

Abstract
Colon cancer development and malignant progression are driven by genetic and epigenetic alterations in tumor cells and by factors from the tumor microenvironment. Cancer cells become reliant on the activity of specific oncogenes and on prosurvival and proliferative signals they receive from the abnormal environment they create and reside in. Accordingly, the response to anticancer therapy is determined by genetic and epigenetic changes that are intrinsic to tumor cells and by the factors present in the tumor microenvironment. Recent advances in the understanding of the involvement of the tumor microenvironment in tumor progression and therapeutic response are optimizing the application of prognostic and predictive factors in colon cancer. Moreover, new targets in the tumor microenvironment that are amenable to therapeutic intervention have been identified. Because stromal cells are with rare exceptions genetically stable, the tumor microenvironment has emerged as a preferred target for therapeutic drugs. In this review, we discuss the role of stromal fibroblasts and macrophages in colon cancer progression and in the response of colon cancer patients to therapy.
Keywords: colon cancer, macrophages, fibroblasts, tumor microenvironment
Tumor Microenvironment and Tumor Heterogeneity
It has been known for a long time that cancers originating from diverse tissues have unique characteristics. In fact, most current anticancer therapies are based on the organ of origin and new drugs are being tested in organ-based clinical trials. However, extensive variations exist between tumors of different patients that arise from the same tissues (interpatient variability), and among primary and metastatic tumors, which are referred to as intertumoral differences.
In addition, tumors are characterized by extensive intratumoral heterogeneity, as cells within a tumor exhibit a high degree of molecular and phenotypic heterogeneity. Several studies established that intratumoral heterogeneity is driven by the coevolution of tumor cells with nonmalignant stromal cells, such as fibroblasts, immune cells, endothelial cells, and the extracellular matrix (ECM; reviewed in Refs. 1–3). However, it has only recently become evident that intratumoral heterogeneity has both prognostic and predictive values and is a key factor driving treatment failure.
The Cancer Genome Atlas, a collaborative effort of cancer biologists and oncologists, discovered nearly 10 million cancer-related mutations,4 with very few mutations present in the majority of tumor cells.5 However, most of these mutations are passenger or bystander mutations that are not required to maintain the transformed phenotype. Because passenger mutations offer no selective growth advantage, they do not constitute appropriate therapeutic targets. Only mutations that actively contribute to tumor initiation and progression, called driver mutations, should guide the selection of patients for targeted therapy. Several recent reports established that classification of colon cancer patients into molecular disease subtypes based on whole-genome expression data is more informative than classification based on a single mutation and has both prognostic and predictive values (see below).3,6,7 Based on this work, it has been suggested that cancers from different organs that have related molecular characteristics and share common driver mutations should be managed similarly.8
Although tumor heterogeneity is primarily the result of genetic instability, an evolving hallmark of cancer, epigenetic changes also contribute to inter- and intratumor heterogeneity.9 Factors in the tumor microenvironment promote tumor heterogeneity, at least in part, by providing an appropriate niche for cancer stem cells (CSCs).10,11 Myofibroblasts have been shown to foster CSC population by promoting Wnt signaling through production of hepatocyte growth factor (HGF).12 We demonstrated that macrophage-derived IL-1 enhances Wnt signaling in colon cancer cells,13–15 underscoring the significance of the tumor microenvironment in generating functional diversity within a tumor. In agreement with preclinical findings, primary colon tumors display heterogeneous activity of Wnt signaling and cells with the highest levels of Wnt signaling display characteristics of CSCs.12,16–18
Intratumoral heterogeneity presents a major challenge for targeted therapeutic strategies.19 Anti-epidermal growth factor receptor (anti-EGFR) antibodies, such as cetuximab and panitumumab, are approved for the treatment of colon cancer patients with wild-type (WT) KRas; however, only a proportion (15%–35%)20–23 of these patients responds to therapy and virtually all patients develop resistance. It has been demonstrated that chronic exposure of cetuximab-responsive cells to the drug results in the emergence of cetuximab-resistant clones that harbor KRas amplification or KRas mutations.24 The authors showed that these resistant lines emerged as a result of expansion of rare preexisting drug-resistant clones or due to acquisition of de novo activating KRas mutations. Indeed, the vast majority of patients who develop resistance to cetuximab have been shown to gain either KRas mutations or amplifications, confirming clinical relevance of these findings. Another study confirmed that rare cells with KRas mutations are present at a low level in WT KRas tumors.25 The authors found mutant KRas DNA in the circulation of 38% of patients whose tumors were initially characterized as WT KRas. However, as we discuss below, it is often factors from the tumor microenvironment, such as HGF, that blunt the response to anti-EGFR therapy.
In addition, anti-EGFR therapy appears to have a negative impact on the outcome in patients who carry a KRas mutation.26 While cetuximab improved median progression-free survival (PFS) in patients with WT KRas, it actually shortened the PFS in patients with MT KRas. Thus, the presence of KRas mutations not only predicts resistance to inhibitors of EGFR signaling but also identifies patients who may be actually harmed by these drugs. Similarly, treatment of BRAF WT multiple myeloma cells with BRAF inhibitors results in activation of the oncogenic RAS-ERK pathway and promotes metastasis,27 confirming that treatment of resistant cancers might actually accelerate disease progression.28
Preclinical models, perhaps with the exception of patient-derived xenografts, do not recapitulate heterogeneity observed in human beings, and current treatment strategies in colon cancer patients do not take into account tumor heterogeneity. However, it is becoming clear that simultaneously blocking of multiple targets, using combinations of drugs, is required to prevent acquired therapeutic resistance and to achieve a durable patient response. This situation resembles the need to treat infectious diseases caused by microbes prone to develop drug resistance, such as HIV or Mycobacterium tuberculosis, simultaneously with multiple antiviral agents or antibiotics in order to prevent development of multidrug resistance. Presently, treatment decisions are based on the most prevalent clone derived from biopsies that are captured from a single tumor location. Although profiling of tumors from multiple regions and serial tumor testing may improve the therapeutic strategy, it is inconvenient for the patients as it entails obtaining multiple biopsies from numerous regions of the tumor. The hope is that the development of minimally invasive methods, such as profiling circulating tumor cells and analyzing circulating tumor DNA, will overcome these obstacles.29–32
While therapeutic resistance can develop due to evolution of tumors, accompanied by the acquisition of advantageous genetic and epigenetic changes, the tumor microenvironment plays a major role in therapeutic response. This review is focused on the role of cancer-associated fibroblasts (CAFs) and macrophages in colon cancer progression and the response of colon cancer patients to therapy (Fig. 1). However, by no means do we assume that the contribution of other stromal components, such as T- and B-cells, endothelial cells, and the ECM, to prognosis and therapeutic response of colon cancer patients is marginal. In fact, TNM (T is for T cells and M is for memory) staging in colon cancer has been recently proposed33 and the immunoscore has been introduced for the classification of malignant tumors.34–36
Figure 1.

Interplay between tumor cells and stroma. Fibroblasts and macrophages secrete a variety of soluble factors that trigger oncogenic signaling in tumor cells (Wnt, STAT3, NF-κB), resulting in enhanced proliferation, migration, and resistance to therapy. Note that some factors (eg, TGFβ, IL-6) can be produced by both macrophages and fibroblasts. In turn, tumor cells can produce factors, such as TGFβ, IL-1β, and HGF (indicated by red arrows), that activate stromal cells.
CAFs have Prognostic and Predictive Values
CAFs or myofibroblasts are often the predominant nonmalignant cell type in the tumor stroma. The origin of CAFs is not well understood; they have been shown to derive from epithelial cells via epithelial–mesenchymal transition (EMT) or by transdifferentiation from resident fibroblasts, adipocytes, mesenchymal cells, or hematopoietic stem cells.37 Tumor-derived factors, including transforming growth factor-β (TGF-β), have been shown to mediate the conversion of normal fibroblasts into CAFs.38 CAFs share characteristics with activated fibroblasts found in injured and fibrotic tissues. However, activated fibroblasts revert to a quiescent phenotype once the process of healing is completed, tumor-associated fibroblasts remain constitutively activated and promote growth and survival of cancer cells.
Common markers to identify (and to quantify) CAFs in patient samples include alpha smooth muscle actin (α-SMA), fibroblast activation protein (FAP), fibroblast-specific protein-1 (FSP-1/S100A4), or platelet-derived growth factor receptor-β.39,40 In addition, secretome of colon CAF has been characterized.41 CAFs stimulate tumor cell proliferation, survival, migration, and invasion through the secretion of growth factors and cytokines, such as HGF, TGF-β, interleukin-6 (IL-6), and stromal cell-derived factor-1α.42 In addition, CAFs secrete ECM and proteases, such as matrix metalloproteases, cathepsins, and plasminogen activators, and thereby induce EMT and promote invasive growth of colon cancer cells.39,42–45
In a recent study, we compared the ability of normal intestinal fibroblasts and fibroblasts isolated from Crohn’s disease (CD), ulcerative colitis (UC), or colon cancer patients to regulate the growth of colon cancer cells. We found that normal fibroblasts induce STAT1 signaling in colon cancer cells and restrain their growth,46 consistent with a tumor-suppressive role of the normal stroma.47 Fibroblasts failed to inhibit growth of STAT1-deficient tumor cells, confirming a crucial role of STAT1 for the cross-talk between tumor cells and fibroblasts.46 Our findings demonstrated that myofibroblasts isolated from CD or UC patients, who have a significantly elevated risk of developing colon cancer, failed to induce STAT1 signaling and to inhibit growth of cancer cells. TNF, a cytokine with a crucial role in CD, reduced the ability of fibroblasts to induce STAT1 signaling in tumor cells. Thus, our data suggest that an initial step in the evolution of tumor-associated stroma is loss of the ability of stromal cells to restrain the growth of the adjacent epithelium. This is ultimately followed by the gain of tumor-promoting activity of CAFs. Proteome profiling confirmed a pro-inflammatory and desmoplastic signature of colon CAFs.48
α-SMA, a marker of myofibroblasts, has been shown to be an independent prognostic factor, comparable to lymph node metastasis, an established and powerful prognostic factor for colorectal cancer (CRC).49,50 α-SMA as a prognostic factor was also superior to other tumor and stromal components, including histology of the tumor invasive front, peritumoral lymphocytic infiltration, and Crohn’s-like lymphoid reaction. It has been suggested that the abundance of myofibroblasts in cancer-associated stroma may serve as a useful indicator of disease recurrence after curative CRC surgery.51 CAFs with increased ability to promote the migration of colon cancer cells have also been shown to have higher expression of α-SMA mRNA than fibroblasts with low promigratory ability.52 Most significantly, patients with the high “CAF signature” had a remarkably poor prognosis. A recent study investigated the expression level of Hsp27 in CAFs and its clinical implications in patients with CRC lung metastases. The authors found that Hsp27 is highly expressed in tumor stroma of CRC lung metastases and that it is co-expressed with α-SMA. Stromal α-SMA and Hsp27 were found to be associated with poor clinical outcome after pulmonary metastasectomy. Moreover, soluble Hsp27 was found to be increased in patients before pulmonary metastasectomy and to decrease after surgery to levels comparable to healthy controls, suggesting that serum Hsp27 may be a potential marker for metastatic disease in CRC.53 Accordingly, expression of FAP, an established marker of CAFs, has been shown to be associated with an aggressive disease and to be an independent negative prognostic factor in colon cancer patients.40,54
It has become clear that the current staging system is not optimal to ascertain the prognosis of stage II colon cancer patients, which leads to both under- and overtreatment of patients. Analysis of 192 CRC patients revealed that tumors with abundant myofibroblasts were associated with shorter disease-free survival for stages II and III CRC.51 Although the abundance of stroma can help to predict outcome of colon cancer patients, it is not merely the number of fibroblasts that is prognostic, but their activation state. Recent studies established that the CAF-derived 5-gene signature (AMIGO2, ULBP2, PDLIM3, SLC7A2, and CCL11) identifies stage II patients’ risk of relapse more precisely than conventional clinicopathological criteria alone.55 This established that the CAF-derived 5-gene classifier may help to identify high-risk stage II patients who would benefit from adjuvant therapy.
Several independent studies have proposed classification of colon cancer patients based on distinct global gene expression profiles,6,7,56 which appears to have both prognostic and predictive significances. All these studies confirmed that the expression of mesenchymal genes correlates with poor outcome in CRC patients (Fig. 2).6,7,56
Figure 2.
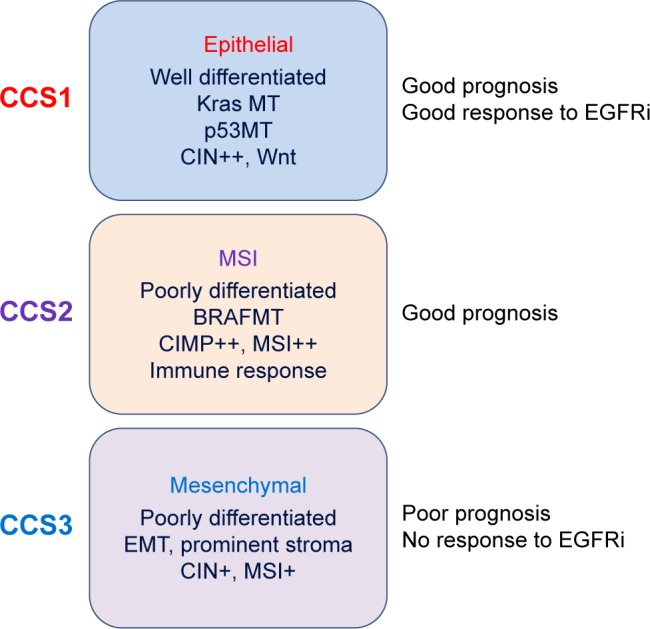
Molecular classification of colon cancer into three main colon cancer subtypes (CCS).6 This approach is more accurate for guiding therapeutic decisions. For example, these classifications established that KRas mutations are not optimal criteria for the selection of patients for an anti-EGFR therapy, as the CCS3 patients fail to respond to EGFR inhibitors irrespective of the KRas status.
Abbreviations: CIN, chromosomal instability; MSI, microsatellite instability; CIMP, CpG island methylator phenotype; EMT, epithelial–mesenchymal transition.
Remarkably, careful evaluation of these classification systems by Calon et al established that the predictive power of these studies is derived from genes expressed in stromal rather than in epithelial cells.57 The authors found that active TGF-β signaling in CAFs increases the frequency of tumor-initiating cells, a common feature of all the CRC subtypes with poor prognosis. Most importantly, they showed that inhibitors of TGF-β signaling block the cross-talk between cancer cells and fibroblasts and prevent metastatic spread.57
Another study confirmed that the CAF signature was associated with poor prognosis in untreated colon cancer patients and also predicted resistance to radiotherapy in rectal cancer.58 Together, these data confirmed that stroma significantly contributes to clinical features of CRC and shapes the therapeutic response.
Therapeutic strategies have traditionally been implemented based on the molecular characterization of malignant cells, such as genomic, genetic and epigenetic alterations, chromosomal instability (CIN), tumor suppressor gene inactivation/oncogene activation, and microsatellite instability (MSI).59–61 In colon cancer, this has led to the establishment and clinical implementation of biomarkers of drug sensitivity such as the status of KRas being used to select patients for anti-EGFR therapy. However, the influence of nonmalignant stroma cells in the tumor microenvironment on therapeutic efficacy is increasingly being appreciated. Indeed, fibroblasts not only have a prognostic value but also predict the response to therapy. Recently, it has been demonstrated that growth factors, such as HGF, FGF, and neuregulin-1, which are predominantly secreted by cells in the tumor microenvironment, inhibit the efficiency of targeted therapeutic agents.62 A study by Liska et al63 confirmed that fibroblast-derived HGF could rescue CRC cells from cetuximab-mediated EGFR inhibition, suggesting that inhibitors of the HGF/MET signaling pathway could improve the response of CRC patients to EGFR inhibitors. Indeed, a recent study revealed that increased serum levels of HGF are associated with resistance to anti-EGFR antibodies in metastatic CRC patients with WT KRas.64 Serum HGF levels could therefore be a promising biomarker for the prediction of the response to anti-EGFR therapy and for the selection of patients for polytherapy with anti-EGFR and anti-HGF/MET drugs.
Finally, therapeutic agents induce significant changes in the microenvironment. Therapy-induced DNA damage response in cancer and stromal cells is associated with the production of a complex network of secreted inflammatory factors, called the DNA damage-associated secretory program or therapy-induced secretome. Thus, therapeutic agents generate their own pro-inflammatory microenvironment, which enhances the survival of drug-sensitive cells and promotes the expansion of drug-resistant clones, contributing to the development of acquired resistance to therapy.65 For example, the analysis of matched CRC samples from patients before and after treatment revealed a significant increase in CAFs. Most significantly, chemotherapy-treated human CAFs enhanced self-renewal of CSCs and promoted tumor growth, which was dependent on therapy-induced secretion of specific cytokines and chemokines, including interleukin-17A (7A).66
It therefore appears that a durable therapeutic response can be achieved only by targeting both tumor cells and their supportive stroma and by “normalizing” the tumor micro-environment. However, despite the abundance of encouraging preclinical data, which confirmed that targeting CAFs impacts tumor progression and the response of tumors to therapy, multiple therapies designed to target CAFs or CAF-derived factors have failed in clinical development. Better understanding of the role of the stroma in tumor progression and in modulating the response of tumor cells to therapy is needed to design therapeutic approaches that involve the tumor microenvironment.
Prognostic and Predictive Values of Tumor-associated Macrophages
Macrophages have an innate ability to recognize and to potentially destroy cancer cells. However, tumor-derived factors can polarize macrophages into tumor-promoting cells that support growth, survival, migration, and metastatic spread of cancer cells. In addition, tumor-associated macrophages (TAMs) are immunosuppressive and inhibit the natural killer (NK) cell and T-cell-mediated immune response to tumors.67 The prognostic and predictive roles of macrophages depend on the cancer type. While macrophages indicate poor prognosis in breast, brain, head and neck, kidney, ovarian, skin, and bladder cancers, their role in the progression of colon cancer appears to be more complex, with reports of both tumor-suppressive and tumor-promoting activities.68 Macrophage-specific inactivation of STAT3 in mice has been shown to trigger chronic intestinal inflammation, coupled to the development of invasive intestinal adenocarcinomas,69 confirming a role of macrophages in the development of colon cancer.
TAMs are among the most abundant immune cell population in the microenvironment of various tumors, including colon cancer.70–73 TAMs are derived from monocytic precursors circulating in the blood,72 and it has been recently demonstrated that circulating monocytes isolated from colon cancer patients display a distinct gene signature that can serve as a biomarker for diagnosis and therapeutic response.74 Chemokines, such as CCL2, CCL5, and CXCL1, play a key role in the recruitment of monocytes to the tumors,75,76 and neutralization of CCL2 reduces colitis-associated colon carcinogenesis.77 In addition, vascular endothelial growth factor (VEGF), platelet-derived growth factor, TGF-β, and macrophage colony-stimulating factor have been shown to play an important role in recruitment, survival, and differentiation of macrophages.71
Depending on the nature of the microenvironment, monocytes can differentiate into either M1 or M2 type macrophages.78–80 M1 macrophages are induced in response to IFNγ and microbial stimuli lipopolysaccharide (LPS) or cytokines such as TNF-α and granulocyte-macrophage colony-stimulating factor.71 In contrast, activation by IL-4, IL-13, CSF-1, IL-10, and glucocorticoids triggers alternative polarization of monocytes into M2 macrophages.81,82 M1 and M2 macrophages represent the extremes of TAM polarization, with a continuum of functional states in between, and most TAMs are of mixed phenotype, expressing simultaneously M1 and M2 markers.
M2 macrophages contribute to tumorigenesis by producing an array of cytokines, chemokines, growth factors, and proteases that have tumor-promoting activity.83,84 Macrophages secrete thymidine phosphorylase that promotes endothelial cell migration85 and several matrix metalloproteinases (MMPs), including MMP-2, MMP-7, and MMP-9,73,86,87 to promote angiogenesis. It has been shown that 75% of colon carcinoma samples have elevated expression of MMP-7 compared to the adjacent normal tissue.88 TAMs are also the source of proteases involved in digestion of the ECM and the basement membrane, such as cathepsin B,89 cathepsin D,90 and urokinase-type plasminogen activator (uPA).91 These proteases are overexpressed in cancer tissues and correlate with poor prognosis of colon cancer patients.
The number of TAMs is generally increased in tumors with increased hypoxic conditions.92 Stabilization of hypoxia-inducible factors 1 and 2 (HIF-1 and -2) in TAMs leads to enhanced expression of VEGF, FGF, and several other chemokines.93,94 The expression of VEGF-A and VEGF-C is significantly increased in colon carcinomas compared to normal tissues,95 and HIF-1α expression in colon cancer correlates with both VEGF expression and microvessel density.96 Depletion of macrophages in an orthotopic syngeneic mouse model of colon cancer prevented liver and peritoneal metastases, confirming a proangiogenic and prometastatic role of macrophages.97
Pro-inflammatory cytokines IL-1β and TNF-α secreted by TAMs promote colon cancer angiogenesis, in part, via enhancement of angiogenin expression in colon cancer cells.98 Increased serum angiogenin concentration in CRC correlates with cancer progression.99 We showed that colon cancer cells activate macrophages to secrete IL-1β, which promotes Wnt signaling in colon cancer cells, driving their growth and therapeutic resistance.13 Activated macrophages have also been shown to stimulate NFκB and promote proliferation and migration of colon cancer cells via production of TNFα, IL-β, and IL-6.100
Myeloid-derived suppressor cells (MDSCs), representing immature myeloid cells, are recruited to the tumor microenvironment, and their number correlate with the clinical outcome and metastatic progression in colon cancer.101 MDSCs promote cancer immune evasion by suppressing functions of T-cells and NK cells and have been shown to play a critical role in the development of colitis-associated colon cancer.102
Consistent with the ability of macrophages to modulate the growth of tumors, to suppress antitumor responses, to promote angiogenesis, and to support invasion and metastasis of tumors, increased density of macrophages is associated with poor prognosis in breast, prostate, bladder, and cervical cancers.103–108 However, there are conflicting data about the role of TAMs in CRC. Infiltration of M1 macrophages has been shown to correlate with improved survival of colon cancer patients, independent of the MSI or CpG island methylator phenotype (CIMP) status.109 Likewise, patients with advanced CRC have a low number of macrophages at the invasive front and in the stroma110 and the presence of VEGF expressing TAMs is indicative of improved survival in stage II and stage III colon cancer patients,111 suggesting that TAMs play a protective role in CRC. Colon cancer patients with a low level of macrophage infiltration had a significantly greater depth of tumor invasion than patients with a high level of macrophage infiltration, confirming that macrophages can exert an antitumoral effect in CRC patients.112 Indeed, it has been shown that a high ratio of macrophages to colon cancer cells inhibits the growth of colon cancer cells in vitro, and using macrophage marker CD68, the authors demonstrated that macrophage infiltration is associated with improved survival of colon cancer patients.113 A high density of CD68+ tumor infiltrating cells has been shown to serve as an independent prognostic marker for a five-year relapse-free survival and an overall survival of CRC patients.68,114
In contrast, analysis of 289 CRC patients concluded that both CAF and M2 macrophages were significantly associated with poor disease-free survival and poor overall survival of colon cancer patients.115 The authors used α-SMA and FSP-1 as markers for CAF and CD163 as a marker for M2 macrophages. Interestingly, the association with poor survival was even more robust when analysis was done with CAF and M2 markers combined compared to when these markers were studied individually. The contradictory results may be attributed to the markers used for the macrophages in these studies. Whereas Herrera et al115 used CD163 as a marker of M2 macrophages, studies from other groups described above used CD68, which is a pan-monocyte/macrophage marker.
Macrophages modulate the response to therapy by producing soluble factors, including IL-6, TNF, IL-1β, and IL-17, which activate prosurvival signaling pathways, such as STAT3, Wnt, and NFκB, in cancer cells. We described that macrophages protect colon cancer cells from TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by promoting Wnt signaling in colon cancer cells.14,116 Inactivation of β or silencing of β in macrophages inhibited their ability to counter TRAIL-induced apoptosis. TRAIL-induced collapse of the mitochondrial membrane potential (ΔΨ) and activation of caspases were prevented by macrophages or by recombinant β. However, macrophages can also enhance the response to therapy. For example, we demonstrated that macrophages and IL-1β actually promote 5-fluorouracil (5FU)-induced apoptosis in colon cancer cells (Fig. 3A; Kaler and Klampfer, unpublished data). The presence of macrophages (Mo) or treatment with IL-1β increased the response to 5FU (Fig. 3A), confirmed by enhanced activation of caspase-7 and cleavage of Poly (ADP-ribose) polymerase (PARP) (Fig. 3B). These findings are in line with a complex role of macrophages in colon cancer.
Figure 3.
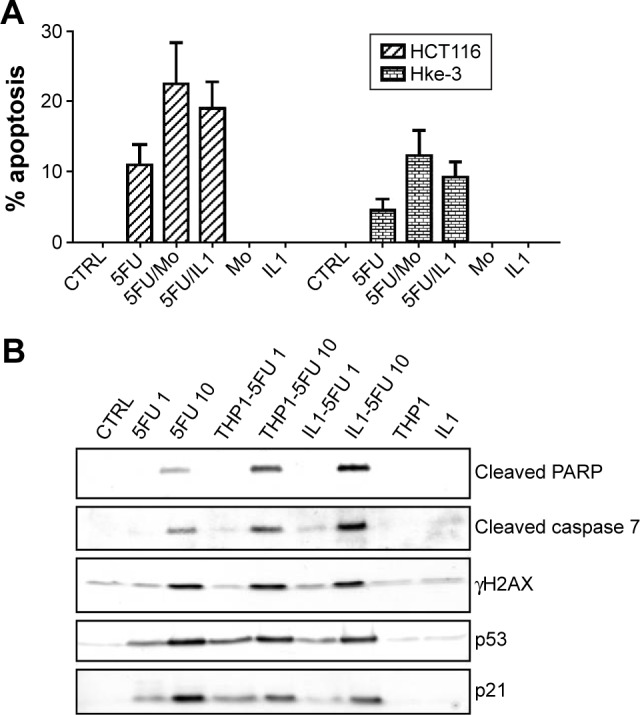
Macrophages promote 5FU-induced apoptosis. (A) HCT116 and HKe-3 cells were treated with 5FU (10 μM) in the absence or the presence of THP1 macrophages (Mo) or IL-1β as indicated and the extent of apoptosis was determined after 48 hours. (B) HCT116 cells were treated with 5FU (1 μM or 10 μM) in the absence or the presence of THP1 macrophages or IL-1β as indicated for 48 hours. Cell lysates were examined for the presence of cleaved PARP, cleaved caspase-7, p53, and p21 by immunoblotting.
Preclinical studies have established that blocking macrophage recruitment into tumors, or altering their function, modulates disease progression and alters the response of tumor cells to therapy.117,118 Because of the critical role of CCL2 and CSF-1 for the recruitment of macrophages to tumors, inhibitors of CSF-1, CSF-1R, and CCL2 have entered clinical trials, testing their efficiency in patients with solid tumors.117 These studies suggest that antagonists of CSF-1, CSF-1R, and CCL2 are well tolerated; however, it remains to be evaluated whether drugs that target TAMs should be used alone or in combination with classical or targeted therapy.
Conclusions
Recent efforts in molecular classification of CRCs have significantly improved management of colon cancer patients. Gene expression-based stratification of colon cancers provides prognostic information and facilitates the rational design of therapeutic strategies for individual patients. Targeted therapeutic agents, directed at specific features of cancer cells, are likely to replace conventional chemotherapy. One of the main barriers in personalized medicine is the identification of biomarkers that predict who will benefit from a particular targeted therapy. It is becoming evident that some of these markers are not intrinsic to tumor cells, but reside in the tumor microenvironment. Therapeutic strategies to normalize the tumor microenvironment or to inhibit signaling between tumor cells and the supporting stroma are being tested in preclinical studies. These new classes of anticancer drugs are redefining traditional treatment and hold great promise to overcome the resistance to classical and targeted therapy.
Acknowledgments
We thank Dr. George Wisniewski and Dr. Marina Manuvakhova for critical reading of the manuscript and their helpful suggestions.
Footnotes
ACADEMIC EDITOR: Barbara Guinn, Editor in Chief
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1,678 words, excluding any confidential comments to the academic editor.
FUNDING: Our work was supported by the Alabama Innovation Fund and by the NIH/NCI grant CA158560 to LK. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: The authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: PK, LK. Analyzed the data: BO, MV, LK. Wrote the first draft of the manuscript: BO, MV, LK. Contributed to the writing of the manuscript: BO, MV, LK. Agree with manuscript results and conclusions: BO, MV, PK, LK. Jointly developed the structure and arguments for the paper: LK. Made critical revisions and approved final version: LK. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.De Sousa EMF, Vermeulen L, Fessler E, Medema JP. Cancer heterogeneity—a multifaceted view. EMBO Rep. 2013;14:686–695. doi: 10.1038/embor.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Junttila MR, de Sauvage FJ. Influence of tumour microenvironment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 3.Linnekamp JF, Wang X, Medema JP, Vermeulen L. Colorectal cancer heterogeneity and targeted therapy: a case for molecular disease subtypes. Cancer Res. 2015;75:245–249. doi: 10.1158/0008-5472.CAN-14-2240. [DOI] [PubMed] [Google Scholar]
- 4.Ledford H. End of cancer-genome project prompts rethink. Nature. 2015;517:128–129. doi: 10.1038/517128a. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Sousa EMF, Wang X, Jansen M, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 7.Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortes J, Calvo E, Vivancos A, Perez-Garcia J, Recio JA, Seoane J. New approach to cancer therapy based on a molecularly defined cancer classification. CA Cancer J Clin. 2014;64:70–74. doi: 10.3322/caac.21211. [DOI] [PubMed] [Google Scholar]
- 9.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. 2007;6:2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 13.Kaler P, Augenlicht L, Klampfer L. Macrophage-derived beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009;28:3892–3902. doi: 10.1038/onc.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaler P, Augenlicht L, Klampfer L. Activating mutations in beta-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS One. 2012;7:e45462. doi: 10.1371/journal.pone.0045462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaler P, Godasi BN, Augenlicht L, Klampfer L. The NF-kappaB/AKT-dependent induction of Wnt signaling in colon cancer cells by macrophages and beta. Cancer Microenviron. 2009;2(1):69–80. doi: 10.1007/s12307-009-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hlubek F, Brabletz T, Budczies J, Pfeiffer S, Jung A, Kirchner T. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. Int J Cancer. 2007;121:1941–1948. doi: 10.1002/ijc.22916. [DOI] [PubMed] [Google Scholar]
- 17.Brabletz T, Jung A, Reu S, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–10361. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Goel S, Huang J, Klampfer L. K-Ras, intestinal homeostasis and colon cancer. Curr Clin Pharmacol. 2015;10(1):73–81. doi: 10.2174/1574884708666131111204440. [DOI] [PubMed] [Google Scholar]
- 20.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 21.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 22.Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 23.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 24.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diaz LA, Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 27.Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez-Laorden B, Viros A, Girotti MR, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7:ra30. doi: 10.1126/scisignal.2004815. [DOI] [PubMed] [Google Scholar]
- 29.Pantel K, Alix-Panabieres C. The potential of circulating tumor cells as a liquid biopsy to guide therapy in prostate cancer. Cancer Discov. 2012;2:974–975. doi: 10.1158/2159-8290.CD-12-0432. [DOI] [PubMed] [Google Scholar]
- 30.Pantel K, Alix-Panabieres C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010;16:398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Schwarzenbach H, Alix-Panabieres C, Muller I, et al. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin Cancer Res. 2009;15:1032–1038. doi: 10.1158/1078-0432.CCR-08-1910. [DOI] [PubMed] [Google Scholar]
- 32.Pantel K, Alix-Panabieres C. The clinical significance of circulating tumor cells. Nat Clin Prac Oncology. 2007;4:62–63. doi: 10.1038/ncponc0737. [DOI] [PubMed] [Google Scholar]
- 33.Broussard EK, Disis ML. TNM staging in colorectal cancer: T is for T cell and M is for memory. J Clin Oncol. 2011;29:601–603. doi: 10.1200/JCO.2010.32.9078. [DOI] [PubMed] [Google Scholar]
- 34.Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galon J, Pages F, Marincola FM, et al. Cancer classification using the immunoscore: a worldwide task force. J Transl Med. 2012;10:205. doi: 10.1186/1479-5876-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galon J, Pages F, Marincola FM, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med. 2012;10:1. doi: 10.1186/1479-5876-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald LT, LaRue AC. Hematopoietic stem cell derived carcinoma-associated fibroblasts: a novel origin. Int J Clin Exp Pathol. 2012;5:863–873. [PMC free article] [PubMed] [Google Scholar]
- 38.Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. [PubMed] [Google Scholar]
- 39.Tommelein J, Verset L, Boterberg T, Demetter P, Bracke M, De Wever O. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front Oncol. 2015;5:63. doi: 10.3389/fonc.2015.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henry LR, Lee HO, Lee JS, et al. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin Cancer Res. 2007;13:1736–1741. doi: 10.1158/1078-0432.CCR-06-1746. [DOI] [PubMed] [Google Scholar]
- 41.Chen SX, Xu XE, Wang XQ, et al. Identification of colonic fibroblast secretomes reveals secretory factors regulating colon cancer cell proliferation. J Proteomics. 2014;110:155–171. doi: 10.1016/j.jprot.2014.07.031. [DOI] [PubMed] [Google Scholar]
- 42.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013;437:397–402. doi: 10.1016/j.bbrc.2013.06.089. [DOI] [PubMed] [Google Scholar]
- 44.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuyada A, Chow A, Wu J, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72:2768–2779. doi: 10.1158/0008-5472.CAN-11-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaler P, Owusu BY, Augenlicht L, Klampfer L. The role of STAT1 for crosstalk between fibroblasts and colon cancer cells. Front Oncol. 2014;4:88. doi: 10.3389/fonc.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torres S, Bartolome RA, Mendes M, et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res. 2013;19:6006–6019. doi: 10.1158/1078-0432.CCR-13-1130. [DOI] [PubMed] [Google Scholar]
- 49.Astler VB, Coller FA. The prognostic significance of direct extension of carcinoma of the colon and rectum. Ann Surg. 1954;139:846–852. doi: 10.1097/00000658-195406000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dukes CE, Bussey HJ. The spread of rectal cancer and its effect on prognosis. Br J Cancer. 1958;12:309–320. doi: 10.1038/bjc.1958.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsujino T, Seshimo I, Yamamoto H, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13:2082–2090. doi: 10.1158/1078-0432.CCR-06-2191. [DOI] [PubMed] [Google Scholar]
- 52.Herrera M, Islam AB, Herrera A, et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin Cancer Res. 2013;19:5914–5926. doi: 10.1158/1078-0432.CCR-13-0694. [DOI] [PubMed] [Google Scholar]
- 53.Schweiger T, Nikolowsky C, Starlinger P, et al. Stromal expression of heat-shock protein 27 is associated with worse clinical outcome in patients with colorectal cancer lung metastases. PLoS One. 2015;10:e0120724. doi: 10.1371/journal.pone.0120724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wikberg ML, Edin S, Lundberg IV, et al. High intratumoral expression of fibroblast activation protein (FAP) in colon cancer is associated with poorer patient prognosis. Tumour Biol. 2013;34:1013–1020. doi: 10.1007/s13277-012-0638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berdiel-Acer M, Berenguer A, Sanz-Pamplona R, et al. A 5-gene classifier from the carcinoma-associated fibroblast transcriptomic profile and clinical outcome in colorectal cancer. Oncotarget. 2014;5:6437–6452. doi: 10.18632/oncotarget.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 58.Isella C, Terrasi A, Bellomo SE, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- 59.Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6:314–321. doi: 10.3978/j.issn.2078-6891.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grady WM, Pritchard CC. Molecular alterations and biomarkers in colorectal cancer. Toxicol Pathol. 2014;42:124–139. doi: 10.1177/0192623313505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dickinson BT, Kisiel J, Ahlquist DA, Grady WM. Molecular markers for colorectal cancer screening. Gut. 2015;64(9):1485–1494. doi: 10.1136/gutjnl-2014-308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17:472–482. doi: 10.1158/1078-0432.CCR-10-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takahashi N, Yamada Y, Furuta K, et al. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2014;110:2716–2727. doi: 10.1038/bjc.2014.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obenauf AC, Zou Y, Ji AL, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368–372. doi: 10.1038/nature14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lotti F, Jarrar AM, Pai RK, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by 7A. J Exp Med. 2013;210:2851–2872. doi: 10.1084/jem.20131195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roxburgh CS, McMillan DC. The role of the in situ local inflammatory response in predicting recurrence and survival in patients with primary operable colorectal cancer. Cancer Treat Rev. 2012;38:451–466. doi: 10.1016/j.ctrv.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 69.Deng L, Zhou JF, Sellers RS, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. 2010;176:952–967. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sickert D, Aust DE, Langer S, Haupt I, Baretton GB, Dieter P. Characterization of macrophage subpopulations in colon cancer using tissue microarrays. Histopathology. 2005;46:515–521. doi: 10.1111/j.1365-2559.2005.02129.x. [DOI] [PubMed] [Google Scholar]
- 71.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory microenvironment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 72.Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–270. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 73.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 74.Hamm A, Prenen H, Van Delm W, et al. Tumour-educated circulating monocytes are powerful candidate biomarkers for diagnosis and disease follow-up of colorectal cancer. Gut. 2015;64:1–11. doi: 10.1136/gutjnl-2014-308988. [DOI] [PubMed] [Google Scholar]
- 75.Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. 1989;169:1485–1490. doi: 10.1084/jem.169.4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bottazzi B, Polentarutti N, Acero R, et al. Regulation of the macrophage content of neoplasms by chemoattractants. Science. 1983;220:210–212. doi: 10.1126/science.6828888. [DOI] [PubMed] [Google Scholar]
- 77.Popivanova BK, Kostadinova FI, Furuichi K, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69:7884–7892. doi: 10.1158/0008-5472.CAN-09-1451. [DOI] [PubMed] [Google Scholar]
- 78.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruffell B, DeNardo DG, Affara NI, Coussens LM. Lymphocytes in cancer development: polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010;21:3–10. doi: 10.1016/j.cytogfr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 82.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. 2011;41:2470–2472. doi: 10.1002/eji.201141988. [DOI] [PubMed] [Google Scholar]
- 84.Sica A, Larghi P, Mancino A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Hotchkiss KA, Ashton AW, Klein RS, Lenzi ML, Zhu GH, Schwartz EL. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res. 2003;63:527–533. [PubMed] [Google Scholar]
- 86.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 87.DeNardo DG, Johansson M, Coussens LM. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008;27:11–18. doi: 10.1007/s10555-007-9100-0. [DOI] [PubMed] [Google Scholar]
- 88.McDonnell S, Navre M, Coffey RJ, Jr, Matrisian LM. Expression and localization of the matrix metalloproteinase pump-1 (MMP-7) in human gastric and colon carcinomas. Mol Carcinog. 1991;4:527–533. doi: 10.1002/mc.2940040617. [DOI] [PubMed] [Google Scholar]
- 89.Campo E, Munoz J, Miquel R, et al. Cathepsin B expression in colorectal carcinomas correlates with tumor progression and shortened patient survival. Am J Pathol. 1994;145:301–309. [PMC free article] [PubMed] [Google Scholar]
- 90.Mayer A, Fritz E, Fortelny R, Kofler K, Ludwig H. Immunohistochemical evaluation of cathepsin D expression in colorectal cancer. Cancer Invest. 1997;15:106–110. doi: 10.3109/07357909709115762. [DOI] [PubMed] [Google Scholar]
- 91.Sato T, Nishimura G, Yonemura Y, et al. Association of immunohistochemical detection of urokinase-type plasminogen activator with metastasis and prognosis in colorectal cancer. Oncology. 1995;52:347–352. doi: 10.1159/000227487. [DOI] [PubMed] [Google Scholar]
- 92.Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer. 1999;79:991–995. doi: 10.1038/sj.bjc.6690158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 94.Knowles HJ, Mole DR, Ratcliffe PJ, Harris AL. Normoxic stabilization of hypoxia-inducible factor-1alpha by modulation of the labile iron pool in differentiating U937 macrophages: effect of natural resistance-associated macrophage protein 1. Cancer Res. 2006;66:2600–2607. doi: 10.1158/0008-5472.CAN-05-2351. [DOI] [PubMed] [Google Scholar]
- 95.Hanrahan V, Currie MJ, Gunningham SP, et al. The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J Pathol. 2003;200:183–194. doi: 10.1002/path.1339. [DOI] [PubMed] [Google Scholar]
- 96.Kuwai T, Kitadai Y, Tanaka S, et al. Expression of hypoxia-inducible factor-1alpha is associated with tumor vascularization in human colorectal carcinoma. Int J Cancer. 2003;105:176–181. doi: 10.1002/ijc.11068. [DOI] [PubMed] [Google Scholar]
- 97.Kruse J, von Bernstorff W, Evert K, et al. Macrophages promote tumour growth and liver metastasis in an orthotopic syngeneic mouse model of colon cancer. Int J Colorectal Dis. 2013;28:1337–1349. doi: 10.1007/s00384-013-1703-z. [DOI] [PubMed] [Google Scholar]
- 98.Etoh T, Shibuta K, Barnard GF, Kitano S, Mori M. Angiogenin expression in human colorectal cancer: the role of focal macrophage infiltration. Clin Cancer Res. 2000;6:3545–3551. [PubMed] [Google Scholar]
- 99.Shimoyama S, Yamasaki K, Kawahara M, Kaminishi M. Increased serum angiogenin concentration in colorectal cancer is correlated with cancer progression. Clin Cancer Res. 1999;5:1125–1130. [PubMed] [Google Scholar]
- 100.Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology. 2010;215:242–249. doi: 10.1016/j.imbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 101.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24:631–644. doi: 10.1016/j.ccr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–1393. [PubMed] [Google Scholar]
- 104.Goede V, Brogelli L, Ziche M, Augustin HG. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer. 1999;82:765–770. doi: 10.1002/(sici)1097-0215(19990827)82:5<765::aid-ijc23>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 105.Goede V, Fleckenstein G, Dietrich M, Osmers RG, Kuhn W, Augustin HG. Prognostic value of angiogenesis in mammary tumors. Anticancer Res. 1998;18:2199–2202. [PubMed] [Google Scholar]
- 106.Hanada T, Nakagawa M, Emoto A, Nomura T, Nasu N, Nomura Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol. 2000;7:263–269. doi: 10.1046/j.1442-2042.2000.00190.x. [DOI] [PubMed] [Google Scholar]
- 107.Salvesen HB, Akslen LA. Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int J Cancer. 1999;84:538–543. doi: 10.1002/(sici)1097-0215(19991022)84:5<538::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 108.Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17:445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 109.Edin S, Wikberg ML, Dahlin AM, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS One. 2012;7:e47045. doi: 10.1371/journal.pone.0047045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakayama Y, Nagashima N, Minagawa N, et al. Relationships between tumor-associated macrophages and clinicopathological factors in patients with colorectal cancer. Anticancer Res. 2002;22:4291–4296. [PubMed] [Google Scholar]
- 111.Khorana AA, Ryan CK, Cox C, Eberly S, Sahasrabudhe DM. Vascular endothelial growth factor, CD68, and epidermal growth factor receptor expression and survival in patients with Stage II and Stage III colon carcinoma: a role for the host response in prognosis. Cancer. 2003;97:960–968. doi: 10.1002/cncr.11152. [DOI] [PubMed] [Google Scholar]
- 112.Funada Y, Noguchi T, Kikuchi R, Takeno S, Uchida Y, Gabbert HE. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncol Rep. 2003;10:309–313. [PubMed] [Google Scholar]
- 113.Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13:1472–1479. doi: 10.1158/1078-0432.CCR-06-2073. [DOI] [PubMed] [Google Scholar]
- 114.Chaput N, Svrcek M, Auperin A, et al. Tumour-infiltrating CD68+ and CD57+ cells predict patient outcome in stage II–III colorectal cancer. Br J Cancer. 2013;109:1013–1022. doi: 10.1038/bjc.2013.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Herrera M, Herrera A, Dominguez G, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;104:437–444. doi: 10.1111/cas.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kaler P, Galea V, Augenlicht L, Klampfer L. Tumor associated macrophages protect colon cancer cells from TRAIL-induced apoptosis through beta-dependent stabilization of Snail in tumor cells. PLoS One. 2010;5:e11700. doi: 10.1371/journal.pone.0011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Strachan DC, Ruffell B, Oei Y, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]