

Abstract
Whole-genome analyses have uncovered that most cancer-relevant genes cluster into 12 signaling pathways. Knowledge of the signaling pathways and associated gene signatures not only allows us to understand the mechanisms of oncogenesis inherent to specific cancers but also provides us with drug targets, molecular diagnostic and prognosis factors, as well as biomarkers for patient risk stratification and treatment. Publicly available genomic data sets constitute a wealth of gene mining opportunities for hypothesis generation and testing. However, the increasingly recognized genetic and epigenetic inter- and intratumor heterogeneity, combined with the preponderance of small-size cohorts, hamper reliable analysis and discovery. Here, we review two methods that are used to infer meaningful biological events from small-size data sets and discuss some of their applications and limitations.
Keywords: cohort size, gene data set, expression profiling, low-incidence cancers, intratumor heterogeneity, intertumor heterogeneity
Introduction
Next-generation sequencing and microarray technologies have generated massive amounts of data that can be mined for disease–gene expression correlates in search for molecular mechanisms, biomarkers, or drug targets. As of August 15, 2015, there were a bit less than 4,000 publicly available Gene Expression Omnibus (GEO) data sets (GDSs) that may be retrieved from GEO alone (the NIH gene expression data set repository at the National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/gds/), several hundreds of which being dedicated to human cancers. Current gene expression arrays encompass some 45k and 22k probesets for protein-encoding and noncoding genes, respectively (eg, Affymetrix’s GeneChip® Human Transcriptome Array 2.0; Illumina’s HumanHT-12 v4 Expression BeadChip), allowing to probe gene expression variation in clinical samples or cell lines at an unprecedented depth. The analytical power of whole-genome analyses, however, remains limited mostly owing to two practical parameters: (i) most cancers are relatively low-incidence diseases (eg, Ewing’s sarcoma affects 1–2 children/year/million1 and subcutaneous panniculitis-like T-cell lymphoma afflicts about 1 person/year/10 million2,3), and most laboratories or even institutions have therefore access to only a limited number of tumor samples and (ii) the cost of the technology remains too high for most low- to mid-budget laboratories, thus forcing investigators to limit the number of tested samples and biological replicates, which in turn yields mostly underpowered studies.
In 2010, McClellan and King highlighted the complex interplay, linking genetic diversity to disease heterogeneity.4 Accordingly, discovery of many disease-associated genetic risk variants requires exceedingly large cohorts in genome-wide association studies, as recently exemplified in a large-size cohort analysis of lung adenocarcinoma, wherein 26 research departments from several countries pulled their resources together to conduct the study.5 The problem posed by the high interindividual allelic variability can be further exacerbated by that of epigenetic diversity (eg, in follicular lymphoma and diffuse large B-cell lymphomas),6 whereby stochastic and/or environmental factors can lead to different epigenetic (and gene expression) landscapes, even in presumably otherwise genetically identical monozygotic twins.7 It is now becoming increasingly appreciated that several cancers exhibit high intratumor variability, including those of the breast,8–10 colon,11,12 head and neck,13 ovary,14 prostate15 and stomach,16 and glioblastoma.17–19 In fact, somatic mutation frequency analysis of more than 3,000 tumor samples encompassing 27 cancer types showed up to three or more orders of magnitude mutation rate variability between tumors (eg, in lung adenocarcinoma and melanoma),20 underscoring the scale of heterogeneity. Furthermore, tumor heterogeneity can be driven in response to chemotherapeutic intervention adding to the complexity of the analysis.21,22 Since heterogeneity can increase through time and/or in cases wherein tumors are exposed to different microenvironments, heterogeneity can be high when comparing metastases to primary tumors, particularly in cases whereby metastases take up to several decades to evolve allowing time for stochastic genotypic or epigenetic changes.23 Thus, for instance, 3%–24% of breast cancer metastases display a different estrogen, progesterone, or HER2 erb-b2 receptor tyrosine kinase 2 receptor status from the primary tumors,24 either due to a receptor switch or due to the fact that the tested metastases arose from sections of the primary tumor not included in the analyses. These intra- and interindividual differences notwithstanding, recurrent alterations in key biological processes often underlie a given disease,4 and for example, only a dozen or so of core signaling pathways appear to drive the tumorigenic phenotype of most cancers.25–27 Whereas cancer genetic and epigenetic diversities offer opportunities for biomarker discovery and risk stratification,28 uncovering genes and pathways associated with specific disease states remains challenging, owing to the sample-size requirements. Fortunately, bioinformatic methods have begun to address this problem, and we briefly summarize subsequently those that proved to be useful in the analysis of small-size cohorts.
How Small are Small-Size Cohorts?
It is common knowledge that most childhood cancer cohorts are relatively small in size. This is not only due to the fact that these diseases are relatively rare in nature but also because less funding is devoted to research on these neoplasms as compared to their adult counterparts. Thus, for example, the combined NIH budget for all types of pediatric sarcomas is only about 1/15th of the budget allocated to breast cancer alone.29 But what about the cohort size of gene data sets of more frequent childhood cancers (eg, leukemia with 88 cases/year/million in 1–4-year-old children30) or adults cancers (80–690 cancers/year/million in men and 73–724 cases/year/million in women for the top 10 cancer types; based on our estimates taking into account the current US population projection31 and cancer frequencies in adults in the US population in 201532)? To address this question, we ran a meta-analysis of all cancer gene data sets deposited to date in GEO. As shown in Figure 1, the majority of data sets contained 50 or less samples and qualified as small-size cohorts (median ; range: 4–192). As may be expected, the largest size data sets were mostly those of cancers with high-incidence and research funding and constituted seven of the 10 largest size data sets (not shown). Interestingly, however, probability density distribution analyses of individual cancers with the highest incidences and research funding, such as those of the breast (representing 29% of all new annual cases in women32; ; range: 4–116), prostate (26% new cases in men32; ; range: 4–171), lung (13%–14% new cases in both genders32; ; range: 4–192), and colorectum (8% new cases in both genders32; ; range: 4–104), show that these too are mostly made up of small-size cohorts (Fig. 2). The problem posed by small-size cohorts affects, therefore, the majority of data sets across the board in both childhood and adult cancers. Larger size data sets can of course be found in the collections of The Cancer Genome Atlas of the National Cancer Institute (NCI)/National Human Genome Research Institute (NHGRI) and of the Wellcome Trust Sanger Institute. These are, however, also subject to two overriding limitations. The first one relates to the aforementioned frequently observed tumor heterogeneity from which one can presume that many large-size cohort data sets are essentially a heterogeneous collection of varying numbers of relatively homogenous smaller size cohorts. Second, although these initiatives have made significant strides at increasing sample size in high-incidence diseases, they still somewhat lag behind in low incidence or so-called orphan diseases, to which many cancers belong.
Figure 1.
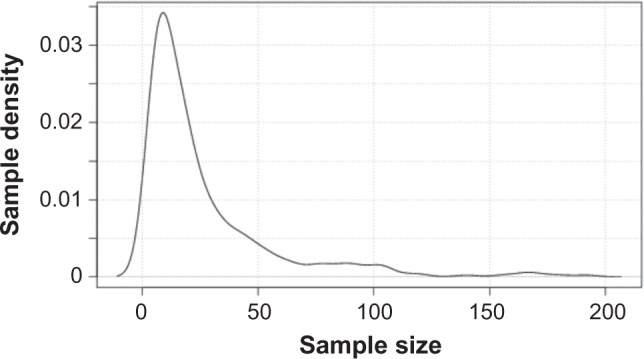
Probability density distribution of all cancer gene data sets in Gene Expression Omnibus (GEO). All cancer data sets were retrieved from GEO (query performed on August 15, 2015) and plotted against sample size (x axis). Gene data sets size refers to the number of tumor samples per data set. The analysis included 368 data sets and 9,845 tumor samples. Only data sets limited to tumor samples were retrieved; those solely listing data on tumor stroma or normal peripheral blood lymphocytes in cancer patients or those that combined several cancer types were omitted from the analysis. There were no other exclusion criteria.
Figure 2.

Probability density distribution of cancer gene data sets of high-incidence adult cancers. All cancer data sets were retrieved from GEO (query performed on August 15, 2015) and plotted against sample size (x axis). These included (A) breast cancer (number of data sets n = 110), (B) prostate cancer (n = 43), (C) lung cancer (n = 25), and (D) colorectal cancer (n = 37).
To illustrate some of the limitations imposed by small-size cohorts, we ran two simple tests comparing gene expression in two publicly available Ewing’s sarcoma gene data sets.33,34 In the first test, we looked at two genes that encode epigenetic modifiers with important roles in tumorigenesis. The first gene, lysine-specific demethylase 1 (to avoid gene and species ambiguity,35,36 NCBI gene IDs are given herein along with the gene symbol; LSD1; GeneID: 23028) was shown to be overexpressed and to serve as a drug target in Ewing’s sarcoma in vitro37 as well as in other neoplasms, such as breast cancer.38 The second gene, enhancer of zeste homolog 2 (EZH2; GeneID: 2146) was also shown to be overexpressed in Ewing’s sarcoma and to be a drug target both in vitro and in vivo.39,40 As proposed by others,41 we computed the bivariate kernel density estimates and run regression analyses in the R environment and compared the probability density distributions for either gene in two equal small-size Ewing’s sarcoma cohorts. As shown in Figure 3A, although LSD1 shows consistently high gene expression in both cohorts, there are a few outliers for EZH2 (Fig. 3B), indicating that the sample size and number of cohorts utilized, while sufficient to analyze LSD1, were borderline in the case of EZH2.
Figure 3.
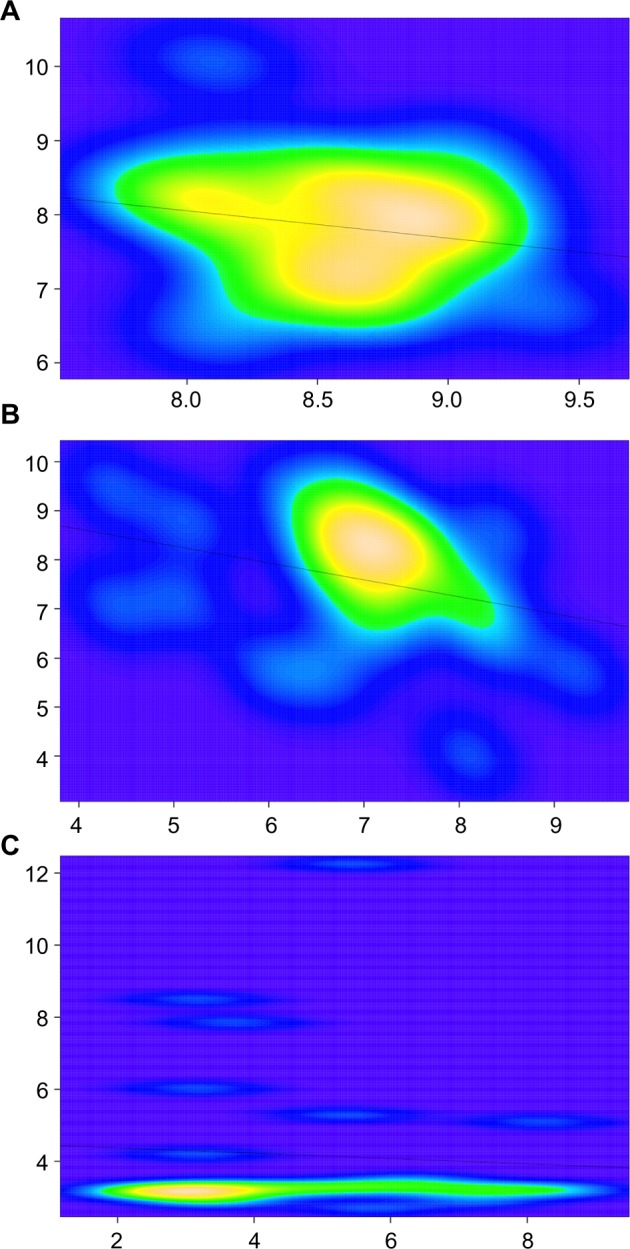
Bivariate kernel density estimates of gene expression consistency across small-size cohorts. Genes tested were LSD1 (A), EZH2 (B), and CXCR4 (C). x and y axes represent Log2 expression values of given genes (xi or yi) across Ewing’s sarcoma data sets either in GDS# GSE7007 (x axis; n = 27) or in ArrayExpress data set E-MEXP-1142 (y axis; n = 27). Lines across graphs depict regression curves as computed in a multiple regression model based on the ordinary least squares method as defined by the equation .
We next ran a test, this time looking at chemokine (C-X-C motif) receptor 4 (CXCR4; GeneID: 7852), a gene encoding a chemokine receptor previously shown to mark metastatic Ewing’s sarcoma and as such associates with about a one-third of all samples, representing the fraction of metastatic tumors.42 Contrary to the tests earlier, here we find the size and number of cohorts to be limiting, as the bivariate kernel density estimates did not fully reproduce the predicted distribution for this gene (Fig. 3C). Although these examples show that analyses in two small-size cohorts may be sufficient for some genes, it is important to note that these were tests for which we already knew the answers. For hypothesis generation through bioinformatics, which is usually one of the main applications of gene data set mining analyses, one would need a method to infer meaningfulness with a much higher degree of confidence.
In statistics, one way of boosting confidence is to increase sample size. One example is that of the so-called sequential analysis, particularly in prospective studies, whereby one adds samples (or recruits patients) until statistical significance is reached or until indications are present that significance is unlikely to be achieved.43 Although the sample size in sequential analysis is unknown prior to the end of the investigation, it tends to be smaller than in other methods wherein the sample size is predetermined, making it particularly suitable for cancers with particularly low incidences. Such a method, however, is impractical in the analysis of publicly available gene data sets, as the size of these is already fixed. In this case, an alternative would be to increase the number of cohorts. Two bioinformatic methods have taken advantage of this option to infer meaningful correlates from small-size cohorts.
Ican and Affiliated Cancer Informatics Methods to Probe Small-Size Cohorts
To address the problem posed by the small size of cancer cohorts, one of the authors developed the first method to reliably infer gene expression significance, and its association to specific patient subsets, from publicly available small-size cohort data sets irrespective of the expression profiling platform.37,42,44 This method, named Intercohort Co-ANalysis or Ican, relies on several innovative tools. First, it utilizes published gene expression levels known to be biologically active in experimentally validated tissues as a benchmark for gene expression significance, thus extracting biological significance from gene expression profiles. This eliminates variability across studies that results from the customary usage by different investigators of different arbitrary cutoffs for gene expression significance. Second, instead of combining small-size cohorts into a larger meta-cohort, each small-size cohort is analyzed individually. This helps avoid conormalization and the averaging out of sample quantiles across cohorts of different variances and distribution functions. The cohort-specific distribution probabilities are in fact used to highlight the high intercohort variability inherent to small-size cohorts. Next, quantile fitting of sample size to specific disease states, say chemoresistant or metastatic tumors, are mined for consistent molecular correlates within individual cohorts. Finally, a subtractive overlay of cohort-restricted associations is carried out to uncover genes whose expression is consistently associated with select sample subsets in all cohorts. In our case, four small-size publicly available data sets, in addition to a fifth nonpublicly available cohort that served for wet laboratory validation, were sufficient to infer gene expression significance. As a case in point, an Ican investigation of Ewing’s sarcoma (a cancer wherein metastasis is the major poor prognosis indicator) yielded several cosegregated chemokine ligand/receptor pairs in Ewing’s sarcoma patient subsets and helped uncover the first two chemokine receptors associated with either metastases or poor prognosis in Ewing’s sarcoma.42 To increase stringency, one may filter the patient-derived data sets through cell line-derived data sets. This can help eliminate genes not necessarily associated with the tumor cells but rather with the tumor stroma or tumor-infiltrating lymphocytes.42 Using such a strategy, we could, for example, zero in on two drugable receptors that represent viable therapeutic strategies for the corresponding Ican patient subsets. Using Ican, another study uncovered a micro-RNA, miR-34a, as a major molecular determinant of chemosensitivity and patient survival.45 The analytical power of Ican can therefore help uncover genes and pathways with clinical significance from underpowered small-size cohorts, as well as from larger cohorts of highly heterogeneous diseases, which represent most diseases.4,28 We surmise that the growing number of gene expression profiling investigations, compounded by the mandatory submission of gene expression data sets to public repositories requested now by most journals, will lead to a large field of Ican applications, and ensuing prognostic factor and biomarker discoveries.
A similar bioinformatics methodology dubbed Integrative Transcriptome Analysis (Itan) was independently developed by research groups at Harvard University and Massachusetts Institute of Technology (MIT).46 In this case, a coanalysis of nine hepatocellular carcinoma (HCC) gene data sets derived from different populations and micro array platforms was sufficient to uncover a novel mechanism of TGF-dependent WNT signaling activation in a subset of HCC patients.46 Contrary to Ican which uses publicly available gene data sets for hypothesis formulation and an additional cohort to experimentally test the hypothesis, Itan uses the larger publicly available gene data sets for training purposes (to avoid data overfitting to any given cohort) and uses the smaller publicly available data sets for testing. The latter was accomplished by subclass mapping, which utilizes hierarchical clustering, k-means clustering, and nonnegative matrix factorization as unsupervised clustering methods to identify tumor subclasses.47 As in Ican, the overriding principle here relies on molecular events consistently associated with particular tumor populations across all tested data sets. Based on this principle, the accuracy of both methods is dependent on the quality and number of data sets included in the analysis.
Limitations of Small-Size Cohort Bioinformatic Methods
Although Ican and Itan can be useful in inferring meaningfulness for any given gene (and corresponding pathway), they remain of limited value when assessing covariance of two or more genes across data sets, for example, to uncover gene networks associated with particular tumor subsets. This is because such analyses rely on Bayesian networks, Boolean networks, or on the mathematics of product moment correlations, and assuming all samples were added to the data set randomly (ie, patients were recruited consecutively without any prior knowledge of their clustering into one or another tumor subset, and no patients were removed from the cohort based on criteria that relate to the query at hand), these analyses are highly dependent on sample size.48 Thus, despite the constraints imposed by data set conormalization procedures, analysis of meta-genes remains here the method of choice. For example, using the same data sets analyzed by Ican, product moment correlation analyses of meta-genes can determine whether a signaling pathway is on or off directly in tumor samples or whether signaling molecules are active within specific pathways.49
Similarly, these methods would be ineffectual in inferring significance of tumor drivers harboring activating mutations and whose gene expression remained unchanged. In these cases, however, Ican and affiliated methods can be used in the analysis of the associated transcriptomes, given that the gene in question impervious to Ican analysis imparts a characteristic downstream gene expression signature, as shown, for instance, for several tumors drivers.50–53 While future studies should give us a better feel about the usefulness of Ican in such cases, this and affiliated methods will certainly find ample application in the field of biomarker discovery in search of markers of diagnosis, prognosis, patient risk stratification, and treatment response.37,42
Finally, though Ican is useful in the analysis of small-size cohorts, it requires multiple cohort data sets to infer differential gene expression significance. Unfortunately, many childhood cancers have very few (if any) gene expression data sets deposited in the public repositories, thus critically limiting the scope of Ican for these cancers. In this regard, an NCI’s Office of Cancer Genomics and Cancer Therapy Evaluation Program initiative, dubbed Therapeutically Applicable Research to Generate Effective Treatments (or TARGET) and which aims at characterizing the transcriptomes and genomes of hard-to-treat childhood cancers, is most welcome. TARGET has already generated data sets for childhood acute lymphoblastic leukemia and for neuroblastoma, and efforts are underway to generate genomic and expression profiling data sets for childhood acute myeloid leukemia, osteosarcoma, and renal tumors.
Conclusions
The majority of gene data sets, including those of high-incidence adult cancers, are represented by small-size cohorts. Bioinformatic methods, such as Ican or Itan, can help analyze underpowered studies, given that several data sets of the same disease type are available.
Although it may still be necessary to experimentally validate findings in additional data sets, particularly in case novel or little-known pathways are uncovered, these methods have proven to be sufficient to uncover with high confidence genes meaningful for a particular biological or pathological state from small-size cohorts. As most cancers are genetically and epigenetically heterogeneous and/or of low incidence, the cancer informatics of small-size cohorts will remain a tool of choice to enable the grasping for the brass ring of meaningful cancer-associated events in genomic and epigenomic data sets.
Appendix
The following source code was used for the kernel density estimate statistical analyses implemented in R1:
if (par1 == “0”) bw <- “nrd0”
if (par1!= “0”) bw <- as.numeric(par1)
par3 <- as.numeric(par3)
mydensity <- array(NA, dim=c(par3,8))
bitmap(file=“density1.png”)
mydensity1<-density(x,bw=bw,kernel=“gaussian”,na.rm=TRUE)
mydensity[,8] = signif(mydensity1$x,3)
mydensity[,1] = signif(mydensity1$y,3)
plot(mydensity1,main=“Gaussian Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
mydensity1
bitmap(file=“density2.png”)
mydensity2<-density(x,bw=bw,kernel=“epanechnikov”,na.rm=TRUE)
mydensity[,2] = signif(mydensity2$y,3)
plot(mydensity2,main=“Epanechnikov Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
bitmap(file=“density3.png”)
mydensity3<-density(x,bw=bw,kernel=“rectangular”,na.rm=TRUE)
mydensity[,3] = signif(mydensity3$y,3)
plot(mydensity3,main=“Rectangular Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
bitmap(file=“density4.png”)
mydensity4<-density(x,bw=bw,kernel=“triangular”,na.rm=TRUE)
mydensity[,4] = signif(mydensity4$y,3)
plot(mydensity4,main=“Triangular Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
bitmap(file=“density5.png”)
mydensity5<-density(x,bw=bw,kernel=“biweight”,na.rm=TRUE)
mydensity[,5] = signif(mydensity5$y,3)
plot(mydensity5,main=“Biweight Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
bitmap(file=“density6.png”)
mydensity6<-density(x,bw=bw,kernel=“cosine”,na.rm=TRUE)
mydensity[,6] = signif(mydensity6$y,3)
plot(mydensity6,main=“Cosine Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
bitmap(file=“density7.png”)
mydensity7<-density(x,bw=bw,kernel=“optcosine”,na.rm=TRUE)
mydensity[,7] = signif(mydensity7$y,3)
plot(mydensity7,main = “Optcosine Kernel”,xlab=xlab,ylab=ylab)
grid()
dev.off()
load(file=“createtable”)
ab<-table.start()
ab<-table.row.start(ab)
ab<-table.element(ab,”Properties of Density Trace”,2,TRUE)
ab<-table.row.end(ab)
ab<-table.row.start(ab)
ab<-table.element(ab,”Bandwidth”,header=TRUE)
ab<-table.element(ab,mydensity1$bw)
ab<-table.row.end(ab)
ab<-table.row.start(ab)
ab<-table.element(ab,”#Observations”,header=TRUE)
ab<-table.element(ab,mydensity1$n)
ab<-table.row.end(ab)
ab<-table.end(ab)
a <- ab
table.save(ab,file=“mytable123.tab”)
b<-table.start()
b<-table.row.start(b)
b<-table.element(b,”Maximum Density Values”,3,TRUE)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Kernel”,1,TRUE)
b<-table.element(b,”x-value”,1,TRUE)
b<-table.element(b,”max. density”,1,TRUE)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Gaussian”,1,TRUE)
b<-table.element(b,mydensity1$x[mydensity1$y==max(mydensity1$y)],1)
b<-table.element(b,mydensity1$y[mydensity1$y==max(mydensity1$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Epanechnikov”,1,TRUE)
b<-table.element(b,mydensity2$x[mydensity2$y==max(mydensity2$y)],1)
b<-table.element(b,mydensity2$y[mydensity2$y==max(mydensity2$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Rectangular”,1,TRUE)
b<-table.element(b,mydensity3$x[mydensity3$y==max(mydensity3$y)],1)
b<-table.element(b,mydensity3$y[mydensity3$y==max(mydensity3$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Triangular”,1,TRUE)
b<-table.element(b,mydensity4$x[mydensity4$y==max(mydensity4$y)],1)
b<-table.element(b,mydensity4$y[mydensity4$y==max(mydensity4$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Biweight”,1,TRUE)
b<-table.element(b,mydensity5$x[mydensity5$y==max(mydensity5$y)],1)
b<-table.element(b,mydensity5$y[mydensity5$y==max(mydensity5$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Cosine”,1,TRUE)
b<-table.element(b,mydensity6$x[mydensity6$y==max(mydensity6$y)],1)
b<-table.element(b,mydensity6$y[mydensity6$y==max(mydensity6$y)],1)
b<-table.row.end(b)
b<-table.row.start(b)
b<-table.element(b,”Optcosine”,1,TRUE)
b<-table.element(b,mydensity7$x[mydensity7$y==max(mydensity7$y)],1)
b<-table.element(b,mydensity7$y[mydensity7$y==max(mydensity7$y)],1)
b<-table.row.end(b)
b<-table.end(b)
a <- b[1]
table.save(b,file=“mytable2a.tab”)
a<-table.start()
a<-table.row.start(a)
a<-table.element(a,”Kernel Density Values”,8,TRUE)
a<-table.row.end(a)
a<-table.row.start(a)
a<-table.element(a,”x-value”,1,TRUE)
a<-table.element(a,”Gaussian”,1,TRUE)
a<-table.element(a,”Epanechnikov”,1,TRUE)
a<-table.element(a,”Rectangular”,1,TRUE)
a<-table.element(a,”Triangular”,1,TRUE)
a<-table.element(a,”Biweight”,1,TRUE)
a<-table.element(a,”Cosine”,1,TRUE)
a<-table.element(a,”Optcosine”,1,TRUE)
a<-table.row.end(a)
if (par2==“yes”) {
for(i in 1:par3) {
a<-table.row.start(a)
a<-table.element(a,mydensity[i,8],1,TRUE)
for(j in 1:7) {
a<-table.element(a,mydensity[i,j],1)
}
a<-table.row.end(a)
}
} else {
a<-table.row.start(a)
a<-table.element(a,”Kernel Density Values are not shown”,8)
a<-table.row.end(a)
}
a<-table.end(a)
table.save(a,file=“mytable1.tab”)
Footnotes
ACADEMIC EDITOR: J.T. Efird, Editor in Chief
PEER REVIEW: Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1784 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
Author Contributions
Conceived and designed the experiments: NBB, IMBB. Analyzed the data: NBB, IMBB. Wrote the first draft of the manuscript: NBB, IMBB. Contributed to the writing of the manuscript: NBB, IMBB. Agree with manuscript results and conclusions: NBB, IMBB. Jointly developed the structure and arguments for the paper: NBB, IMBB. Made critical revisions and approved final version: NBB, IMBB. Both authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Valery PC, Laversanne M, Bray F. Bone cancer incidence by morphological subtype: a global assessment. Cancer Causes Control. 2015;26(8):1127–39. doi: 10.1007/s10552-015-0607-3. [DOI] [PubMed] [Google Scholar]
- 2.Bennani-Baiti B, Yadav S, Flynt L, Bennani-Baiti N. Value of positron emission tomography in diagnosing subcutaneous panniculitis-like T-cell lymphoma. J Clin Oncol. 2015;33(10):1216–7. doi: 10.1200/JCO.2014.59.8193. [DOI] [PubMed] [Google Scholar]
- 3.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756–67. doi: 10.1182/blood-2010-05-231548. [DOI] [PubMed] [Google Scholar]
- 4.McClellan J King MC. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 5.Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450(7171):893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De S, Shaknovich R, Riester M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 2013;9(1):e1003137. doi: 10.1371/journal.pgen.1003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martelotto LG, Ng CK, Piscuoglio S, Weigelt B, Reis-Filho JS. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014;16(3):210. doi: 10.1186/bcr3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng CK, Martelotto LG, Gauthier A, et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015;16:107. doi: 10.1186/s13059-015-0657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–9. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kogita A, Yoshioka Y, Sakai K, et al. Inter- and intra-tumor profiling of multi-regional colon cancer and metastasis. Biochem Biophys Res Commun. 2015;458(1):52–6. doi: 10.1016/j.bbrc.2015.01.064. [DOI] [PubMed] [Google Scholar]
- 12.Sugihara Y, Taniguchi H, Kushima R, et al. Laser microdissection and two-dimensional difference gel electrophoresis reveal proteomic intra-tumor heterogeneity in colorectal cancer. J Proteomics. 2013;78:134–47. doi: 10.1016/j.jprot.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Mroz EA, Tward AD, Hammon RJ, Ren Y, Rocco JW. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. 2015;12(2):e1001786. doi: 10.1371/journal.pmed.1001786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoogstraat M, de Pagter MS, Cirkel GA, et al. Genomic and transcriptomic plasticity in treatment-naive ovarian cancer. Genome Res. 2014;24(2):200–11. doi: 10.1101/gr.161026.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah RB, Bentley J, Jeffery Z, DeMarzo AM. Heterogeneity of PTEN and ERG expression in prostate cancer on core needle biopsies: implications for cancer risk stratification and biomarker sampling. Hum Pathol. 2015;46(5):698–706. doi: 10.1016/j.humpath.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Stahl P, Seeschaaf C, Lebok P, et al. Heterogeneity of amplification of HER2, EGFR, CCND1 and MYC in gastric cancer. BMC Gastroenterol. 2015;15:7. doi: 10.1186/s12876-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szerlip NJ, Pedraza A, Chakravarty D, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 2012;109(8):3041–6. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snuderl M, Fazlollahi L, Le LP, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–7. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakobsen JN, Sorensen JB. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69(2):289–99. doi: 10.1007/s00280-011-1791-9. [DOI] [PubMed] [Google Scholar]
- 22.Yip C, Davnall F, Kozarski R, et al. Assessment of changes in tumor heterogeneity following neoadjuvant chemotherapy in primary esophageal cancer. Dis Esophagus. 2015;28(2):172–9. doi: 10.1111/dote.12170. [DOI] [PubMed] [Google Scholar]
- 23.Yadav S, Bennani-Baiti N. Renal cell carcinoma metastasis to the thyroid: how long is long enough? Acta Oncol. 2014;53(9):1277–8. doi: 10.3109/0284186X.2014.952388. [DOI] [PubMed] [Google Scholar]
- 24.Van Poznak C, Somerfield MR, Bast RC, et al. Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2015;33(24):2695–704. doi: 10.1200/JCO.2015.61.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 28.Merlo LM, Maley CC. The role of genetic diversity in cancer. J Clin Invest. 2010;120(2):401–3. doi: 10.1172/JCI42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennani-Baiti IM. Epigenetic and epigenomic mechanisms shape sarcoma and other mesenchymal tumor pathogenesis. Epigenomics. 2011;3(6):715–32. doi: 10.2217/epi.11.93. [DOI] [PubMed] [Google Scholar]
- 30.United States Cancer Statistics . 1999–2011 Incidence and Mortality Web-Based Report. US Cancer Statistics Working Group; Centers for Disease Control and Prevention (CDC); Atlanta, GA: the National Cancer Institute (NCI); Bethesda, MD: 2014. [Google Scholar]
- 31.United States Census Bureau . US population. Washington, DC: 2015. [Google Scholar]
- 32.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 33.Schaefer KL, Eisenacher M, Braun Y, et al. Microarray analysis of Ewing’s sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur J Cancer. 2008;44(5):699–709. doi: 10.1016/j.ejca.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 34.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11(5):421–9. doi: 10.1016/j.ccr.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 35.Bennani-Baiti B, Bennani-Baiti IM. Gene symbol precision. Gene. 2012;491(2):103–9. doi: 10.1016/j.gene.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 36.Bennani-Baiti B, Toegel S, Viernstein H, Urban E, Noe CR, Bennani-Baiti IM. Inflammation modulates RLIP76/RALBP1 electrophile-glutathione conjugate transporter and housekeeping genes in human blood-brain barrier endothelial cells. PLoS One. 2015;10(9):e0139101. doi: 10.1371/journal.pone.0139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennani-Baiti IM, Machado I, Llombart-Bosch A, Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol. 2012;43(8):1300–7. doi: 10.1016/j.humpath.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Bennani-Baiti IM. Integration of ERalpha-PELP1-HER2 signaling by LSD1 (KDM1A/AOF2) offers combinatorial therapeutic opportunities to circumventing hormone resistance in breast cancer. Breast Cancer Res. 2012;14(5):112. doi: 10.1186/bcr3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter GH, Plehm S, Fasan A, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci USA. 2009;106(13):5324–9. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staege MS, Hutter C, Neumann I, et al. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004;64(22):8213–21. doi: 10.1158/0008-5472.CAN-03-4059. [DOI] [PubMed] [Google Scholar]
- 41.Lucy D, Aykroyd RG, Pollard AM. Non-parametric calibration for age estimation. Appl Stat. 2002;51(2):183–96. [Google Scholar]
- 42.Bennani-Baiti IM, Cooper A, Lawlor ER, et al. Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing’s sarcoma. Clin Cancer Res. 2010;16(14):3769–78. doi: 10.1158/1078-0432.CCR-10-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ildstad ST, Centor RM, Davis E, et al. Small Clinical Trials: Issues and Challenges. National Academy Press; 2001. [PubMed] [Google Scholar]
- 44.Liersch-Lohn B, Slavova N, Buhr HJ, Bennani-Baiti IM. Differential protein expression and oncogenic gene network link tyrosine kinase Ephrin B4 receptor to aggressive gastric and gastroesophageal junction cancers. Int J Cancer. 2015 doi: 10.1002/ijc.29865. [DOI] [PubMed] [Google Scholar]
- 45.Nakatani F, Ferracin M, Manara MC, et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J Pathol. 2012;226(5):796–805. doi: 10.1002/path.3007. [DOI] [PubMed] [Google Scholar]
- 46.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69(18):7385–92. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoshida Y, Brunet JP, Tamayo P, Golub TR, Mesirov JP. Subclass mapping: identifying common subtypes in independent disease data sets. PLoS One. 2007;2(11):e1195. doi: 10.1371/journal.pone.0001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gayen AK. The frequency distribution of the product-moment correlation coefficient in random samples of any size drawn from non-normal universes. Biometrika. 1951;38(1–2):219–47. [PubMed] [Google Scholar]
- 49.Bennani-Baiti IM, Aryee DN, Ban J, et al. Notch signalling is off and is uncoupled from HES1 expression in Ewing’s sarcoma. J Pathol. 2011;225(3):353–63. doi: 10.1002/path.2966. [DOI] [PubMed] [Google Scholar]
- 50.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46(12):1258–63. doi: 10.1038/ng.3141. [DOI] [PubMed] [Google Scholar]
- 51.Jaworski M, Ittrich C, Hailfinger S, et al. Global gene expression in Ha-ras and B-raf mutated mouse liver tumors. Int J Cancer. 2007;121(6):1382–85. doi: 10.1002/ijc.22801. [DOI] [PubMed] [Google Scholar]
- 52.Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120–31. doi: 10.1182/blood-2014-03-564583. [DOI] [PubMed] [Google Scholar]
- 53.Sathyan KM, Nalinakumari KR, Kannan S. H-Ras mutation modulates the expression of major cell cycle regulatory proteins and disease prognosis in oral carcinoma. Mod Pathol. 2007;20(11):1141–8. doi: 10.1038/modpathol.3800948. [DOI] [PubMed] [Google Scholar]
REFERENCE
- 1.Wessa P. Kernel Density Estimation (v1.0.12) in Free Statistics Software (v1.1.23-r7) Office for Research Development and Education; 2015. [Google Scholar]