

Abstract
Background
Chromosome instability syndromes are a group of inherited conditions associated with chromosomal instability and breakage, often leading to immunodeficiency, growth retardation and increased risk of malignancy.
Case presentation
We performed exome sequencing on a girl with a suspected chromosome instability syndrome that manifested as growth retardation, microcephaly, developmental delay, dysmorphic features, poikiloderma, immune deficiency with pancytopenia, and myelodysplasia. She was homozygous for a previously reported splice variant, c.4444 + 3A > G in the POLE1 gene, which encodes the catalytic subunit of DNA polymerase E.
Conclusion
This is the second family with POLE1-deficency, with the affected individual demonstrating a more severe phenotype than previously described.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0177-y) contains supplementary material, which is available to authorized users.
Keywords: POLE1, FILS syndrome, Immunodeficiency, Dysmorphism, Primordial dwarfism
Background
Chromosome instability syndromes are a group of inherited conditions associated with chromosomal instability and breakage which includes LIG4 [1,2], Seckel type 1 [3,4], Bloom syndrome [5], Nijmegen breakage syndrome [6-8], and Fanconi anemia. These genetic conditions are characterized by pre and postnatal growth retardation, microcephaly, dysmorphic features and bone marrow failure [9-11]. There are other conditions with overlapping phenotypes including microcephaly, such as Rad50 deficiency [12,13], Cernunnos-XLF syndrome [14] and Warsaw breakage syndrome [15-18] which can complicate molecular diagnosis. FILS syndrome (facial dysmorphism, immunodeficiency, livedo, and short stature) is a recently described condition caused by variants in POLE1, encoding the catalytic subunit of polymerase E. We describe the second family with a homozygous variant in POLE1, and a more severely affected individual, suggesting a broader phenotypic spectrum for this condition.
Case presentation
Patient CMH812 is a female infant born to healthy non consanguineous Palestinian parents, weighing 1745 g and measuring 38.1 cm at birth. The pregnancy was complicated by subchorionic bleeding in the first trimester, fetal abnormalities on ultrasound including intrauterine growth restriction, short long bones, suspected skull abnormalities and oligohydramnios. TORCH titers were negative. Amniocentesis revealed normal 46,XX karyotype. She was delivered at 37 weeks gestation by elective C-section secondary to breech presentation. Dysmorphic features noted included malar and mandibular hypoplasia (Figure 1A, B). Initial clinical suspicion was for primordial dwarfism such as Seckel type 1 syndrome, however her microcephaly was not as severe. Over several months, lacy reticular pigmentation was noted of the face and extremities. She had recurrent pruritic papular eruptions and skin findings progressed to include appearance of poikiloderma (Figure 1C, D). Erupted teeth were found to be small and dysplastic. She developed a feeding aversion necessitating a gastrostomy tube. Growth remained poor postnatally (Figure 2). Her motor milestones were delayed but social development was normal.
Figure 1.
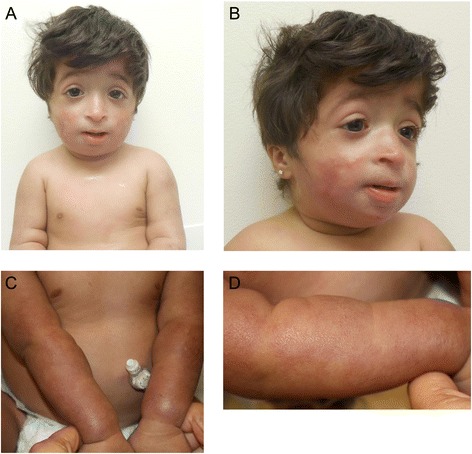
Clinical characteristics of CMH812. Several congenital abnormalities were found, including (A) microcephaly, facial dysmorphism (small and bird-like face, malar and mandibular hypoplasia, prominent nasal bridge and columella, downslanting palpebral fissures, small mouth and low set, posteriorly rotated ears) (B) short stature with shortened long bones but no evidence of dysplasia or craniosynostosis. No major anomalies were found on imaging of her abdomen, brain, brain vasculature or heart. Genitalia and pubertal development were normal. No malabsorption or pituitary or thyroid insufficiency was found. (C-D) Skin findings in CMH812, showing one hypo- and three hyperpigmented patches on the skin. Biopsy of the skin was performed but not diagnostic. Microscopic examination displayed focal parakeratosis and mild spongiosis.
Figure 2.
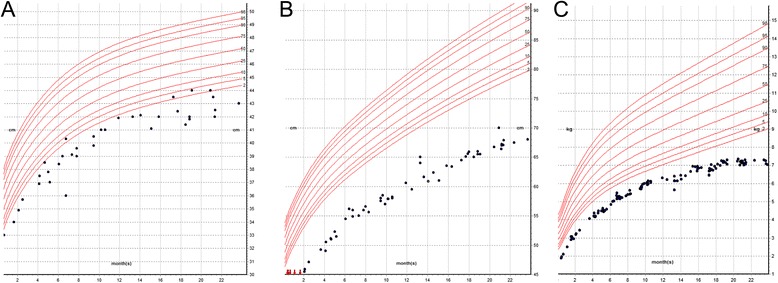
Growth charts of CMH812. The patient is a female with severe intrauterine and postnatal growth retardation head circumference (A) [<3rd centile]; weight (B) [<2nd centile], length (C) [<2nd centile], based on CDC growth chart for girls, age 0–20 months. Growth remained poor postnatally and she was briefly started on growth hormone, which was discontinued due to suspicion for a disorder of DNA repair/instability. At age 15 months she was ~5 SD below the mean for weight and length and ~3-4 SD below the mean for head circumference.
She suffered chronic rhinosinusitis and pulmonary infections with purulent otitis media. At age 20 months she was admitted to the hospital with pancytopenia, splenomegaly, hepatitis and acute CMV infection. Laboratory data showed mild bone marrow myelodysplasia, normal total B, T, and NK cells, low class switched and non switched memory B cells, and high memory T cells. She had high IgA, normal total IgG and low IgM, IgG2 and IgG4. There was no serologic response to pneumococcal vaccine (Table 1). Lymphocyte response to mitogens was normal but absent to pertussis and candida antigens [11]. Hepatitis and pancytopenia resolved following the CMV infection. Extensive molecular and cytogenetic testing was negative, including array-CGH, telomere length studies, chromosome breakage, and gene sequencing with deletion/duplication analysis for the following genes: ATM, ATR, BLM, CENPJ, CEP152, CEP63, ERCC6, ERCC8, LIG4, LMNA, MRE11A, NBN, PCNT, RBBP8, RECQL4, SHOX, WRN, and ZMPSTE24.
Table 1.
Comparison of clinical features of POLE1 patients and other chromosome instability syndromes
| Clinical features | TAR syndrome | Ataxia telangiectasia | RAD50 deficiency | Fanconi anemia | Seckel syndrome | NBS | RS/SC syndrome | Warsaw syndrome | Cernnos-XLF syndrome | Bloom syndrome | LIG4 syndrome2 | FILS syndrome* | CMH812 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIM # | 274000 | 208900 | 613078 | 227650 | 210600 | 251260 | 268300 /269000 | 613398 | 611291 | 210900 | 606593 | 615139 | 615139 |
| Microcephaly | − | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν |
| Malar hypoplasia | − | n.a | ν | − | n.a | n.a | ν | ν | − | ν | n.a | ν | ν |
| Sloping head | ν | − | n.a | − | ν | ν | ν | ν | ν | − | ν | ν | ν |
| Palpebral fissures, upslanting | − | − | n.a | − | − | ν | − | ν | − | − | ν | ν | − |
| Palpebral fissures, down-slanting | − | − | ν | − | ν | − | ν | ν | ν | − | − | ν | ν |
| Epicanthic folds | − | − | ν | − | − | ν | ν | ν | ν | − | ν | ν | ν |
| Micrognathia | ν | − | n.a | − | ν | ν | ν | ν | ν | − | − | ν | − |
| External ear abnormalities | − | − | n.a | ν | ν | ν | ν | ν | ν | ν | − | ν | ν |
| Long/Large nose | ν | − | ν | − | − | ν | ν | ν | ν | ν | ν | ν | − |
| Long philtrum | − | − | n.a | − | − | ν | ν | ν | ν | − | − | ν | ν |
| Cleft palate/high arched palate | − | − | n.a | − | ν | ν | ν | ν | − | − | − | − | − |
| Polydactily | − | − | n.a | ν | − | ν | − | − | − | ν | − | − | − |
| Clinodactily | − | − | n.a | − | ν | ν | ν | ν | − | ν | ν | − | ν |
| Syndactily | |||||||||||||
| Growth retardation | − | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν |
| Short stature | ν | ν | n.a | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν |
| Bone disease or anomalies | ν | − | n.a | ν | ν | − | ν | n.a | ν | − | − | ν | ν |
| Thumb apasia/radial ray anormalities | ν | − | n.a | ν | ν | − | −/ν | n.a | − | − | ν | − | |
| Skin abnormalities | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν |
| Mental retardation | ν | ν | ν | ν | ν | ν | ν | ν | ν | − | ν | − | − |
| Developemental delay | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | − | ν |
| Malignancy | ν | ν | − | ν | ν | ν | ν | n.a | ν | ν | ν | − | − |
| Recurrent infections | − | ν | − | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν |
| Genital abnormalities | − | − | − | ν | ν | ν | ν | n.a | − | − | ν | − | − |
| Ataxia | − | ν | ν | − | ν | n.a | − | n.a | − | n.a | n.a | − | − |
| Brain anomalie/degeneration | CA, CH, aCC, S | CA, CH,T, S | ND | ν | CA, CH, PG, S | ND | S | n.a | PG | n.a | n.a | n.a | − |
| Endocrine | − | AZ, D, DP, HH | − | HH | D, DP | POF | n.a | n.a | − | AZ, D RF | − | − | − |
| Immunologic Features | |||||||||||||
| Pancytopenia | − | n.a | n.a | ν | ν | − | n.a | n.a | ν | − | ν | − | ν |
| Thrombocytopenia | ν | n.a | − | ν | − | − | n.a | n.a | − | − | − | − | ν |
| CID | − | ν | − | − | n.a | − | n.a | n.a | ν | n.a | ν | 2/14 | ν |
| SCID | − | ν | − | − | ν | − | n.a | n.a | ν | n.a | ν | n.a | - |
| Neutropenia | − | n.a | n.a | ν | n.a | ν | n.a | n.a | ν | N | ν | n.a | -* |
| B cell lymphocytopenia | − | ν | N | ν | − | − | n.a | n.a | ν | − | 2/17 | ν | ν |
| T cell lymphocytopenia | − | ν | N | ν | ν | ν | n.a | n.a | ν | ν | N | ν | ν |
| IgA | n.a | ↓ | N | N | ↓ | n.a | n.a | n.a | ↓ | ↓ | ↓ | N | ↑ |
| IgE | n.a | ↓ | N | n.a | ↓ | n.a | n.a | n.a | n.a | N | n.a | N | N |
| IgG | n.a | ↓ | N | ↓ | ↓ | n.a | n.a | n.a | ↓ | ↓ | n.a | ↓ | ↓** |
| IgM | n.a | ↓ | N | ↓ | ↓ | n.a | n.a | n.a | ↓ | ↓ | ↓ | ↓ | ↓ |
| Anti-pseudomonae | n.a | n.a | N | n.a | ↓ | n.a | n.a | n.a | ν | n.a | n.a | ν | ν |
| polysaccaride IgG | |||||||||||||
| Auto-immunity | n.a | ν | − | n.a | ↓ | ν | n.a | n.a | 1/17 | n.a | 0/17 | − | - |
| Sister chromatide | n.a | − | n.a | n.a | ↑ or N | ν | ν | ν | n.a | ν | n.a | N | N |
| DNA breakage studies | n.a | ν | ν | ν | ν | ν | ν | ν | ν | ν | n.a | − | N |
| Radiosensitivity | n.a | ν | ν | ν | ν | ν | ν | ν | ν | ν | ν | − | n.a. |
| Gene | RBM8A | ATM | RAD50 | FANC | ATR | NBS1 | ESCO2 | DDX11 | NHEJ1 | BLM | LIG4 | POLE1 | POLE1 |
| Mode of Inheritance | AR | AR | AR | AR | AR | AR | AR | AR | AR | AR | AR | AR | AR |
Legend
n.a; not reported/applicable.
−; negative.
ν; positive.
N; normal range.
↓; decreased.
↑; increased.
NBS: Nijmegen breakage syndrome.
TAR: Thrombocytopenia-absent radius syndrome.
RS: Roberts syndrome.
SC: SC phocomelia syndrome.
aCC; absence of corpus collosum.
AR; autosomal recessive.
AZ; azoospermia.
CA; cerebellar ataxia.
CH; cerebellar hypoplasia.
CID; combined immunodeficiency.
D; diabetes.
DP; delayed puberty.
HH; hypergonadotropic hypogonadism.
NBS; Nijmegen breakage syndrome.
ND; neurodegenerative.
OA; oculomotor apraxia.
PG; polygyria.
POF; primary ovarian failure.
RF; reduced fertility.
S; seizures.
SCID; severe combined immunodeficiency.
T; Tremor.
*consanguineous family reported.
£ Ataxia-telangiectasia-like disorder.
- * Transient pancytopenia associated with CMV infection.
↓** IgG2 and IgG 4 ↓. Total IgG N.
Trio-exome sequencing was performed on CMH812 and her healthy parents (CMH813 & CMH814) following informed consent, and with methods as previously published [19-22]. Variants were filtered to 1% minor allele frequency, then prioritized by the American College of Medical Genetics (ACMG) categorization [23,24], OMIM identity and phenotypic assessment. This individual was homozygous for a splice-site variant, c.4444 + 3A > G, in intron 34 of the POLE1 [11]. The parents were both heterozygous carriers (Additional file 1: Table S1). Homozygosity mapper was used to identify intervals of homozygosity and identity by descent segments (Additional file 2: Figure S1) [25].
The c.4444 + 3A > G variant was previously reported in consanguineous French family with three generations of affected members [11]. FILS phenotype was variable but included macrocephaly, recurrent respiratory infections, livedo and telangiectasia, bone dysplasia, short stature, and decreased IgM and IgG. The phenotype was considered similar to that of Bloom syndrome but with normal sister chromatid exchange. Table 1 compares the clinical and cellular features of CMH812 to those of individuals with inherited chromosomal instability and breakage syndromes, as well as the first reported FILS family. Features closely matched those reported in FILS with exceptions of microcephaly and intrauterine growth restriction. Although the POLE1 variant identified in the present case is the same as previously reported, CMH812 seems to have had more significantly impaired growth and immunity, raising the hypothesis that rare variant(s) in other POLƐ subunits or MMR genes may act as phenotypic modifiers. However, no rare variant were detected in MMR genes, POLE1 interacting proteins or other DNA breakage/instability syndrome genes.
The c.4444 + 3A > G POLE1 variant confers abnormal splicing whereby exon 34 is deleted [11] leading to significant decrease in the POLE1 subunit [11]. T- lymphocytes from affected individuals showed a proliferation defect as well as impaired cell cycle progression. The primary function of polymerase Ɛ1 is to synthesize DNA at the leading strand during replication [26,27], however, it is also involved in other cellular processes, including cell cycle progression and DNA repair/recombination [26,27]. Exonucleolytic proofreading and the MMR pathway act to maintain high-fidelity DNA replication and to protect against mutagenesis [28]. Somatic and germline heterozygous missense variants in POLƐ1 have been associated with an increased cancer risk [28-32]. Functional studies in yeast showed that heterozygosity for a pathogenic allele can cause complete MMR deficiency, and that subsequent loss of heterozygosity is not required for the development of POLE-related tumors [28]. Taken together, these findings suggest that POLE1 carriers are likely to be at increased risk for malignancy due to MMR deficiency.
Conclusions
In summary, we report a second family with POLE1-related disease. The clinical and immunologic features of our patient are reminiscent of LIG4 syndrome, possibly representing the more severe end of an ill-defined clinical spectrum. For this reason, POLE1 deficiency may be a more apt description of this disorder. This report illustrates the cost-effectiveness of trio-exome sequencing as a powerful diagnostic method considering that this family underwent an extensive diagnostic odyssey, with no molecular basis identified prior exome.
Consent statement
The project was approved by the research ethics committee of the Children’s Mercy Hospitals. Written informed consent was obtained from the patient’s legal guardians for publication of this case report. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgments
We are grateful to the patient and her family. We thank the other staff of the Center for Pediatric Genomic Medicine, Children’s Mercy Kansas City, and Dr. Geneviève de Saint Basile and her team for discussions. This work was supported by the Marion Merrell Dow Foundation, Children’s Mercy - Kansas City, Patton Trust, W.T. Kemper Foundation, Pat & Gil Clements Foundation, Claire Giannini Foundation, and Black & Veatch.
Bioinformatics
The accession numbers for the reference sequences are listed as follows: POLE1 NM_006231.2:,HGMD 290829429, ClinVarRCV000034317.25
1000 Genomes Project, http://www.1000genomes.org/
NHBLI Exome Sequencing Project (ESP) Exome Variant Server,
http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SNPdb, http://www.ncbi.nlm.nih.gov/projects/SNP
UCSC Genome Browser, http://genome.ucsc.edu/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
ClinSeq, http://genome.gov/20519355
RefSeq, http://www-ncbi-nlm-nih-gov.proxy2.library.mcgill.ca/RefSeq
SIFT, sift.jcvi.org/
Abbreviations
- ACMG
American College of Medical Genetics
- aCGH
Array of comparative genomic hybridization
- DNA
Deoxyribonucleic acid
- FILS
Facial dysmorphism immunodeficiency livedo and short stature
- IUGR
Intrauterine growth restriction
- LIG4
DNA ligase IV
- Mb
Megabase pair
- MMR
DNA mismatch repair
- NK
Natural Killer
- OMIM
Online Mendelian Inheritance in Man
- NICU
Neonatal intensive-care unit
- PCR
Polymerase chain reaction
- POLE1
DNA polymerase E1
- TAR
Thrombocytopenia absent radii syndrome
- TORCH
Toxoplasmosis, other infections, rubella, cytomegalovirus (CMV), and herpes simplex virus (HSV). The “other infections” usually include syphilis, hepatitis B, coxsackie virus, Epstein-Barr virus, varicella-zoster virus, and human parvovirus
- SD
Standard deviation
- SNP
Single nucleotide polymorphism
- XLF
XRCC4-like factor
Additional files
Trio-Exome sequencing data.
Homozygosity Mapping Analysis. Homozygosity mapper was used to identify intervals of homozygosity. The homozygosity scores are plotted against the physical position. Red and black bars represent the excess (red) or the shortage (black) of homozygosity (A) Eighteen loci of > 1 Mb of homozygosity were identified on seven chromosomes; loci ranged between 1.06 and 6.37 Mb in length. The length of identical by descent segments in genomes of CMH812 parents suggests that they are not closely related. However, they shared several relatively small identical segments of genome which is explained by the shared ancestry broken into pieces by the recombination events and Mendelian laws of inheritance. POLE1 lies in a 1.1 Mb region of homozygosity on chromosome 12 (chr12:132635257–133702440), which includes 20 genes (B). It is reasonable to hypothesize that the POLE1 variant, if not the result of a combination of recent origin and chance, owes its origin to a founder who lived several hundred years ago. Consistent with the rarity of the c.4444 + 3A > G variant and the small physical distances between each haplotype marker in relationship to POLE1 gene (1.1 Mb); the size of the shared haplotype may have been broken into smaller segments due to genetic drift. This assumption could explain that the same variant (c.4444 + 3A > G) has been found in two apparently unrelated FILS families.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The authors have made the following declarations about their contributions: Conceived and designed the experiments: CJS, EF, IT, GT. Performed the experiments: CJS, EF, IT, NK, NM, LZ. Contributed reagents/materials/analysis tools: NM, EF, DZ, GT. Wrote the paper: IT, CJS, NPS, JJ. Reviewed the manuscript: DZ, EF, KC, MS, HW, NM, NR, SFK. Contributed to the recruitment and clinical investigations of the patient for the study: DZ, HW, KC, JJ, MS, NK, NPS, NS. All authors read and approved the final manuscript.
Contributor Information
Isabelle Thiffault, Email: ithiffault@cmh.edu.
Carol Saunders, Email: csaunders@cmh.edu.
Janda Jenkins, Email: jljenkins@cmh.edu.
Nikita Raje, Email: nraje@cmh.edu.
Kristi Canty, Email: kmcanty@cmh.edu.
Mukta Sharma, Email: msharma@cmh.edu.
Lauren Grote, Email: lgrote@cmh.edu.
Holly I Welsh, Email: hwelsh@cmh.edu.
Emily Farrow, Email: egfarrow@cmh.edu.
Greyson Twist, Email: gtwist@cmh.edu.
Neil Miller, Email: nmiller@cmh.edu.
David Zwick, Email: dzwick58@gmail.com.
Lee Zellmer, Email: lazellmer@cmh.edu.
Stephen F Kingsmore, Email: sfkingsmore@cmh.edu.
Nicole P Safina, Email: nsafina@cmh.edu.
References
- 1.Rucci F, Notarangelo LD, Fazeli A, Patrizi L, Hickernell T, Paganini T, et al. Homozygous DNA ligase IV R278H mutation in mice leads to leaky SCID and represents a model for human LIG4 syndrome. Proc Natl Acad Sci U S A. 2010;107(7):3024–9. doi: 10.1073/pnas.0914865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8(6):1175–85. doi: 10.1016/S1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 3.Ogi T, Walker S, Stiff T, Hobson E, Limsirichaikul S, Carpenter G, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet. 2012;8(11) doi: 10.1371/journal.pgen.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33(4):497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 5.Dubowitz V. Familial low birthweight dwarfism with an unusual facies and a skin eruption. J Med Genet. 1965;2(1):12–7. doi: 10.1136/jmg.2.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curry CJ, O’Lague P, Tsai J, Hutchison HT, Jaspers NG, Wara D, et al. ATFresno: a phenotype linking ataxia-telangiectasia with the Nijmegen breakage syndrome. Am J Hum Genet. 1989;45(2):270–5. [PMC free article] [PubMed] [Google Scholar]
- 7.Cerosaletti KM, Lange E, Stringham HM, Weemaes CM, Smeets D, Solder B, et al. Fine localization of the Nijmegen breakage syndrome gene to 8q21: evidence for a common founder haplotype. Am J Hum Genet. 1998;63(1):125–34. doi: 10.1086/301927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93(3):477–86. doi: 10.1016/S0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am J Med Genet A. 2005;137A(3):283–7. doi: 10.1002/ajmg.a.30869. [DOI] [PubMed] [Google Scholar]
- 10.Gruhn B, Seidel J, Zintl F, Varon R, Tonnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. 2007;2:5. doi: 10.1186/1750-1172-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pachlopnik Schmid J, Lemoine R, Nehme N, Cormier-Daire V, Revy P, Debeurme F, et al. Polymerase epsilon1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”) J Exp Med. 2012;209(13):2323–30. doi: 10.1084/jem.20121303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Med Genet A. 2009;84(5):605–16. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577–87. doi: 10.1016/S0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 14.Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–99. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Sommers JA, Khan I, de Winter JP, Brosh RM., Jr Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem. 2012;287(2):1007–21. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Lelij P, Oostra AB, Rooimans MA, Joenje H, de Winter JP. Diagnostic overlap between fanconi anemia and the cohesinopathies: Roberts syndrome and Warsaw breakage syndrome. Anemia. 2010;2010:565268. doi: 10.1155/2010/565268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Med Genet A. 2010;86(2):262–6. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capo-Chichi JM, Bharti SK, Sommers JA, Yammine T, Chouery E, Patry L, et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum Mutat. 2013;34(1):103–7. doi: 10.1002/humu.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soden S, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of acuity – guided exome and genome sequencing for etiologic diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014. In press. [DOI] [PMC free article] [PubMed]
- 20.Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;65):65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10(4):294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 24.ACMG . Points to Consider in the Clinical Application of Genomic Sequencing. In: Genetics ACoM, editor. American College of Medical Genetics 14 edn. Bethesda, MD: American College of Medical Genetics; 2012. pp. 759–61. [DOI] [PubMed] [Google Scholar]
- 25.Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue)):W593-599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang D, Pospiech H, Kesti T, Syvaoja JE. Structural organization and splice variants of the POLE1 gene encoding the catalytic subunit of human DNA polymerase epsilon. Biochem J. 1999;339(Pt 3):657–65. doi: 10.1042/0264-6021:3390657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang D, Knuuti R, Palosaari H, Pospiech H, Syvaoja JE. cDNA and structural organization of the gene Pole1 for the mouse DNA polymerase epsilon catalytic subunit. Biochim Biophys Acta. 1999;1445(3):363–71. doi: 10.1016/S0167-4781(99)00058-5. [DOI] [PubMed] [Google Scholar]
- 28.Kane DP, Shcherbakova PV. A common cancer-associated DNA polymerase epsilon mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014;74(7):1895–901. doi: 10.1158/0008-5472.CAN-13-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–44. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230(2):148–53. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22(14):2820–8. doi: 10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou Y, Liu FY, Liu H, Wang F, Li W, Huang MZ, et al. Frequent POLE1 p.S297F mutation in Chinese patients with ovarian endometrioid carcinoma. Mutat Res. 2014;761:49–52. doi: 10.1016/j.mrfmmm.2014.01.003. [DOI] [PubMed] [Google Scholar]