotide changes were analysed in eight affected and four unaffected individuals from three unrelated families by direct sequencing method. In addition, exon 4 of VSX1 was sequenced in 105 unrelated ethnically matched normal controls to validate the pathogenicity of nucleotide variations.
Bioinformatics analysis
In order to predict the effect of nucleotide change at the protein level, we used in silico prediction servers Polyphen–2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.bii.a-star.edu.sg), Mutation Assessor (http://mutationassessor.org/v1/) and PROVEAN (http://provean.jcvi.org/index.php). Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and multiple sequence alignment programs were used to check the evolutionary conservation of VSX1 protein in other vertebrates. The effect of amino acid changes in the stability of VSX1 was assessed by using the MUpro (version 1.0, http://mupro.proteomics.ics.uci.edu/) prediction server (AAMSPSM).
Haplotype analysis
To examine the disease and mutation associated haplotypes of the eight affected and four unaffected individuals from two unrelated KC families, we analysed four intragenic SNPs (rs12480307, rs6138482, rs56157240), and (IVS3–24C > T) flanking VSX1 by direct sequencing. Haplotypes were constructed manually.
Results
In this study, we analysed 20 affected individuals of eight families with a clinical diagnosis of KC for mutations in the VSX1 gene (Table 1). A novel missense coding variant (p. Leu268His) was found in five patients from two unrelated KC families (KC_01,KC_02). Another novel heterozygous missense change (p. Ser251Thr) was identified in a third KC family (KC_03) with two affected siblings and their affected father. The clinical features of the affected individuals are summarized in Table 2.
Table 1.
Summary of genotype and phenotype characteristics in the study subjects
| Family ID | Individuals | Age at diagnosis/Sex | Keratoconus | Segregation of VSX1 nucleotide changes | |
|---|---|---|---|---|---|
| Coding variants | SNPs | ||||
| KC–01 | I:1 | 40/F | No | - | rs56157240, |
| I:2 | 35/M | Yes/RE | L268H | rs12480307, rs56157240, IVS3–24C > T | |
| II:1 | 22/M | Yes/RE | L268H | rs12480307, rs56157240, IVS3–24C > T | |
| II:2a | 19/M | Yes/BE | L268H | rs12480307, rs56157240 | |
| KC–02 | I:1 | 42/F | No | - | rs12480307, rs6138482, IVS3–24C > T |
| I:2 | 37/M | Yes/LE | L268H | rs12480307, rs56157240, rs6138482, | |
| II:1a | 16/M | Yes/BE | L268H | rs12480307, rs56157240, rs6138482, | |
| II:2 | 12/F | No | - | rs12480307, | |
| KC–03 | I:1 | 45/F | No | - | rs56157240, rs6138482 |
| I:2 | 50/M | Yes/BE | S251T | IVS3–24C > T | |
| II:1 | 20/F | Yes/BE | S251T | IVS3–24C > T | |
| II:2a | 18/M | Yes/BE | S251T | rs56157240 | |
| KC–04 | I:1 | 53/M | No | - | rs12480307, rs56157240 |
| I:2 | 44/F | Yes/BE | - | IVS3–24C > T | |
| II:1a | 29/M | Yes/LE | - | rs56157240 | |
| II:2 | 26/F | No | - | rs56157240, IVS3–24C > T | |
| KC–05 | I:1 | 60/M | No | - | rs6138482, rs56157240, IVS3–24C > T |
| I:2 | 47/M | Yes/LE | - | rs56157240, | |
| II:1 | 31/M | Yes/LE | - | IVS3–24C > T, rs6138482 | |
| II:2a | 27/F | Yes/LE | - | rs56157240, rs6138482 | |
| KC–06 | I:1 | 43/F | Yes/BE | - | rs12480307, rs56157240, |
| I:2 | 50/M | No | - | rs6138482 | |
| II:1 | 29/M | No | - | rs12480307, rs6138482 | |
| II:2a | 24/F | Yes/BE | - | rs56157240, rs6138482 | |
| KC–07 | I:1 | 53/F | Yes/BE | - | rs12480307, rs6138482, IVS3–24C > T |
| I:2 | 61/M | No | - | rs12480307, rs6138482, | |
| II:1a | 34/M | Yes/RE | - | rs6138482, IVS3–24C > T | |
| KC–08 | I:1 | 39/F | No | - | rs12480307, rs6138482, rs56157240 |
| I:2 | 45/M | Yes/RE | - | rs6138482, rs56157240 | |
| II:1a | 18/M | Yes/RE | - | rs12480307, rs56157240 | |
| II:2 | 16/F | Yes/RE | - | rs6138482, rs56157240 | |
RE: Right eye, LE: Left eye, BE: Both eye, M-Male, F-Female, The symbol (−) denotes the absence. Symbol (a) indicates the probands
Table 2.
Clinical features of affected individuals from KC families with VSX1 coding variants
| Family ID | BCVA | Scisorrs’ retinoscopic reflex | Location of corneal thinning | Corneal topography | Thinnest | Fleisher's ring | Apical corneal scarring | KC |
|---|---|---|---|---|---|---|---|---|
| Pachymetry | ||||||||
| (μm) | ||||||||
| KC–01_II:2 | 0.15/ 0.1 | + | RE: Central | RE, LE: Infero-superior, asymmetry Inferior cone, steepening corresponds with anterior, and posterior elevation. | RE: 447 | BE: + | RE: + | Both eyes |
| LE: Inferior | LE : 450 | |||||||
| KC–01_II:1 | 0/ 0 | + | RE: Central | RE: Infero-superior, asymmetry, Infero nasal cone. | RE: 482 | RE: + | - | RE: + |
| LE: Normal | LE: 493 | LE: Normal | ||||||
| LE: Normal | ||||||||
| KC–01_I:2 | 0.47/0 | + | RE: Infero temporal cone | RE: Inferior-Superior, asymmetry, Infero temporal cone, central cone with skewing of axis of 40° | RE: 404 | RE: + | - | RE: + |
| LE: 440 | LE: Normal | |||||||
| LE: Normal | ||||||||
| LE: Normal | ||||||||
| KC–02_II:1 | 0.15/0.15 | - | RE: Central | RE: Posterior elevation | RE: 490 | BE: + | - | Both eyes |
| LE: Central | LE: Central corresponds with thinnest pachymetry and posterior elevation | LE: 414 | ||||||
| KC–02_I:2 | 0/ 0 | - | RE: Normal | RE: Normal | RE: 506 | - | - | RE: Normal |
| LE: Infero- temporal | LE: Posterior elevation, | LE: 488 | ||||||
| Infero-temporal cone | LE: + | |||||||
| KC–03_II:2 | 0.47/ 0.15 | - | RE: Central | RE: Advanced KC, | RE: 333 | + | - | Both eyes |
| LE: Central | central cone with gross posterior elevation | LE: 443 | ||||||
| LE: Central cone | ||||||||
| KC–03_II:1 | 0/0 | RE: Central | RE: Posterior elevation, | RE: 526 | - | - | Both eyes | |
| LE: Inferior | Central cone LE: Infero nasal cone | LE: 532 | ||||||
| KC–03_I:2 | 0.15/0.15 | - | RE: Central | BE: Inferior cone, inferior- superior asymmetry with similar involvement in both eyes | RE: 419 | - | - | Both eyes |
| LE: Central | LE: 421 |
RE: Right eye, LE: Left eye, BE: Both eye, BCVA- Best corrected visual acuity, KC-Keratoconus, The symbols + and - represent present and absent, respectively. M-Male, F-Female
Mutation screening of VSX1 gene
Direct sequencing analysis of patients from KC families (KC–01_II: 2, KC–02_II:1) (Fig. 1A,B) showed a novel heterozygous c.803 T > A (p. Leu268His) change in exon 4 (Fig. 2A) of the VSX1 gene. In these families KC was observed in two generations suggesting an autosomal dominant inheritance. This missense substitution had co-segregated in five affected individuals (KC–01_II:1,I:2, KC–02_I:2) with the disease phenotype (Fig. 1A,B). Amino acid conservation analysis revealed that leucine at position 268 was highly conserved in nine vertebrate orthologs and other species (Fig. 2C). This nucleotide change was not present in the 105 normal controls and the unaffected family members.
Fig. 1.
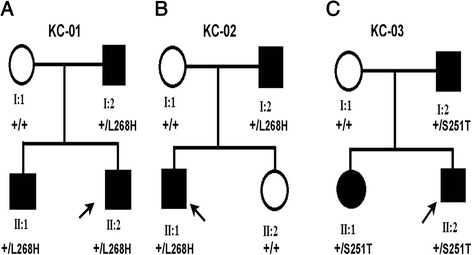
Pedigrees of the KC families with novel coding variants in VSX1 Legend: A, B, C denotes three unrelated families. Squares and circles indicate males and females, respectively. Black symbols indicate affected members and open symbols indicate unaffected individuals. The black arrow indicates the proband, the sign ‘+’ represents the wild type and the mutations identified are p. Leu268His, p. Ser251Thr
Fig. 2.
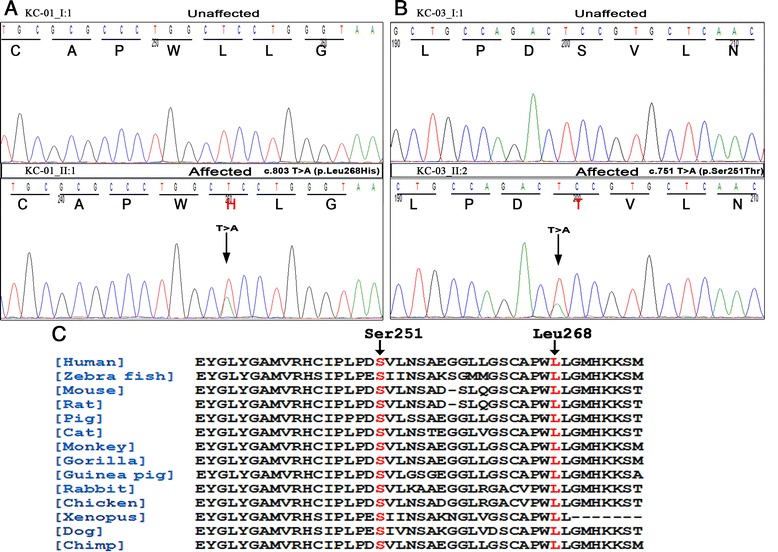
Novel VSX1 coding variants in KC families A, B: Comparison of DNA sequence chromatogram of an unaffected individual (top) with an affected (bottom). A: Patients DNA from KC–01, KC–02 revealed heterozygous T-to-A (c.803 T > A) transition (black arrow) in exon 4 of VSX1, resulting in a leucine 268 (CTC) to histidine (CAC) change (p. Leu268His). B: Affected individuals from family KC–03 shows a heterozygous T > A (c.751 T > A) nucleotide change in exon 4 of VSX1, which altered the serine 251 (TCC) to theronine (ACC) amino acid change (p. Ser251Thr). C: Amino acid sequence alignment of the human VSX1 protein (amino acids from 234–274) with other species. The Ser 251 and Leu 268 are shown in red
In another two generation KC family (KC–03) (Fig. 1C), mutation screening of VSX1 revealed a transition at exon 4 and c.751 T > A was found in three affected (II:2, II:1 I:2) individuals (Fig. 2B). The heterozygous T > A substitution at codon 251 (Ser251Thr) converts a highly conserved amino acid serine (TCC) into threonine (ACC). The unaffected mother (I:1) and 105 controls showed wild type alleles of VSX1. None of these identified missense variations have so far been reported in public databases, including NHLBI ESP (http://evs.gs.washington.edu/EVS/), dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and 1000 Genomes (http://www.1000genomes.org/).
Clinical findings of the three unrelated KC families
Family KC–01
There are three affected and one unaffected member (Fig. 1A). The proband (II:2) is a 20 year old male with a refraction of–0.5 D spherical and–2.25 D cylinder in his right eye;–1.25 D cylinder in his left eye. Corneal topography (Pentacam, Oculus Inc) revealed grade 1 KC in both eyes with an inferior cone with inferior-superior asymmetry. He had a thinnest pachymetry of 447 μm and 450 μm in his right and left eye respectively (Fig. 3A). He underwent corneal collagen crosslinking in both eyes and was stable at the end of the first year of follow up. The proband’s male sibling (II:1) had a refraction of–1 D spherical with–3 D cylinder in his right eye and–2 D spherical in the left eye. Topography mapping determined grade 1 KC with inferior-superior asymmetry and an inferonasal cone in the right eye with a mean keratometry of 43.6 D and a thinnest pachymetry of 482 μm. He had a normal corneal topography in the left eye. The right eye of the patient I:2 showed grade 1 KC. His refraction in the right eye was–4.25 D spherical with–4.5 D cylinder and in the left eye it was–2.75 D spherical with–3.25 D cylinder. His topography showed a central cone with inferior-superior asymmetry and skewing of 40° in his right eye and normal corneal topography in the left eye.
Fig. 3.

Corneal topography of the KC probands with VSX1 coding variants. A. Pentacam image of patient II:2 from KC–01, both eyes show an area of inferior steepening on the sagittal curvature map with gross inferior-superior asymmetry, more in the right eye. This area of steepening corresponds to areas of abnormal elevation on both the anterior and posterior elevation maps with values suggestive of Keratoconus (KC) with an inferior cone. B. In the second family (KC–02), patient II:1 has an area of central steepening on the sagittal curvature map. This area of steepening corresponds to areas of abnormal elevation on both the anterior and posterior elevation maps with values suggestive of KC with a central cone in the left eye (OS); right eye (OD) showing the posterior elevation, suggestive of early KC. C. The sagittal curvature on Scheimpflug imaging of patient II:2 from the third family (KC–03) with the left eye showing (OS) an area of central steepening. The anterior and posterior curvature maps show areas of abnormal elevation with values suggestive of KC with a central cone. The corneal thickness map also shows an area of central thinning which is corresponding to the areas of abnormal elevation. The right eye (OD) shows a fairly central area of steepening with features suggestive of advanced KC. There is gross posterior elevation with significant corneal thinning (thinnest pachymetry of 370 μm) in the central 3 mm zone
Family KC–02
Consists of four individuals with two having the characteristic features of KC (Fig. 1B). The patient (II:1) is a 16 year old male, with a posterior elevation on topography mapping in the right eye with a grade 2–3 KC in the left eye. Refraction in the right eye was –0.75 D sphere with–1.5 D cylinder and was–8 D sphere with–5 D cylinder in the left eye; mean keratometry was 45.3 D in the right and 53.3 D in the left eye. He had a corneal astigmatism of 2.5 D and 3.6 D and a thinnest pachymetry of 490 μm and 434 μm in the right and left eye respectively (Fig. 3B). The patient underwent corneal collagen crosslinking in the left eye and was stable at follow up two years later. The patient’s father (I:2) had 0.5 D Spherical with–1D cylinder in the right eye and plano refraction in the left eye. His topography evaluation showed normal corneal topography in the right eye while the left eye showed corneal thinning inferiorly, which corresponded with the posterior elevation. Grade 1 KC was noted with a mean keratometry of 43.3 D and pachymetry of 488 μm in the left eye.
Family KC–03
This family had one unaffected parent and three individuals with the clinical features of KC (Fig. 1C). The proband (II:2) is an 18 yr old male. Refraction in his right eye was–10 D sphere with–2.25 D cylinder and–3.75 D cylinder in the left eye. The right eye showed advanced KC with a fairly central cone while the left eye showed a grade 2 KC with a mean keratometry of 50.2 D and a thinnest pachymetry of 443 μm (Fig. 3C). He underwent corneal collagen crosslinking in both eyes and was stable on follow up. His sister (II:1) at initial presentation had a normal topography. At the second year follow up (20 years of age), the left eye showed progression to grade 1 keratoconus while the right eye remained stable. Both eyes of the patient I:2 exhibited grade 2 KC; his refraction in the right eye was–6 D spherical with a cylinder of–3.5 D and in the left eye, a sphere of–7.5 D with cylinder of–5 D. His topography scans in both eyes showed inferior steepening with a significant inferior-superior asymmetry.
In silico analysis of the VSX1 missense variants
In silico prediction algorithms of SIFT, Polyphen–2, Provean, and Mutation assessor suggested that the missense change p. Leu268His might negatively affect the function of the coding protein. On the other hand, p. Ser251Thr showed a neutral effect on the functional properties of the protein according to the prediction server results (Table 3). Analysis of amino acid mutation stability for p. Leu268His using the Amino Acid Mutation Stability Prediction Server showed a decrease in the stability of VSX1 protein structure.
Table 3.
The functional classification and score of VSX1 variants are predicted by using various bioinformatics tools
| c. DNA position | Protein change | Location of protein | Polyphen–2 humDiv | SIFT | Mutation assessor | PROVEAN |
|---|---|---|---|---|---|---|
| c.751 T > A | p. Ser251Thr | CVC domain | Benign (0.9) | Tolerated | Neutral | Neutral |
| (0.17) | (0.485) | –2.467 | ||||
| c.803 T > A | p. Leu268His | CVC domain | Possibly damaging for function (1.0) | Deleterious | Functional effect on protein | Deleterious |
| (0.05) | –6.831 | |||||
| (1.905) |
Prediction servers are Polymorphism Phenotyping v2 (PolyPhen–2), Sorting Tolerant From Intolerant (SIFT), Protein Variation Effect Analyzer (PROVEAN), CVC (Chx10/Vsx–1, and ceh–10)
Polyphen–2 scores: 0: benign, 1 possibly damaging for function; 2: Probably damaging for function
SIFT scores: Intolerant or deleterious: score ≤0.05, Tolerant: score >0.05
Mutation Assessor scores: 0–1: no functional effect, 2–3: functional effect on protein function
Provean scores: Cut off threshold = −2.5,
-Variants with a score equal to or below −2.5 are considered “deleterious,”-Variants with a score above −2.5 are considered “neutral
Haplotype analysis
We carried out haplotype analysis to examine whether the missense change, p. Leu268His, was due to a founder effect or was likely to have arisen independently in two unrelated families with KC. The haplotype of affected and unaffected individuals were constructed and compared between families using polymorphic SNPs (rs12480307, rs6138482, rs56157240), and (IVS3–24C > T) markers flanking the VSX1 gene (Table 4). The haplotype analysis showed that three SNP markers (rs12480307, rs6138482, rs56157240) were shared by five affected individuals (KC–01, KC–02), both families from an endogamous community. (Fig. 4A, B). The affected, and unaffected individuals from the third family (KC–03) showed a different haplotype (Fig. 4C).
Table 4.
Details of SNP markers used for the haplotype analysis
| db SNP ID | Physical position | VSX1 transcript ID | cDNA change | Protein change | Allele frequencya | Population frequencya |
|---|---|---|---|---|---|---|
| rs12480307 | chr20: 25078910 | NM_014588 | c.546A > G | p.A182A | A : 0.748 | A : 75 % |
| G : 0.252 | G : 25 % | |||||
| rs56157240 | chr20: 25078745 | NM_014588 | c.627 + 84 T > A | - | T: 0.252 | T: 25 % |
| A: 0.748 | A: 75 % | |||||
| rs6138482 | chr20: 25078806 | NM_014588 | c.627 + 23G > A | - | G: 0.735 | G: 74 % |
| A: 0.265 | A: 26 % | |||||
| (IVS3-24C) | chr20: 25078976 | NM_014588 | c.504-24C > T | - | C:0.999 | Not available |
| T:0.001 |
aAllele and population frequencies were determined by 1000 Genomes Project Phase 1, HapMap, and ESP for human
Fig. 4.
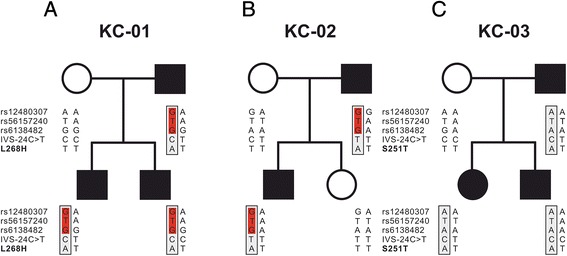
Pedigrees and haplotype analysis of VSX1 containing L268H and S251T variants in KC families. Legend: A,B,C The haplotype segregating with L268H and S251T are presented on white and gray backgrounds, respectively. Sharing of three common SNPs markers (KC–01,KC–02) families were shown in red
Discussion
Various mutation detection studies have reported that VSX1 coding variants are associated with KC and PPCD in different ethnic groups (Table 5) [16, 22, 24–26]. In this study, we screened 20 patients of eight unrelated families with KC for mutations in the VSX1 gene. Among these, five patients from two families had a novel coding variant (p. Leu268His) while another variant (p. Ser251Thr) was identified in a KC family with three affected individuals. The VSX1 consists of a paired-like homeodomain (HD) with a highly conserved CVC domain in the C-terminal, essential for the proliferation, and survival of retinal progenitor cells and bipolar interneurons [32]. The probands from the KC families (KC–01_II: 2, KC–02_II:1) had a heterozygous c.803 T > A nucleotide change associated with clinical features of bilateral KC with a typical sign of Fleisher’s ring. The leucine 268 amino acid residue was located in the C-terminal region of the CVC domain of VSX1 protein. So far only two mutations have been reported in this region in familial KC patients and PPCD [16, 24]. This amino acid change (p. Leu268His) in the CVC domain implied drastic modifications in the physicochemical properties, since leucine is a neutral non-polar amino acid while histidine is a basic-polar residue. It may cause abnormal protein folding that may affect DNA binding properties of the VSX1 transcriptional modulation activity. Moreover, this coding variant (p. Leu268His) leads to the replacement of highly conserved amino acid leucine by histidine in the VSX1 protein, perhaps implicating the functional consequence of this region. Interestingly, CVC domain change (p. His244Arg) has been associated with PPCD along with signs of bipolar cell dysfunction and macular degeneration [33]. According to SIFT, Polyphen-2, Provean, and Mutation assessor, the (p. Leu268His) mutation is found to have a deleterious effect on protein function, attributing a pathogenic nature to this missense mutation in VSX1. Furthermore, in our study, this potentially damaging mutation was detected in two families consisting of five affected individuals with a dominant inheritance of KC. This is consistent with previous findings that missense mutations (p. Arg166Trp, p. His244Arg) in VSX1 were identified in patients with dominant inheritance of KC phenotype [24]. Another study has demonstrated the coding variant p. Gln175His in the homeodomain of VSX1 in an Indian family of KC with incomplete penetrance [25].
Table 5.
Summary of VSX1 coding variants identified in patients with KC and PPCD
| Coding variants | Clinical significance | Phenotype | Unrelated | Ethnic groups | References |
|---|---|---|---|---|---|
| Controls | |||||
| p. Leu17Pro | Pathogenicb | KC | - | Italian | [26] |
| p. Leu17Val | Non-pathogenic | KC | + | Korean | [23] |
| p. Pro58Leu | Pathogenicbc | KC | - | Caucasian | [22] |
| p. Asp144Glu | Unknown | PPCD | - | Italian, Ashkenazi Jewish, British, European | [16, 26, 34–37] |
| p. Leu159Met | Unknown | KC | - | Caucasian | [16, 38] |
| p. Asn151Ser | Pathogenicb | KC | - | Korean | [39] |
| p. Gly160Asp | Non-pathogenic | PPCD | - | Italian, European | [16, 26, 40] |
| p. Gly160Val | Non-Pathogenic | KC | + | Korean | [23, 39] |
| p. Val199Leu | Non pathogenic | KC | + | Korean | [23] |
| p. Arg166Trp | Unknown | KC | + | Caucasian, Iranian | [16, 24] |
| p. Gln175His | Unknown | KC | - | Indian | [25] |
| p. Arg217His | Non- Pathogenic | KC | + | Indian, Pakistan, European | [40, 41] |
| p. Gly239Arg | Pathogenicbc | KC | - | Italian | [42] |
| p. His244Arg | Unknown | KC | + | Caucasian, Iranian | [16, 24, 38, 43] |
| p. Ser251Thr a | Unknown | KC | - | Indian | Present study |
| p. Pro247Arg | Non-pathogenic | KC | + | Italian | [16, 26, 35] |
| p. Leu268His a | Pathogenicbc | KC | - | Indian | Present study |
Coding variants of the VSX1 gene have been reported in presenta and other studies based on original reportb and bioinformatics predictionsc
KC: Keratoconus, PPCD: Posterior polymorphous corneal dystrophy, The symbols “ + ” and “-” represent present and absent, respectively
The probands from KC families showing the variable clinical phenotype, which has been observed in earlier studies as well [34]. On the other hand, it is difficult to establishing a genotype–phenotype correlation in the study subjects due to the presence of inter and even intra-familial clinical variability. Another novel c.751 T > A missense variant was identified in a proband (KC–03_II:2) who exhibited bilateral KC. It is interesting to note that this (p. Ser251Thr) coding variant introduces a missense change that leads to the replacement of highly conserved serine by threonine in the CVC domain of VSX1, probably highlighting the functional importance of this region of the protein. Though serine and threonine have similar properties, threonine is less polar than serine due to the presence of an extra non-polar methyl group. In this context, it may affect the interaction with neighbouring residues that may lead to improper polypeptide folding, thus affecting the protein’s wild-type function. Although this variant was absent in 105 normal controls, in silico studies suggest that p. Ser251Thr might be a benign or neutral variant that may not affect the protein function. At this stage, it is difficult to conclude about the pathogenic nature of variants p. Leu268His and p. Ser251Thr. While some previous studies have concluded that missense substitutions in the VSX1 may or may not be a disease-causing variant [16, 35], others have reported that the mutations were actually non-pathogenic or showed polymorphism [35]. Recent studies have shown that the absence of VSX1 mutations in a large number of unrelated KC patients suggesting a multiple gene involvement with environmental interaction playing a significant role in the pathogenesis of the disease [36, 37]. Haplotype analysis demonstrated a sharing of common SNPs around the missense change (p. Leu268His) in two unrelated KC families, suggesting the possibility of a founder effect, which requires further investigation. The disease causative variants identified in this study were compared to the reported literature of 1–3 % is high (2 probands out of 8, 25 %), probably due to the families belonging to an endogamous community and coincidental selection of study population with high VSX1 mutations. Further screening of the coding variant (p. Ser251Thr) on a large cohort of familial KC cases may reveal the exact pathological role of the VSX1 gene. In the eight families who were analysed for VSX1 mutation screening, we were able to identify a novel missense change (p. Leu268His) in two families and a variant of unknown significance (p. Ser251Thr) in a third family. Screening for other candidate genes including SOD1, ZEB1, TGFB1, FLG in KC families could determine the underlying genetic mechanism of the disease in VSX1 mutation negative patients.
Conclusions
In summary, we add one novel missense variation in the coding region of VSX1 to the existing repertoire of VSX1 coding variants observed in Indian patients with the characteristic phenotype of KC. Another variant p. Ser251Thr that was identified may be a benign polymorphism or a variant of unknown significance. Further biophysical studies are necessary to evaluate the precise molecular mechanism of VSX1 caused by this variant. The variation p. Leu268His may be involved in the pathogenesis of KC and therefore help in the genetic counselling of patients and their family.
Acknowledgements
We are grateful to the patients who participated in this study. The study was financially supported by Indian Council of Medical Research, New Delhi, India, and Naryana Nethralaya foundation, Bangalore, India. We would like to thank reviewers for their helpful comments on the manuscript.
Footnotes
Govindasamy Kumaramanickavel and Jeyabalan Nallathambi contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RS, RN, and SGN in recruitment of study subjects and collection of clinical details, VR carried out the molecular genetic studies. JN in research design, data analysis, and drafting of the manuscript. AG, GK, and CJ helped in the acquisition of genetic data and critical reading of the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Rohit Shetty, Email: drrohitshetty@yahoo.com.
Rudy M.M.A. Nuijts, Email: rudy.nuijts@mumc.nl
Soumya Ganesh Nanaiah, Email: soumya.kaveri@gmail.com.
Venkata Ramana Anandula, Email: ramanagene@gmail.com.
Arkasubhra Ghosh, Email: arkasubhra@narayananethralaya.com.
Chaitra Jayadev, Email: drchaitra@hotmail.com.
Natasha Pahuja, Email: dr.natashapahuja@gmail.com.
Govindasamy Kumaramanickavel, Email: gkumarmvel@gmail.com.
Jeyabalan Nallathambi, Email: drnallathambi@narayananethralaya.com.
References
- 1.Kang PC, Klintworth GK, Kim T, Carlson AN, Adelman R, Stinnett S, et al. Trends in the indications for penetrating keratoplasty, 1980–2001. Cornea. 2005;24(7):801–3. doi: 10.1097/01.ico.0000157407.43699.22. [DOI] [PubMed] [Google Scholar]
- 2.Cosar CB, Sridhar MS, Cohen EJ, Held EL, Alvim Pde T, Rapuano CJ, et al. Indications for penetrating keratoplasty and associated procedures, 1996–2000. Cornea. 2002;21(2):148–51. doi: 10.1097/00003226-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Zadnik K, Barr JT, Edrington TB, Everett DF, Jameson M, McMahon TT, et al. Baseline findings in the Collaborative Longitudinal Evaluation of Keratoconus (CLEK) Study. Invest Ophthalmol Vis Sci. 1998;39(13):2537–46. [PubMed] [Google Scholar]
- 4.Rabinowitz YS. Keratoconus. Surv Ophthalmol. 1998;42(4):297–319. doi: 10.1016/S0039-6257(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 5.Goldich Y, Marcovich AL, Barkana Y, Mandel Y, Hirsh A, Morad Y, et al. Clinical and corneal biomechanical changes after collagen cross-linking with riboflavin and UV irradiation in patients with progressive keratoconus: results after 2 years of follow-up. Cornea. 2012;31(6):609–14. doi: 10.1097/ICO.0b013e318226bf4a. [DOI] [PubMed] [Google Scholar]
- 6.Jafri B, Lichter H, Stulting RD. Asymmetric keratoconus attributed to eye rubbing. Cornea. 2004;23(6):560–4. doi: 10.1097/01.ico.0000121711.58571.8d. [DOI] [PubMed] [Google Scholar]
- 7.Edwards M, McGhee CN, Dean S. The genetics of keratoconus. Clin Experiment Ophthalmol. 2001;29(6):345–51. doi: 10.1046/j.1442-9071.2001.d01-16.x. [DOI] [PubMed] [Google Scholar]
- 8.Parker J, Ko WW, Pavlopoulos G, Wolfe PJ, Rabinowitz YS, Feldman ST. Videokeratography of keratoconus in monozygotic twins. J Refract Surg. 1996;12(1):180–3. doi: 10.3928/1081-597X-19960101-31. [DOI] [PubMed] [Google Scholar]
- 9.Falls HFAA. Dominantly inherited keratoconus: report of a family. J Genet Hum. 1969;17:317–24. [PubMed] [Google Scholar]
- 10.Hamilton JB. The significance of heredity in ophthalmology preliminary survey of hereditary eye diseases in Tasmania. Br J Ophthalmol. 1938;22(2):83–108. doi: 10.1136/bjo.22.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler JHM, Afshari NA, Allingham RR, Liu Y. The genetics of Keratoconus: a review. Reproductive Sys Sexual Disord. 2012;6:001. doi: 10.4172/2161-038X.S6-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdon KP, Coster DJ, Charlesworth JC, Mills RA, Laurie KJ, Giunta C, et al. Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum Genet. 2008;124(4):379–86. doi: 10.1007/s00439-008-0555-z. [DOI] [PubMed] [Google Scholar]
- 13.Hutchings H, Ginisty H, Le Gallo M, Levy D, Stoesser F, Rouland JF, et al. Identification of a new locus for isolated familial keratoconus at 2p24. J Med Genet. 2005;42(1):88–94. doi: 10.1136/jmg.2004.022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brancati F, Valente EM, Sarkozy A, Feher J, Castori M, Del Duca P, et al. A locus for autosomal dominant keratoconus maps to human chromosome 3p14–q13. J Med Genet. 2004;41(3):188–92. doi: 10.1136/jmg.2003.012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hameed A, Khaliq S, Ismail M, Anwar K, Ebenezer ND, Jordan T, et al. A novel locus for Leber congenital amaurosis (LCA4) with anterior keratoconus mapping to chromosome 17p13. Invest Ophthalmol Vis Sci. 2000;41(3):629–33. [PubMed] [Google Scholar]
- 16.Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G, et al. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet. 2002;11(9):1029–36. doi: 10.1093/hmg/11.9.1029. [DOI] [PubMed] [Google Scholar]
- 17.Czugala M, Karolak JA, Nowak DM, Polakowski P, Pitarque J, Molinari A, et al. Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur J Hum Genet. 2012;20(4):389–97. doi: 10.1038/ejhg.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan T, Liu C, Ma Z, Ding S. The point mutation and polymorphism in keratoconus candidate gene TGFBI in Chinese population. Gene. 2012;503(1):137–9. doi: 10.1016/j.gene.2012.04.061. [DOI] [PubMed] [Google Scholar]
- 19.Udar N, Atilano SR, Brown DJ, Holguin B, Small K, Nesburn AB, et al. SOD1: a candidate gene for keratoconus. Invest Ophthalmol Vis Sci. 2006;47(8):3345–51. doi: 10.1167/iovs.05-1500. [DOI] [PubMed] [Google Scholar]
- 20.Droitcourt C, Touboul D, Ged C, Ezzedine K, Cario-Andre M, de Verneuil H, et al. A prospective study of filaggrin null mutations in keratoconus patients with or without atopic disorders. Dermatology. 2011;222(4):336–41. doi: 10.1159/000328408. [DOI] [PubMed] [Google Scholar]
- 21.Muszynska DLJ, Dash D, Heon E, Hughes A, Willoughby C. Identification and characterization of a novel missense homeodomain mutation in ZEB1 resulting in keratoconus. Invest Ophthalmol Vis Sci. 2011;52:1077. [Google Scholar]
- 22.Vincent AL, Jordan C, Sheck L, Niederer R, Patel DV, McGhee CN. Screening the visual system homeobox 1 gene in keratoconus and posterior polymorphous dystrophy cohorts identifies a novel variant. Mol Vis. 2013;19:852–60. [PMC free article] [PubMed] [Google Scholar]
- 23.Jeoung JW, Kim MK, Park SS, Kim SY, Ko HS, Wee WR, et al. VSX1 gene and keratoconus: genetic analysis in Korean patients. Cornea. 2012;31(7):746–50. doi: 10.1097/ICO.0b013e3181e16dd0. [DOI] [PubMed] [Google Scholar]
- 24.Saee-Rad S, Hashemi H, Miraftab M, Noori-Daloii MR, Chaleshtori MH, Raoofian R, et al. Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol Vis. 2011;17:3128–36. [PMC free article] [PubMed] [Google Scholar]
- 25.Paliwal P, Tandon R, Dube D, Kaur P, Sharma A. Familial segregation of a VSX1 mutation adds a new dimension to its role in the causation of keratoconus. Mol Vis. 2011;17:481–5. [PMC free article] [PubMed] [Google Scholar]
- 26.Bisceglia L, Ciaschetti M, De Bonis P, Campo PA, Pizzicoli C, Scala C, et al. VSX1 mutational analysis in a series of Italian patients affected by keratoconus: detection of a novel mutation. Invest Ophthalmol Vis Sci. 2005;46(1):39–45. doi: 10.1167/iovs.04-0533. [DOI] [PubMed] [Google Scholar]
- 27.Semina EV, Mintz-Hittner HA, Murray JC. Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics. 2000;63(2):289–93. doi: 10.1006/geno.1999.6093. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi T, Huang J, Deeb SS. RINX (VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics. 2000;67(2):128–39. doi: 10.1006/geno.2000.6248. [DOI] [PubMed] [Google Scholar]
- 29.Krumeich JH, Daniel J, Knulle A. Live-epikeratophakia for keratoconus. J Cataract Refract Surg. 1998;24(4):456–63. doi: 10.1016/S0886-3350(98)80284-8. [DOI] [PubMed] [Google Scholar]
- 30.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174(2):247–50. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 32.Chow RL, Volgyi B, Szilard RK, Ng D, McKerlie C, Bloomfield SA, et al. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc Natl Acad Sci U S A. 2004;101(6):1754–9. doi: 10.1073/pnas.0306520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valleix S, Nedelec B, Rigaudiere F, Dighiero P, Pouliquen Y, Renard G, et al. H244R VSX1 is associated with selective cone ON bipolar cell dysfunction and macular degeneration in a PPCD family. Invest Ophthalmol Vis Sci. 2006;47(1):48–54. doi: 10.1167/iovs.05-0479. [DOI] [PubMed] [Google Scholar]
- 34.Paliwal P, Singh A, Tandon R, Titiyal JS, Sharma A. A novel VSX1 mutation identified in an individual with keratoconus in India. Mol Vis. 2009;15:2475–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Eran P, Almogit A, David Z, Wolf HR, Hana G, Yaniv B, et al. The D144E substitution in the VSX1 gene: a non-pathogenic variant or a disease causing mutation? Ophthalmic Genet. 2008;29(2):53–9. doi: 10.1080/13816810802008242. [DOI] [PubMed] [Google Scholar]
- 36.Aldave AJ, Yellore VS, Salem AK, Yoo GL, Rayner SA, Yang H, et al. No VSX1 gene mutations associated with keratoconus. Invest Ophthalmol Vis Sci. 2006;47(7):2820–2. doi: 10.1167/iovs.05-1530. [DOI] [PubMed] [Google Scholar]
- 37.Verma A, Das M, Srinivasan M, Prajna NV, Sundaresan P. Investigation of VSX1 sequence variants in South Indian patients with sporadic cases of keratoconus. BMC Res Notes. 2013;6:103. doi: 10.1186/1756-0500-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang YG, Picornell Y, Su X, Li X, Yang H, Rabinowitz YS. Three VSX1 gene mutations, L159M, R166W, and H244R, are not associated with keratoconus. Cornea. 2008;27(2):189–92. doi: 10.1097/ICO.0b013e31815a50e7. [DOI] [PubMed] [Google Scholar]
- 39.Mok JW, Baek SJ, Joo CK. VSX1 gene variants are associated with keratoconus in unrelated Korean patients. J Hum Genet. 2008;53(9):842–9. doi: 10.1007/s10038-008-0319-6. [DOI] [PubMed] [Google Scholar]
- 40.Dash DP, George S, O’Prey D, Burns D, Nabili S, Donnelly U, et al. Mutational screening of VSX1 in keratoconus patients from the European population. Eye (Lond) 2010;24(6):1085–92. doi: 10.1038/eye.2009.217. [DOI] [PubMed] [Google Scholar]
- 41.Tanwar M, Kumar M, Nayak B, Pathak D, Sharma N, Titiyal JS, et al. VSX1 gene analysis in keratoconus. Mol Vis. 2010;16:2395–401. [PMC free article] [PubMed] [Google Scholar]
- 42.De Bonis P, Laborante A, Pizzicoli C, Stallone R, Barbano R, Longo C, et al. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol Vis. 2011;17:2482–94. [PMC free article] [PubMed] [Google Scholar]
- 43.Dehkordi FA, Rashki A, Bagheri N, Chaleshtori MH, Memarzadeh E, Salehi A, et al. Study of VSX1 mutations in patients with keratoconus in southwest Iran using PCR-single-strand conformation polymorphism/heteroduplex analysis and sequencing method. Acta Cytol. 2013;57(6):646–51. doi: 10.1159/000353297. [DOI] [PubMed] [Google Scholar]