

Abstract
Rapid biosynthesis is key to the success of bacteria and viruses. Highly expressed genes in bacteria exhibit a strong codon bias corresponding to the differential availability of tRNAs. However, a large clade of lambdoid coliphages exhibits relatively poor codon adaptation to the host translation machinery, in contrast to other coliphages that exhibit strong codon adaptation to the host. Three possible explanations were previously proposed but dismissed: (1) the phage-borne tRNA genes that reduce the dependence of phage translation on host tRNAs, (2) lack of time needed for evolving codon adaptation due to recent host switching, and (3) strong strand asymmetry with biased mutation disrupting codon adaptation. Here, we examined the possibility that phages with relatively poor codon adaptation have poor translation initiation which would weaken the selection on codon adaptation. We measured translation initiation by: (1) the strength and position of the Shine–Dalgarno (SD) sequence, and (2) the stability of the secondary structure of sequences flanking the SD and start codon known to affect accessibility of the SD sequence and start codon. Phage genes with strong codon adaptation had significantly stronger SD sequences than those with poor codon adaptation. The former also had significantly weaker secondary structure in sequences flanking the SD sequence and start codon than the latter. Thus, lambdoid phages do not exhibit strong codon adaptation because they have relatively inefficient translation initiation and would benefit little from increased elongation efficiency. We also provided evidence suggesting that phage lifestyle (virulent versus temperate) affected selection intensity on the efficiency of translation initiation and elongation.
Introduction
Bacterial species and viruses need to replicate themselves rapidly in order to successfully compete against others. Translation is a key limiting factor in biosynthesis and microbial species typically evolve features to improve translation efficiency. Codon usage in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae strongly depends on the availability of their cognizant tRNA species (Ikemura, 1981a, b, 1982, 1992; Xia, 1998), especially in highly expressed genes (Coghlan & Wolfe, 2000; Comeron & Aguadé, 1998; Duret & Mouchiroud, 1999; Xia, 2007). Similarly, codon usage in bacteriophages (phages) is strongly shaped by the tRNA pool of their host (Chithambaram et al., 2014a, b). Experimental modification to improve or disrupt codon adaptation generally leads to a predictable change in the protein production rate (Haas et al., 1996; Ngumbela et al., 2008; Robinson et al., 1984; Sørensen et al., 1989). In fact, gene-specific codon usage indices (Sharp & Li, 1987; Sun et al., 2013; Wright, 1990; Xia, 2007) are excellent predictors of translation efficiency (Coghlan & Wolfe, 2000).
In this context it is puzzling that a large cluster of 16 E. coli lambdoid phages (Clade B in Fig. 1), consisting of 10 siphophages, four podophages and two myophages, exhibits poor codon adaptation in Y-ending codons in their protein-coding genes, whereas eight E. coli podophages in Clade A (Fig. 1) uniformly exhibit strong codon adaptation (Chithambaram et al., 2014b). The same pattern remains if one measures codon adaptation by using the Codon Adaptation Index (CAI) (Sharp & Li, 1987) or its improved version (Xia, 2007) when E. coli highly expressed genes are used as a reference set, or by the index of translation elongation (ITE) that takes into account the effect of background mutation bias (Xia, 2014). Thus, genes in Clade B phages have significantly weaker codon adaptation than those in Clade A phages.
Fig. 1. Partial phylogenetic tree showing two clades of phages (A and B), with Clade A exhibiting stronger codon adaptation to host E. coli than Clade B. Modified from Chithambaram et al. (2014b).

Three possible explanations for poor codon adaptation in Clade B phages to the host tRNA pool have been proposed but dismissed on the basis of empirical evidence (Chithambaram et al., 2014a, b). The first invokes the differential presence of phage genome-encoded tRNA genes, which vary from zero to 20 in different E. coli phages (Chithambaram et al., 2014a). A large number of phage-encoded tRNA genes would reduce the dependence of phage codon decoding on host tRNAs and allow the phage codon usage to deviate from host codon usage (Limor-Waisberg et al., 2011; Prabhakaran et al., 2014). Indeed, the degree of codon adaptation decreases with increasing number of phage-encoded tRNA genes (Chithambaram et al., 2014a). It was also reported that selective enrichment of host tRNA by human immunodeficiency virus type 1 can also decrease the likelihood of the virus acquiring a codon usage similar to the host (van Weringh et al., 2011). However, the difference in phage-encoded tRNA genes is minimal between the two clades in Fig. 1. Five Clade B phages (enterobacteria phages 933W, Min27, VT2-Sakai, and Stx2-converting phages II and 86) have three phage-encoded tRNA genes and one Clade B phage (enterobacteria phage ϕP27) has two phage-encoded tRNA genes. All other Clade B phages, as well as all Clade A phages, do not have phage-encoded tRNA genes. Those six Clade B phages carrying two or three tRNA genes do not have codon adaptation better or worse than other Clade B phages based on a t-test of the ITE (Xia, 2014) between the two groups [t = 0.0879, degrees of freedom (d.f.) = 14, P = 0.9312, two-tailed test].
The second explanation attributes poor codon adaptation to lack of evolutionary time if phages have recently switched hosts. Take, for example, E. coli phage PRD1 which exhibits poor codon adaptation to the host (Xia, 2014). As the closest relatives of phage PRD1 all parasitize Gram-positive bacteria that have different codon usage from that of E. coli, phage PRD1 most likely has only recently switched to E. coli and consequently has had little time to evolve codon adaptation to the new E. coli host. However, this explanation is also inapplicable to the differential codon adaptation between Clade A and Clade B phages because both have diverse lineages parasitizing E. coli and should have evolved in the E. coli host for a long time. An associated possibility is that Clade B phages may have a more diverse host range than Clade A. If Clade B hosts happen to have diverse codon usage, then good codon adaptation to one host would mean poor codon adaptation to other hosts. Thus, parasitizing different hosts with different codon usage would interfere with codon adaptation to one particular host such as E. coli. However, we were not able to find conclusive evidence that Clade B phages have a more diverse host range than Clade A phages. Both clades can parasitize hosts such as Yersinia pestis, Salmonella enterica and Klebsiella pneumonia, in addition to E. coli. Furthermore, highly expressed protein-coding genes in all these four host species have almost identical codon usage. Thus, switching among these hosts should not interfere with phage codon adaptation to E. coli.
The third explanation invokes strand asymmetry and associated mutation bias often observed in circular microbial and mitochondrial genomes (Marín & Xia, 2008; Xia, 2012a, c). Highly expressed E. coli genes prefer CCU over CCC codons and UUC over UUU codons. However, phage CCY codons are mainly found in C-rich segments of the phage genome with over-represented CCC codons that are not preferred by E. coli highly expressed genes. Similarly, UUY codons are mainly found in T-rich genomic segments of the phage genome with over-represented UUU codons that are not preferred by E. coli highly expressed genes. However, whilst this explanation works well for ssDNA phages (Chithambaram et al., 2014b), it does not seem sufficient to explain the poor codon adaptation in the dsDNA phages in Clade B relative to those in Clade A (Fig. 1).
Here, we proposed a hypothesis invoking differential translation initiation between the two clades of phages, based on the recent recognition that codon adaptation depends on translation initiation efficiency (Supek & Šmuc, 2010; Tuller et al., 2010; Xia, 2014; Xia et al., 2007). If translation initiation is highly efficient, then translation elongation will become rate-limiting and the selection for increasing translation efficiency will drive codon adaptation. If translation initiation is not efficient, then the selection for increasing translation efficiency will not reach codon usage because elongation is not rate-limiting. Thus, if translation initiation is more efficient in Clade A phages than in Clade B phages, then the selection for translation elongation efficiency will be stronger on Clade A phages than on Clade B phages, leading to differential codon adaptation.
To test the hypothesis that Clade A and Clade B phages have different translation initiation efficiencies, we need to measure the translation initiation efficiency. In bacterial species, translation initiation efficiency depends strongly on three factors: (1) the nature of the start codon (Hartz et al., 1991; Ma et al., 2002; O’Donnell & Janssen, 2001; Osterman et al., 2013; Ringquist et al., 1992), (2) the base-pairing potential and position of the Shine–Dalgarno (SD) sequence (de Smit & van Duin, 1994; Hui & de Boer, 1987; Olsthoorn et al., 1995; Osterman et al., 2013; Shine & Dalgarno, 1974), and (3) the stability of the secondary structure of sequences flanking the SD sequence and start codon (de Smit & van Duin, 1990, 1994; Milón et al., 2012; Milón & Rodnina, 2012; Nivinskas et al., 1999; Osterman et al., 2013), with higher translation initiation generally associated with weaker secondary structure. dsDNA phages are known to have reduced secondary structure near the start codon (Zhou & Wilke, 2011), presumably to avoid having the SD sequence and start codon embedded in the secondary structure.
Pairing between the SD sequence and anti-SD (aSD) sequence on the small subunit ribosomal RNA is important for start codon localization (Hui & de Boer, 1987; Vimberg et al., 2007), although such pairing is not always essential in translating E. coli messages (Fargo et al., 1998; Melançon et al., 1990) or in Chlamydomonas reinhardtii chloroplasts (Fargo et al., 1998). Some leaderless genes with an AUG start codon can be translated efficiently in E. coli (Giliberti et al., 2012; Krishnan et al., 2010; O’Donnell & Janssen, 2002; Vesper et al., 2011) or in the halophilic archaeon Halobacterium salinarum (Sartorius-Neef & Pfeifer, 2004). However, translation initiation of most E. coli genes appears to benefit from a well-positioned SD sequence, especially genes that follow the first gene in a multigene operon (Osterman et al., 2013). In general, the effects of the SD sequence and the stability of the secondary structure flanking the SD sequence and start codon have become so well established that they serve as key design principles for computational tools optimizing translation initiation, such as RBSdesigner (Na & Lee, 2010), RBScalculator (Salis, 2011) and UTRdesigner (Seo et al., 2013). As protein-coding genes in both Clade A and Clade B phages use AUG as the start codon, we tested the difference in the second and third factors between the two groups of phages. We predict that Clade A phage genes had stronger well-positioned SD sequences than those in Clade B phages and that Clade A phage genes also had weaker secondary structure in sequences flanking the SD and start codon than those in Clade B phages. These predictions were strongly supported by our empirical analysis of host and phage genomic sequences.
Given that Clade A phages exhibit better adaptation in translation initiation and elongation than Clade B phages, one would naturally ask if the former is under stronger selection for translation efficiency than the latter. One relevant observation is that all eight phage species in Clade A were virulent and all 16 phage species in Clade B were temperate (with a lysogenic phase). In contrast to virulent phages that are almost always engaged in translation once they enter the host cell, protein-coding genes in a prophage are not under any purifying selection and the evolutionary success of temperate phages does not necessarily rely on rapid biosynthesis. Thus, selection for more efficient translation may be stronger in the virulent phages than in the temperate phages, leading to more efficient translation initiation and better codon adaptation in the virulent (Clade A) phages than the temperate (Clade B) phages. This hypothesis is consistent with coliphages. To test its generality, we analysed phages infecting the Gram-positive Staphylococcus aureus, which has more sequenced phage genomes than any other Gram-positive bacterial species. The lifestyle hypothesis was consistent with the empirical evidence.
Results
Our first objective was to explain why Clade A phages exhibited better codon adaptation to the E. coli host than Clade B phages and our hypothesis was that translation initiation was more efficient in the former than the latter so that codon adaptation would increase the protein production rate more in the former than in the latter. Our specific predictions were that: (1) the proportion of SD-containing genes (PSD) is higher in Clade A phages than in Clade B phages, (2) the length of SD–aSD pairing [mean number of consecutively matched sites (MSD)] should be closer to the optimal in Clade A phages than in Clade B phages, with the optimal SD length being 6 nt (Komarova et al., 2002; Schurr et al., 1993; Vimberg et al., 2007), and (3) the minimum folding energy (MFE) in 40 nt upstream of the start codon (MFE40nt) and the MFE at sites from four sites upstream of the start codon to 37 sites downstream of the start codon (MFE−4+37) are less negative in sequences flanking the start codon in Clade A phages than in Clade B phages.
Comparison of SD sequence features between Clade A and Clade B phages
PSD was highly significantly greater in the eight Clade A phages (mean PSD = 94.20 %) than in the 16 Clade B phages (mean PSD = 68.27 %), as we had predicted (Table 1, t-test assuming unequal variances: t = 10.9900, d.f. = 21, P<0.0001, two-tailed test). We used the t-test with unequal variances because the two variances are significantly different from each other according to an F-test (F = 8.2400, d.f.numerator = 15, d.f.denominator = 7, P = 0.0045). However, a regular t-test assuming equal variance also strongly rejected the null hypothesis of equal PSD between Clade A and Clade B phages (t = 8.3340, d.f. = 22, P<0.0001, two-tailed test).
Table 1. SD sequence features (PSD and MSD) in Clade A and Clade B phages.
| Phage | GenBank accession no. | No. of coding sequences | PSD | MSD |
| Clade A | ||||
| T7 | NC_001604 | 60 | 96.670 | 5.879 |
| T3 | NC_003298 | 55 | 90.910 | 6.020 |
| K1F | NC_007456 | 43 | 88.370 | 5.921 |
| K1E | NC_007637 | 62 | 95.160 | 5.763 |
| K1-5 | NC_008152 | 52 | 96.150 | 5.600 |
| BA14 | NC_011040 | 52 | 94.230 | 5.878 |
| EcoDS1 | NC_011042 | 53 | 94.340 | 6.140 |
| 13 a | NC_011045 | 55 | 96.360 | 5.943 |
| Clade B | ||||
| VT2-Sakai | NC_000902 | 83 | 62.650 | 5.000 |
| 933W | NC_000924 | 80 | 70.000 | 5.036 |
| λ | NC_001416 | 73 | 69.860 | 4.922 |
| N15 | NC_001901 | 60 | 80.000 | 5.000 |
| HK022 | NC_002166 | 57 | 56.140 | 4.875 |
| HK97 | NC_002167 | 61 | 68.850 | 5.024 |
| ϕP27 | NC_003356 | 58 | 67.240 | 5.359 |
| SFV | NC_003444 | 53 | 69.810 | 4.676 |
| Stx2-I | NC_003525 | 166 | 47.590 | 4.873 |
| BP-4795 | NC_004813 | 85 | 63.530 | 5.019 |
| Stx1-phage | NC_004913 | 84 | 80.950 | 5.162 |
| Stx2-II | NC_004914 | 89 | 75.280 | 5.239 |
| Stx2-86 | NC_008464 | 81 | 74.070 | 5.150 |
| cdtI | NC_009514 | 60 | 71.670 | 4.977 |
| Min27 | NC_010237 | 83 | 71.080 | 4.915 |
| Stx2-1717 | NC_011357 | 77 | 63.640 | 5.082 |
MSD was smaller than the optimal 6 nt (Schurr et al., 1993; Vimberg et al., 2007) for both Clade A and Clade B phages, which simplifies our statistical analysis. That is, we only need to test whether MSD is significantly greater in Clade A phages than in Clade B phages, which is equivalent to testing which mean MSD is closer to the optimal MSD. The mean MSD was greater for the eight Clade A phages (5.8930) than the 16 Clade B phages (5.0190), the difference being statistically highly significant (t-test assuming equal variance, t = 12.5160, d.f. = 22, P<0.0001, two-tailed test). The variance in MSD was nearly identical between the two groups. In short, both PSD and MSD supported our hypothesis that translation initiation was more efficient in Clade A phages than in Clade B phages.
Comparison of secondary structure stability between Clade A and Clade B phages
The secondary structure formed from the 40 bases upstream of the start codon may bury the SD sequence and consequently interfere with the SD–aSD pairing. Our hypothesis predicted that Clade A phages should have a weaker secondary structure (less negative MFE40nt) than Clade B phages. The empirical evidence strongly supported this prediction (Table 2), with MFE40nt significantly weaker in Clade A phages (mean MFE40nt = −5.1770) than in Clade B phages (mean MFE40nt = −6.4610, t = 6.7879, d.f. = 22, P<0.0001, two-tailed test).
Table 2. Secondary structure stability (MFE40nt and MFE−4+37) in Clade A and Clade B phages.
| Phage | GenBank accession no. | No. of coding sequences | MFE40nt | MFE−4+37 |
| Clade A | ||||
| 13 a | NC_011045 | 55 | −5.3735 | −4.7076 |
| EcoDS1 | NC_011042 | 53 | −5.5532 | −5.6291 |
| K1-5 | NC_008152 | 52 | −5.2631 | −3.8735 |
| K1E | NC_007637 | 62 | −4.8619 | −4.1960 |
| K1F | NC_007456 | 43 | −5.8679 | −6.5579 |
| T3 | NC_003298 | 55 | −5.1473 | −4.6391 |
| T7 | NC_001604 | 60 | −5.0047 | −4.2622 |
| BA14 | NC_011040 | 52 | −4.3469 | −4.2873 |
| Clade B | ||||
| ϕP27 | NC_003356 | 58 | −5.7564 | −5.2038 |
| SFV | NC_003444 | 53 | −6.5808 | −6.0811 |
| 933W | NC_000924 | 80 | −6.2833 | −6.4413 |
| Min27 | NC_010237 | 83 | −6.4707 | −6.0010 |
| VT2-Sakai | NC_000902 | 83 | −5.9863 | −6.2927 |
| Stx2-I | NC_003525 | 166 | −6.4645 | −6.6228 |
| BP-4795 | NC_004813 | 85 | −7.0353 | −5.9579 |
| cdtI | NC_009514 | 60 | −7.0232 | −6.1000 |
| HK022 | NC_002166 | 57 | −6.2793 | −4.6179 |
| HK97 | NC_002167 | 61 | −6.6669 | −4.7044 |
| λ | NC_001416 | 73 | −6.9789 | −5.7668 |
| N15 | NC_001901 | 60 | −7.0329 | −5.6890 |
| Stx1-phage | NC_004913 | 84 | −6.0289 | −5.6139 |
| Stx2-II | NC_004914 | 89 | −6.3666 | −5.9200 |
| Stx2-1717 | NC_011357 | 77 | −6.6052 | −5.8212 |
| Stx2-86 | NC_008464 | 81 | −5.8188 | −5.5885 |
The secondary structure formed around the start codon may interfere with the accessibility of the start codon, which is crucially important for translation initiation in bacterial species (Nakamoto, 2006). Our hypothesis predicted that Clade A phages should have less negative MFE−4+37 (weaker secondary structure) at this region than Clade B phages, which is again supported by the empirical evidence (Table 2). The mean MFE−4+37 was −4.7690 for the eight Clade A phages and −5.7760 for the 16 Clade B phages, the difference being statistically significant ( t = 3.4170, d.f. = 22, P = 0.0025, two-tailed test). Thus, the differences in secondary structure stability between Clade A and Clade B phages were also consistent with the interpretation that genes in Clade A phages have more efficient translation initiation than those in Clade B phages.
Relationship between SD features and secondary structure stability
If efficient translation initiation is evolutionary beneficial, then both SD features (PSD and MSD) and the MFE of sequences flanking the start codon will be subject to the same selection and consequently are expected to have correlated changes. That is, a gene that requires high translation initiation efficiency is expected to have both high PSD and MSD, and weak MFE40nt and MFE−4+37. However, if there is an optimal rate of translation with a rate too high or too low being not as good, then an increase in PSD and MSD may result in selection increasing the stability of the secondary structure to maintain the optimal rate. In that case, we may observe a negative correlation between SD features (PSD and MSD) and structural features (MFE40nt and MFE−4+37).
We observed a highly significant positive correlation between PSD and two secondary structure features (MFE40nt and MFE−4+37, Fig. 2). A strong positive correlation was also observed for MSD and the two MFE measures (Fig. 3). This suggested selection operating to maximize translation initiation efficiency in phages instead of stabilizing it at one particular level.
Fig. 2. A high PSD is associated with weak secondary structure around the SD sequence and start codon measured by MFE in E. coli phages at two locations: (a) 40 bases upstream of the start codon (MFE40nt) and (b) from four bases upstream to 37 bases downstream of the start codon (MFE−4+37).

Fig. 3. A large MSD (strong SD with more base pairs with aSD) is associated with weak secondary structure around the SD sequence and start codon measured by MFE in E. coli phages at two locations: (a) 40 bases upstream of the initiation codon (MFE40nt) and (b) from four bases upstream to 37 bases downstream of the initiation codon (MFE−4+37).

As some phage species share a common ancestry, a more appropriate characterization of the relationship between SD features (PSD and MSD) and secondary structure features (MFE40nt and MFE−4+37) should be carried out with the method of phylogeny-based independent contrasts (Felsenstein, 1985). We performed the contrasts using dambe (Xia, 2013b),which implemented the method with extensions (Xia, 2013a) and the tree from a previous study (Chithambaram et al., 2014b). The four positive associations (Figs 2 and 3) were still significant (P<0.05).
Virulent phages exhibit better translation adaptation than temperate phages
Our second objective was to understand why genes in Clade A phages exhibited better adaptation in translation initiation and elongation than those in Clade B phages. All eight coliphages in Clade A were virulent and all 16 coliphages in Clade B were temperate, suggesting phage lifestyle as a contributing factor. Virulent phages are almost always engaged in translation once they enter the host cell. In contrast, protein-coding genes in a prophage are under little purifying selection and the evolutionary success of temperate phages may not necessarily rely on rapid biosynthesis. Thus, selection for more efficient translation may be stronger in the virulent phages than in temperate phages, leading to more efficient translation initiation and better codon adaptation in virulent phages (Clade A) than temperate phages (Clade B). This hypothesis was consistent with coliphages, not only for phages in Clades A and B, but also for the other 28 phages in fig. 8 of Chithambaram et al. (2014b) not included in Clades A and B. To further test the generality of this lifestyle hypothesis, we analysed 35 phages (six virulent and 29 temperate) infecting the Gram-positive Staphylococcus aureus.
There are notable differences between the two host species that are relevant to understanding phage adaptation. First, S. aureus genes tend to have SD sequences more often than E. coli genes. If we operationally define a SD sequence as a sequence (1) of at least 4 nt long, (2) located within 30 nt upstream of the initiation AUG and (3) having perfect base-pairing with the last 13 nt at the 3′ end of 16S rRNA, then PSD is significantly higher (χ2 = 136.69, d.f. = 1, P<0.0001) in the 2767 S. aureus genes (0.9256) than in the 4321 E. coli genes (0.8287). Second, the secondary structure of the sequences flanking SD and start codon is significantly weaker in S. aureus genes than in E. coli genes, with mean MFE40nt equal to −2.9000 for S. aureus and −5.5543 for E. coli (t = 35.8074, d.f. = 7086, P<0.0001), and mean MFE−4+37 equal to −3.1422 for S. aureus and −4.9700 for E. coli (t = 28.0094, d.f. = 7086, P<0.0001). This suggested that the translation machinery in S. aureus had a more stringent requirement for the 5′ end of mRNA than E. coli.
The Staphylococcus phages, both virulent and temperate, had high PSD values similar to their host, but did not differ either in PSD or MSD between the six virulent and the 29 temperate phages. However, there were significant differences in MFE40nt, MFE−4+37 and codon adaptation measured by ITE between virulent and temperate Staphylococcus phages in the predicted direction. The secondary structure flanking the SD sequence was weaker in virulent Staphylococcus phages than in temperate phages, with mean MFE40nt being −2.9494 in the former and −3.3560 in the latter (t = 3.0723, d.f. = 33, P = 0.0042, two-tailed test). Similar differences were observed in sequences flanking the start codon, with mean MFE−4+37 being −2.5402 in the former and −2.8792 in the latter (t = 2.6714, d.f. = 33, P = 0.0116, two-tailed test).
For measuring codon adaptation, we used ITE which has the advantage over CAI in that ITE incorporates background mutation bias (Xia, 2014). ITE is computed by four different methods in dambe (Xia, 2013b) that differ in treating synonymous codon families, and we use the second method that breaks compound synonymous codon families (e.g. sixfold Arg, Ser and Leu for the standard code) into two separate four- and twofold codon families. ITE was significantly greater in the virulent Staphylococcus phages than in the temperate phages, with the mean ITE being 0.5065 for the former and 0.4899 for the latter (t = 11.1861, d.f. = 33, P<0.0001, two-tailed test). As shown in Fig. 4, high ITE values of the coding sequences in virulent Staphylococcus phages were associated with weak secondary structure at the 5′ end of mRNA, measured by MFE40nt and MFE−4+37.
Fig. 4. Virulent Staphylococcus phages (♦) have genes with relatively weaker secondary structure in sequences flanking the SD sequence and start codon as well as better codon adaptation to the host than temperate phages (⋄). (a) MFE40nt. (b) MFE−4+37.

Discussion
Evolution of codon usage and translation elongation efficiency has recently been recognized to depend on translation initiation efficiency (Supek & Šmuc, 2010; Tuller et al., 2010; Xia, 2014; Xia et al., 2007). In short, an mRNA with low translation initiation efficiency is not expected to increase protein production with optimized codon usage. In contrast, protein production for an mRNA with high translation initiation efficiency may become limited by translation elongation and such an mRNA can increase protein production with optimized codon usage. This implies little selection for codon optimization for genes with low translation initiation efficiency, but strong selection for codon optimization for genes with high translation initiation efficiency.
We have extended this hypothesis to explain why two clades of E. coli phages differ greatly in codon adaptation to their hosts (Chithambaram et al., 2014b). In particular, why Clade A phages exhibit stronger codon adaptation than Clade B phages. Our hypothesis that genes in Clade A phages have higher translation initiation efficiency than those in Clade B phages is highly consistent with our empirical results (Tables 1 and 2), with the Clade A phages having both a higher PSD and MSD than the Clade B phages. Higher PSD has also been observed in highly expressed genes than lowly expressed genes in E. coli (Ma et al., 2002).
Our finding of a positive correlation between strong SD sequences and weak secondary structure (Figs 2 and 3) suggests that natural selection may operate simultaneously to optimize these features to increase translation initiation efficiency. One may suggest that the presence of a SD sequence, which is typically purine-rich, may itself result in a change in the two MFE measures, so that the positive correlations in Figs 3 and 4 have little to do with simultaneous selection on both SD features and secondary structure features. This suggestion is not true. If we replace a 6mer in the sequences flanking the start codon by a typical SD sequence such as AGGAGG, the resulting MFE40nt and MFE−4+37 may increase or decrease, but overall do not become significantly weaker. Thus, the presence of a stronger SD sequence in the Clade A phage genes cannot explain its weaker MFE40nt and MFE−4+37.
Given the seemingly obvious benefit of efficient translation, one would naturally ask what has prevented the Clade B phages from acquiring more efficient translation initiation, i.e. higher PSD and MSD and weaker MFE40nt and MFE−4+37. As we have mentioned earlier, both clades have evolved and diverged into multiple lineages in the E. coli host, so lack of evolutionary time may not be the right answer.
Our results are consistent with the lifestyle hypothesis. That is, selection for more efficient translation may be stronger in the virulent phages than in the temperate phages, leading to more efficient translation initiation and better codon adaptation in the virulent (Clade A) phages than the temperate (Clade B) phages. Whilst this interpretation of phage lifestyle on translation is consistent with the coliphages, we have shown the interpretation is also consistent with the Staphylococcus phages (six virulent and 29 temperate phages). The secondary structure of sequences flanking the SD sequence and start codon is significantly weaker in the virulent than in the temperate Staphylococcus phages (Fig. 4). The ITE is also highly significantly greater in the virulent than in the temperate Staphylococcus phages (Fig. 4).
A previous study (Chithambaram et al., 2014a) used correlation in relative synonymous codon usage (rRSCU) between phage and host as a measure of phage codon adaptation, and found temperate phages to have higher rRSCU values than virulent phages. This seems to contradict the conclusion that Clade A phages (all being virulent) exhibit better codon adaptation than Clade B phages (all being temperate). However, rRSCU, like the effective number of codons (Sun et al., 2013; Wright, 1990), is strongly affected by mutation bias when RSCU for the host is computed from all E. coli genes. When host highly expressed genes are used for computing RSCU, the difference in rRSCU between the virulent and temperate phages becomes smaller. If one uses wi (equation 2 in Xia, 2014) from the host as host RSCU (wi is essentially RSCU corrected for mutation bias), then rRSCU is significantly greater for the virulent phages than for the temperate phages.
Another relevant observation is that all eight phage species in Clade A have all their genes on the same DNA strand and all 16 phage species in Clade B have their genes distributed on both DNA strands. Strong strand asymmetry can affect both synonymous and non-synonymous substitutions (Chithambaram et al., 2014b; Marín & Xia, 2008; Xia, 2012b, c). If two DNA strands have dramatically different mutation bias, then mutation bias in one strand that is in the same direction as codon adaptation is necessarily accompanied by mutation bias in the other strand going against codon adaptation.
Methods
Genomic data.
The genomes of E. coli and S. aureus, as well as their phages, were retrieved from GenBank. Coding sequences were extracted and their codon usage analyzed by dambe (Xia, 2013b). Only coding sequences with at least 33 codons were included to alleviate stochastic fluctuations of codon usage. All phage genomes were scanned for tRNAs by using the tRNAscan-SE Search Server (Schattner et al., 2005). Phage data compilation consisting of clade, phage name, phage family, phage GenBank accession number, phage genome length, number of coding sequences in each phage genome, ITE and the number of tRNA genes encoded in each phage genome are included in Table S1 (available in the online Supplementary Material).
Identification of SD sequences.
We also extracted 30 nt upstream of the start codon (Upstream30) from each gene in phage and host genomes, and the last 20 nt of the E. coli small subunit (SSU) rRNA using dambe (Xia, 2013b), to identify SD sequences. As we show below, it is not appropriate to define the SD sequence simply as an AGGAGG motif within a fixed distance range upstream of the start codon. The SD sequence on the mRNA and the aSD sequence on the SSU rRNA pair to position the anticodon of the initiation tRNA at the start codon (Fig. 5a). The optimal location of the SD sequence in the literature is often measured by the distance from the SD sequence to the start codon (e.g. D1 and D2 in Fig. 5a) or from the middle of the SD sequence to the start codon (Osterman et al., 2013). However, this approach is probably incorrect as illustrated in Fig. 5(a). Both SD1 and SD2 position the tRNA anticodon properly at the start codon AUG, but their associated D1 and D2 are different (Fig. 5a). A correct distance measure should take into consideration the relative position of both mRNA and the rRNA 3′ tail. One such distance is the distance from the end of the SSU rRNA to the beginning of the start codon (DtoAUG, Fig. 5a).
Fig. 5. (a) Schematic representation of the SD sequence on mRNA pairing with the aSD sequence on the SSU rRNA. (b–d) The free 3′ end of SSU rRNA (b), the frequency distribution of 4577 putative matches of at least four bases between the rRNA 3′ tail and the upstream 30 nt of coding sequences (c), and the number of times each nucleotide site at the rRNA 3′ tail participated in the SD–aSD matches (d).
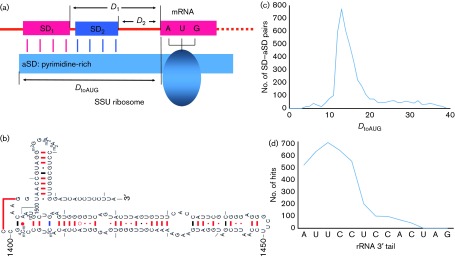
Based on the E. coli SSU rRNA secondary structure (Woese et al., 1980; Yassin et al., 2005), there are 13 nt at the 3′ end of the rRNA (referred to as the rRNA 3′ tail hereafter) that are free to base-pair with the SD sequence (Fig. 5b). We searched each Upstream30 sequence against the rRNA 3′ tail for matches with a length of at least four consecutive bases. The frequency distribution of DtoAUG from 4577 such matches peaked at DtoAUG = 13 and decreased rapidly towards DtoAUG = 10 and DtoAUG = 20 (Fig. 5c). We thus operationally defined a SD sequence as a sequence four bases or longer that can pair with the rRNA 3′ tail leading to a DtoAUG within the range of 10–20. Note that a SD sequence such as AGGAGG would need a space of five bases between the end of the SD sequence and the beginning of the start codon in order to have a DtoAUG = 13. A SD sequence such as AGGAG would need to have six bases between the end of SD and the beginning of the start codon in order to have a DtoAUG = 13.
Although the rRNA 3′ tail has 13 bases free (Fig. 5b), the sites that are involved in SD–aSD base pairing mainly belong to the first six sites (Fig. 5d). However, 754 putative SDs (including 156 GUGA, 166 GAGGU, 169 AGGU and 263 UGAU) in Upstream30 sequences in E. coli genes involve the second A from the 3′ end of SSU rRNA. This is consistent with the experimental observation that mutations at that site are moderately deleterious (Yassin et al., 2005).
We computed two indices for each phage: (1) percentage of SD-containing genes (PSD) and (2) mean number of consecutively matched sites (MSD). Previous studies have shown that highly expressed E. coli genes are more likely to have a SD sequence than lowly expressed genes (Ma et al., 2002) and that MSD is important for gene expression (Osterman et al., 2013).
Measuring stability of local mRNA secondary structure.
The stability of local secondary structure formed in mRNA is generally measured by MFE (kJ mol−1). The more negative the MFE value, the greater the stability of the secondary structure. We computed MFE using dambe, which implements the functionality of the Vienna RNA package (Hofacker, 2003). The settings used were: folding temperature 37 °C, with no lonely pairs and with no G/U pairs at the end of helices. Changing these settings did not affect the relative magnitude of MFE.
Translation initiation greatly depends on the secondary structure of sequences flanking the start codon (de Smit & van Duin, 1990, 1994; Nivinskas et al., 1999; Xia & Holcik, 2009; Xia et al., 2011). Burying either the SD sequence or the start codon in a stable secondary structure would affect its accessibility and decreases protein production dramatically in E. coli (Osterman et al., 2013). For this reason we measured the stability of the secondary structure for two associated regions: (1) 40 bases upstream of the start codon where the presence of a hairpin strongly inhibits translation (Osterman et al., 2013), and (2) the region −4 to +37, which has been previously studied and considered as a key contributor to translation initiation (Kudla et al., 2009; Osterman et al., 2013; Xia, 2014). MFEs for the two regions were designated MFE40nt and MFE−4+37, respectively. The two regions are related, respectively, to the accessibility of the SD sequence and the start codon.
Phage lifestyle classification.
The classification of phages into temperate and virulent categories was based on three publications (Deschavanne et al., 2010; Lima-Mendez et al., 2007; McNair et al., 2012). For E. coli and S. aureus phages, phage name, family, GenBank accession number of their sequenced genomes and their lifestyle identification (virulent/temperate) are available as Tables S2 and S3.
Acknowledgements
This study was funded by a Discovery Grant from the Natural Science and Engineering Research Council of Canada. We thank S. Aris-Brosou, N. Corradi, A. Golshani and J. Wang for discussion and comments. The manuscript was substantially improved by the comments from two anonymous reviewers.
Footnotes
Three supplementary tables are available with the online Supplementary Material.
References
- Chithambaram S., Prabhakaran R., Xia X. ( 2014a). Differential codon adaptation between dsDNA and ssDNA phages in Escherichia coli . Mol Biol Evol 31, 1606–1617. 10.1093/molbev/msu087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chithambaram S., Prabhakaran R., Xia X. ( 2014b). The effect of mutation and selection on codon adaptation in Escherichia coli bacteriophage. Genetics 197, 301–315. 10.1534/genetics.114.162842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A., Wolfe K. H. ( 2000). Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae . Yeast 16, 1131–1145. [DOI] [PubMed] [Google Scholar]
- Comeron J. M., Aguadé M. ( 1998). An evaluation of measures of synonymous codon usage bias. J Mol Evol 47, 268–274. 10.1007/PL00006384 [DOI] [PubMed] [Google Scholar]
- de Smit M. H., van Duin J. ( 1990). Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Natl Acad Sci U S A 87, 7668–7672. 10.1073/pnas.87.19.7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smit M. H., van Duin J. ( 1994). Translational initiation on structured messengers. Another role for the Shine–Dalgarno interaction. J Mol Biol 235, 173–184. 10.1016/S0022-2836(05)80024-5 [DOI] [PubMed] [Google Scholar]
- Deschavanne P., DuBow M. S., Regeard C. ( 2010). The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol J 7, 163. 10.1186/1743-422X-7-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L., Mouchiroud D. ( 1999). Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis . Proc Natl Acad Sci U S A 96, 4482–4487. 10.1073/pnas.96.8.4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargo D. C., Zhang M., Gillham N. W., Boynton J. E. ( 1998). Shine–Dalgarno-like sequences are not required for translation of chloroplast mRNAs in Chlamydomonas reinhardtii chloroplasts or in Escherichia coli . Mol Gen Genet 257, 271–282. 10.1007/s004380050648 [DOI] [PubMed] [Google Scholar]
- Felsenstein J. ( 1985). Phylogenies and the comparative method. Am Nat 125, 1–15. 10.1086/284325 [DOI] [Google Scholar]
- Giliberti J., O’Donnell S., Van Etten W. J., Janssen G. R. ( 2012). A 5′-terminal phosphate is required for stable ternary complex formation and translation of leaderless mRNA in Escherichia coli . RNA 18, 508–518. 10.1261/rna.027698.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J., Park E.-C., Seed B. ( 1996). Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr Biol 6, 315–324. 10.1016/S0960-9822(02)00482-7 [DOI] [PubMed] [Google Scholar]
- Hartz D., McPheeters D. S., Gold L. ( 1991). Influence of mRNA determinants on translation initiation in Escherichia coli . J Mol Biol 218, 83–97. 10.1016/0022-2836(91)90875-7 [DOI] [PubMed] [Google Scholar]
- Hofacker I. L. ( 2003). Vienna RNA secondary structure server. Nucleic Acids Res 31, 3429–3431. 10.1093/nar/gkg599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui A., de Boer H. A. ( 1987). Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli . Proc Natl Acad Sci U S A 84, 4762–4766. 10.1073/pnas.84.14.4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. ( 1981a). Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol 146, 1–21. 10.1016/0022-2836(81)90363-6 [DOI] [PubMed] [Google Scholar]
- Ikemura T. ( 1981b). Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol 151, 389–409. 10.1016/0022-2836(81)90003-6 [DOI] [PubMed] [Google Scholar]
- Ikemura T. ( 1982). Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Biol 158, 573–597. 10.1016/0022-2836(82)90250-9 [DOI] [PubMed] [Google Scholar]
- Ikemura T. ( 1992). Correlation between codon usage and tRNA content in microorganisms. In Transfer RNA in Protein Synthesis, pp. 87–111. Edited by Hatfield D. L., Lee B. J., Pirtle R. M. Boca Raton, FL: CRC Press. [Google Scholar]
- Komarova A. V., Tchufistova L. S., Supina E. V., Boni I. V. ( 2002). Protein S1 counteracts the inhibitory effect of the extended Shine–Dalgarno sequence on translation. RNA 8, 1137–1147. 10.1017/S1355838202029990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K. M., Van Etten W. J., III, Janssen G. R. ( 2010). Proximity of the start codon to a leaderless mRNA’s 5′ terminus is a strong positive determinant of ribosome binding and expression in Escherichia coli . J Bacteriol 192, 6482–6485. 10.1128/JB.00756-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G., Murray A. W., Tollervey D., Plotkin J. B. ( 2009). Coding-sequence determinants of gene expression in Escherichia coli . Science 324, 255–258. 10.1126/science.1170160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Mendez G., Toussaint A., Leplae R. ( 2007). Analysis of the phage sequence space: the benefit of structured information. Virology 365, 241–249. 10.1016/j.virol.2007.03.047 [DOI] [PubMed] [Google Scholar]
- Limor-Waisberg K., Carmi A., Scherz A., Pilpel Y., Furman I. ( 2011). Specialization versus adaptation: two strategies employed by cyanophages to enhance their translation efficiencies. Nucleic Acids Res 39, 6016–6028. 10.1093/nar/gkr169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Campbell A., Karlin S. ( 2002). Correlations between Shine–Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol 184, 5733–5745. 10.1128/JB.184.20.5733-5745.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín A., Xia X. ( 2008). GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: new substitution models incorporating strand bias. J Theor Biol 253, 508–513. 10.1016/j.jtbi.2008.04.004 [DOI] [PubMed] [Google Scholar]
- McNair K., Bailey B. A., Edwards R. A. ( 2012). phacts, a computational approach to classifying the lifestyle of phages. Bioinformatics 28, 614–618. 10.1093/bioinformatics/bts014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melançon P., Leclerc D., Destroismaisons N., Brakier-Gingras L. ( 1990). The anti-Shine–Dalgarno region in Escherichia coli 16S ribosomal RNA is not essential for the correct selection of translational starts. Biochemistry 29, 3402–3407. 10.1021/bi00465a037 [DOI] [PubMed] [Google Scholar]
- Milón P., Rodnina M. V. ( 2012). Kinetic control of translation initiation in bacteria. Crit Rev Biochem Mol Biol 47, 334–348. 10.3109/10409238.2012.678284 [DOI] [PubMed] [Google Scholar]
- Milón P., Maracci C., Filonava L., Gualerzi C. O., Rodnina M. V. ( 2012). Real-time assembly landscape of bacterial 30S translation initiation complex. Nat Struct Mol Biol 19, 609–615. 10.1038/nsmb.2285 [DOI] [PubMed] [Google Scholar]
- Na D., Lee D. ( 2010). RBSDesigner: software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics 26, 2633–2634. 10.1093/bioinformatics/btq458 [DOI] [PubMed] [Google Scholar]
- Nakamoto T. ( 2006). A unified view of the initiation of protein synthesis. Biochem Biophys Res Commun 341, 675–678. 10.1016/j.bbrc.2006.01.019 [DOI] [PubMed] [Google Scholar]
- Ngumbela K. C., Ryan K. P., Sivamurthy R., Brockman M. A., Gandhi R. T., Bhardwaj N., Kavanagh D. G. ( 2008). Quantitative effect of suboptimal codon usage on translational efficiency of mRNA encoding HIV-1 gag in intact T cells. PLoS One 3, e2356. 10.1371/journal.pone.0002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivinskas R., Malys N., Klausa V., Vaiskunaite R., Gineikiene E. ( 1999). Post-transcriptional control of bacteriophage T4 gene 25 expression: mRNA secondary structure that enhances translational initiation. J Mol Biol 288, 291–304. 10.1006/jmbi.1999.2695 [DOI] [PubMed] [Google Scholar]
- O’Donnell S. M., Janssen G. R. ( 2001). The initiation codon affects ribosome binding and translational efficiency in Escherichia coli of cI mRNA with or without the 5′ untranslated leader. J Bacteriol 183, 1277–1283. 10.1128/JB.183.4.1277-1283.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell S. M., Janssen G. R. ( 2002). Leaderless mRNAs bind 70S ribosomes more strongly than 30S ribosomal subunits in Escherichia coli . J Bacteriol 184, 6730–6733. 10.1128/JB.184.23.6730-6733.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsthoorn R. C. L., Zoog S., van Duin J. ( 1995). Coevolution of RNA helix stability and Shine–Dalgarno complementarity in a translational start region. Mol Microbiol 15, 333–339. 10.1111/j.1365-2958.1995.tb02247.x [DOI] [PubMed] [Google Scholar]
- Osterman I. A., Evfratov S. A., Sergiev P. V., Dontsova O. A. ( 2013). Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res 41, 474–486. 10.1093/nar/gks989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakaran R., Chithambaram S., Xia X. ( 2014). Aeromonas phages encode tRNAs for their overused codons. Int J Comput Biol Drug Des 7, 168–182. 10.1504/IJCBDD.2014.061645 [DOI] [PubMed] [Google Scholar]
- Ringquist S., Shinedling S., Barrick D., Green L., Binkley J., Stormo G. D., Gold L. ( 1992). Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol 6, 1219–1229. 10.1111/j.1365-2958.1992.tb01561.x [DOI] [PubMed] [Google Scholar]
- Robinson M., Lilley R., Little S., Emtage J. S., Yarranton G., Stephens P., Millican A., Eaton M., Humphreys G. ( 1984). Codon usage can affect efficiency of translation of genes in Escherichia coli . Nucleic Acids Res 12, 6663–6671. 10.1093/nar/12.17.6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H. M. ( 2011). The ribosome binding site calculator. Methods Enzymol 498, 19–42. 10.1016/B978-0-12-385120-8.00002-4 [DOI] [PubMed] [Google Scholar]
- Sartorius-Neef S., Pfeifer F. ( 2004). In vivo studies on putative Shine–Dalgarno sequences of the halophilic archaeon Halobacterium salinarum . Mol Microbiol 51, 579–588. 10.1046/j.1365-2958.2003.03858.x [DOI] [PubMed] [Google Scholar]
- Schattner P., Brooks A. N., Lowe T. M. ( 2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33 (Web Server issue), W686–W689. 10.1093/nar/gki366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr T., Nadir E., Margalit H. ( 1993). Identification and characterization of E. coli ribosomal binding sites by free energy computation. Nucleic Acids Res 21, 4019–4023. 10.1093/nar/21.17.4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S. W., Yang J. S., Kim I., Yang J., Min B. E., Kim S., Jung G. Y. ( 2013). Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng 15, 67–74. 10.1016/j.ymben.2012.10.006 [DOI] [PubMed] [Google Scholar]
- Sharp P. M., Li W. H. ( 1987). The Codon Adaptation Index – a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15, 1281–1295. 10.1093/nar/15.3.1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J., Dalgarno L. ( 1974). The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A 71, 1342–1346. 10.1073/pnas.71.4.1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen M. A., Kurland C. G., Pedersen S. ( 1989). Codon usage determines translation rate in Escherichia coli . J Mol Biol 207, 365–377. 10.1016/0022-2836(89)90260-X [DOI] [PubMed] [Google Scholar]
- Sun X. Y., Yang Q., Xia X. ( 2013). An improved implementation of effective number of codons (nc). Mol Biol Evol 30, 191–196. 10.1093/molbev/mss201 [DOI] [PubMed] [Google Scholar]
- Supek F., Šmuc T. ( 2010). On relevance of codon usage to expression of synthetic and natural genes in Escherichia coli . Genetics 185, 1129–1134. 10.1534/genetics.110.115477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T., Waldman Y. Y., Kupiec M., Ruppin E. ( 2010). Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A 107, 3645–3650. 10.1073/pnas.0909910107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weringh A., Ragonnet-Cronin M., Pranckeviciene E., Pavon-Eternod M., Kleiman L., Xia X. ( 2011). HIV-1 modulates the tRNA pool to improve translation efficiency. Mol Biol Evol 28, 1827–1834. 10.1093/molbev/msr005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesper O., Amitai S., Belitsky M., Byrgazov K., Kaberdina A. C., Engelberg-Kulka H., Moll I. ( 2011). Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli . Cell 147, 147–157. 10.1016/j.cell.2011.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimberg V., Tats A., Remm M., Tenson T. ( 2007). Translation initiation region sequence preferences in Escherichia coli . BMC Mol Biol 8, 100. 10.1186/1471-2199-8-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese C. R., Magrum L. J., Gupta R., Siegel R. B., Stahl D. A., Kop J., Crawford N., Brosius J., Gutell R., et al. ( 1980). Secondary structure model for bacterial 16S ribosomal RNA: phylogenetic, enzymatic and chemical evidence. Nucleic Acids Res 8, 2275–2294. 10.1093/nar/8.10.2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. ( 1990). The ‘effective number of codons’ used in a gene. Gene 87, 23–29. 10.1016/0378-1119(90)90491-9 [DOI] [PubMed] [Google Scholar]
- Xia X. ( 1998). How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics 149, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. ( 2007). An improved implementation of codon adaptation index. Evol Bioinform Online 3, 53–58. [PMC free article] [PubMed] [Google Scholar]
- Xia X. ( 2012a). DNA replication and strand asymmetry in prokaryotic and mitochondrial genomes. Curr Genomics 13, 16–27. 10.2174/138920212799034776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. ( 2012b). Position weight matrix, gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica (Cairo) 2012, 917540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. ( 2012c). Rapid evolution of animal mitochondria. In Evolution in the Fast Lane: Rapidly Evolving Genes and Genetic Systems, pp. 73–82. Edited by Singh R. S., Xu J., Kulathinal R. J. Oxford: Oxford University Press; 10.1093/acprof:oso/9780199642274.003.0008 [DOI] [Google Scholar]
- Xia X. ( 2013a). Comparative Genomics. Berlin: Springer; 10.1007/978-3-642-37146-2 [DOI] [Google Scholar]
- Xia X. ( 2013b). dambe5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30, 1720–1728. 10.1093/molbev/mst064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. ( 2014). A major controversy in codon–anticodon adaptation resolved by a new codon usage index. Genetics 10.1534/genetics.114.172106 [Epub ahead of print]. 10.1534/genetics.114.172106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Holcik M. ( 2009). Strong eukaryotic IRESs have weak secondary structure. PLoS One 4, e4136. 10.1371/journal.pone.0004136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Huang H., Carullo M., Betrán E., Moriyama E. N. ( 2007). Conflict between translation initiation and elongation in vertebrate mitochondrial genomes. PLoS One 2, e227. 10.1371/journal.pone.0000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., MacKay V., Yao X., Wu J., Miura F., Ito T., Morris D. R. ( 2011). Translation initiation: a regulatory role for poly(A) tracts in front of the AUG codon in Saccharomyces cerevisiae . Genetics 189, 469–478. 10.1534/genetics.111.132068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassin A., Fredrick K., Mankin A. S. ( 2005). Deleterious mutations in small subunit ribosomal RNA identify functional sites and potential targets for antibiotics. Proc Natl Acad Sci U S A 102, 16620–16625. 10.1073/pnas.0508444102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Wilke C. O. ( 2011). Reduced stability of mRNA secondary structure near the translation-initiation site in dsDNA viruses. BMC Evol Biol 11, 59. 10.1186/1471-2148-11-59 [DOI] [PMC free article] [PubMed] [Google Scholar]