

Abstract
Aims
The aim of the phase Ib, two part SAWYER study (BO25341; NCT01292603) was to investigate the pharmacokinetics and safety of subcutaneous (s.c.) rituximab compared with intravenous (i.v.) rituximab, both in combination with fludarabine and cyclophosphamide (FC), as first line treatment for patients with chronic lymphocytic leukaemia (CLL).
Methods
During part 1 (dose-finding), CLL patients received rituximab i.v. followed by a single dose of rituximab s.c. at one of three fixed doses (1400, 1600 or 1870 mg) in cycle 6. The primary objective was to identify a fixed s.c. dose that would achieve comparable rituximab serum trough concentrations (Ctrough) to those achieved with the standard 4 weekly 500 mg m–2 rituximab i.v. dose.
Results
Fifty-five patients received a fixed dose of rituximab s.c., 16 received 1400 mg, 17 received 1600 mg and 22 received 1870 mg. The 1600 mg dose was predicted to achieve non-inferior Ctrough to standard rituximab i.v. treatment. The rituximab s.c. safety profile was comparable with rituximab i.v., except that local administration-related reactions, mainly mild/moderate injection site reactions, occurred more frequently with rituximab s.c., which was not unexpected. Subcutaneous administration was preferred to i.v. administration by >90% of patients and nurses (n = 112).
Conclusions
SAWYER part 1 data predict that rituximab s.c. 1600 mg will achieve non-inferior Ctrough concentrations to rituximab i.v. 500 mg m–2, administered 4 weekly. This fixed s.c. dose is currently undergoing formal non-inferiority assessment in SAWYER part 2. In future, CLL treatment regimens comprising rituximab s.c. and oral FC could substantially reduce i.v. chair time.
Keywords: chronic lymphocytic leukaemia, cyclophosphamide, fludarabine, pharmacokinetics, rituximab, subcutaneous
What is Already Known about this Subject
Rituximab plus fludarabine and cyclophosphamide is the standard treatment for chronic lymphocytic leukaemia (CLL).
Intravenous (i.v.) rituximab administration is inconvenient for patients and burdensome on healthcare resources.
This study investigates a new rituximab formulation for subcutaneous (s.c.) administration that could shorten administration times and improve patient convenience.
What this Study Adds
Rituximab s.c. 1600 mg is predicted to achieve non-inferior Ctrough compared with 4 weekly rituximab i.v. 500 mg m–2.
Safety profiles for rituximab s.c. and i.v. were similar and >90% of patients preferred s.c. administration.
CLL treatment comprising rituximab s.c. and oral chemotherapy could substantially reduce i.v. chair time.
Introduction
Rituximab (MabThera®, Rituxan®), a chimeric anti-CD20 monoclonal antibody, in combination with fludarabine and cyclophosphamide (FC) has improved both progression free and overall survival as first line treatment for physically fit patients with chronic lymphocytic leukaemia (CLL) 1.
Rituximab is currently administered as an intravenous (i.v.) infusion over 2.5–4 h, which is inconvenient for patients and burdensome on healthcare resources. Subcutaneous (s.c.) administration of rituximab could simplify drug delivery, shorten administration times and improve patient convenience, as previously shown for trastuzumab and alemtuzumab 2,3.
The existing rituximab i.v. formulation is unsuitable for s.c. administration as the volumes required to deliver an efficacious dose would exceed tolerable levels. S.c. administration of rituximab was made possible by hyperconcentrating rituximab (12-fold compared with the i.v. formulation) and including rHuPH20 as an excipient. rHuPH20 transiently degrades interstitial hyaluronan at the injection site, allowing administration of larger volumes and facilitating drug entry into the circulation 4–8.
Together with SparkThera (NCT00930514) and SABRINA (NCT01200758), SAWYER is part of the rituximab s.c. clinical development programme. These studies are designed to demonstrate pharmacokinetic non-inferiority of selected rituximab s.c. doses vs. established i.v. dosing regimens for follicular lymphoma (FL) and CLL (pharmacokinetic bridging), and confirm that rituximab's anti-lymphoma activity is not impaired by a change in administration route (clinical bridging). The underlying hypothesis is that a rituximab s.c. dose which achieves serum trough concentrations (Ctrough) at least as high as those achieved with rituximab i.v., would result in at least the same degree of target site saturation and hence the same degree of efficacy. Fixed dosing is possible for rituximab due to its wide therapeutic window 9,10, and this non-inferiority design was chosen to ensure that under-dosing was prevented in all patient subgroups. A similar approach was used in the development of trastuzumab s.c. 11–13.
SparkThera was a phase Ib study in patients with FL receiving maintenance therapy. A 1400 mg fixed dose achieved non-inferior Ctrough relative to rituximab i.v. 375 mg m–2 for both 2 and 3 monthly FL regimens 14. Pharmacokinetic results from the phase III SABRINA study subsequently showed that Ctrough with rituximab s.c. 1400 mg was non-inferior to that achieved with 3 weekly rituximab i.v. 375 mg m–2 as induction therapy for FL 15. Furthermore, overall and complete response rates in SABRINA Stage 1 suggested that s.c. administration did not impair rituximab's anti-lymphoma activity.
Here we report part 1 of the open label, multicentre, phase Ib SAWYER study (NCT01292603) in patients with previously untreated CLL. Of note, the SAWYER study was required in addition to the SparkThera study as it was necessary to bridge individually different rituximab doses and dosing intervals. Part 1 was designed to predict the rituximab s.c. fixed dose that would provide non-inferior Ctrough concentrations to rituximab i.v. Confirmation of the pharmacokinetic non-inferiority of the selected dose, compared with rituximab i.v. 500 mg m−2, is currently ongoing in SAWYER part 2.
Methods
Study design and treatment
Figure1 shows the single group, sequential treatment design. Eligible patients were enrolled at any point during their rituximab plus FC treatment prior to commencement of cycle 5. All patients received FC plus rituximab i.v. 375 mg m–2 in cycle 1 and rituximab i.v. 500 mg m–2 in cycles 2–4, at 4 weekly intervals, either on-study or prior to enrolment. In cycle 5, all patients received rituximab i.v. 500 mg m–2 plus FC; in cycle 6, rituximab i.v. was replaced by a single fixed (i.e. not body surface area [BSA]-adjusted) s.c. dose. Cycle 6 was selected for the analysis of rituximab s.c. pharmacokinetics as it allowed sample collection for more than 28 days. The first patients received the initial fixed dose of rituximab s.c. 1870 mg, predicted from the SparkThera study. This was higher than the 1400 mg s.c. dose which demonstrated non-inferiority to the 375 mg m–2 regimens employed in FL induction and maintenance 14,15, and reflects the higher i.v. dose used in the established 4 weekly CLL regimen (500 mg m–2). The protocol allowed dose adjustments in subsequent patients based on ongoing pharmacokinetic analyses. FC could be administered intravenously or orally, but for each patient the route had to remain the same throughout treatment. Recommended i.v. doses were fludarabine 25 mg m–2 and cyclophosphamide 250 mg m–2 (both on days 1–3 for up to six cycles). Oral FC was given in accordance with local practice and guidelines. Recommended regimens included fludarabine 24 mg m–2 plus cyclophosphamide 150 mg m–2 (both on days 1–5 for up to six cycles) or fludarabine 30–40 mg m–2 plus cyclophosphamide 200–250 mg m–2 (both on days 1–3 for up to six cycles). Follow-up visits were at 28 days, 56 days and 3 months from last treatment, then every 3 months until 3 years from last treatment and every 6 months until 4 years from last treatment.
Figure 1.
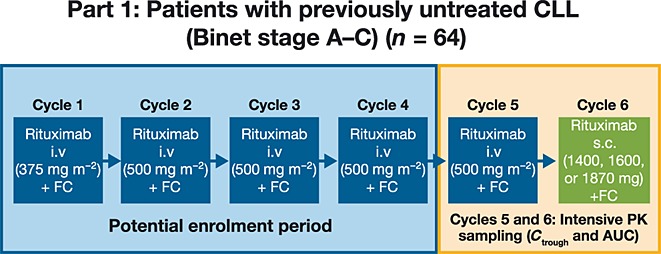
Study design. AUC, area under the plasma concentration–time curve; CLL, chronic lymphocytic leukaemia; Ctrough, trough serum concentration; FC, fludarabine and cyclophosphamide; i.v., intravenous; PK, pharmacokinetic; s.c., subcutaneous
The study was conducted in accordance with U.S. Food and Drug Administration regulations, the International Conference on Harmonization ‘Guideline for Good Clinical Practice’ Tripartite Guideline and applicable local laws. The protocol for SAWYER was approved by the relevant institutional review boards or ethics committees at the participating study centres. All patients provided written informed consent.
Patient eligibility
Eligible patients were aged ≥18 years with previously untreated CD20-positive CLL (Binet stage A, B or C) diagnosed per International Workshop CLL (iwCLL) criteria 16. Patients had an Eastern Cooperative Oncology Group performance status of 0–1 and life expectancy >6 months. Patients whose disease had transformed to an aggressive B-cell malignancy or with a history of another malignancy (unless treated with curative intent and in remission for ≥5 years) were not eligible. Other exclusion criteria included a Cumulative Illness Rating Scale score >6, inadequate liver or renal function (creatinine clearance <70 ml min–1) or disease requiring prolonged use of glucocorticoids dosed at >20 mg day–1. Patients were also excluded if they had experienced a grade 3 or 4 infusion-related reaction during up to four previous rituximab i.v. treatment cycles prior to enrolment.
Rituximab pharmacokinetic analysis
Blood samples for rituximab pharmacokinetic analyses were drawn during cycle 5 (day 1 pre-dose and post-dose, and days 2, 5, 11 and 15) and cycle 6 (pre-dose and days 2, 3, 5, 11, 15, 29 and 57). For patients enrolled before cycle 5, no pharmacokinetic sampling was performed in cycles 1–4. Rituximab concentrations were measured using a validated enzyme-linked immunosorbent assay (ELISA). The lower limit of quantification was 500 ng ml–1 14.
In SAWYER part 1, pharmacokinetic data collected after administration of one of three s.c. doses were used to refine a pre-existing population pharmacokinetic model that was developed based on clinical data from non-Hodgkin's lymphoma (NHL) and CLL populations, including data from the SparkThera trial 14. The base model was a two compartment model with time-dependent clearance 17. Clearance was presented as a sum of a non-specific time-independent clearance and time-dependent clearance that exponentially decreased with time, possibly due to depletion of the rituximab target (B cells). Data from SAWYER part 1 were integrated into this base model using parametric, non-linear, mixed effects modelling with nonmem software version 7.2.0 (ICON Development Solutions, Ellicott City, Maryland, USA) 18. The model was refined to describe the absorption phase of rituximab following s.c. administration. Model refinement was data driven and based on selected goodness-of-fit indicators. Due to the small study size, only a limited covariate analysis was performed. Exploratory covariate diagnostics were conducted by graphical exploration of all measured covariate effects and the model was revised, as necessary. The model was evaluated by graphical evaluation, precision of parameter estimates and visual predictive check plots.
The final population pharmacokinetic model was then used to predict Ctrough and area under the curve (AUC) in cycle 6 for incremental rituximab s.c. doses in a virtual population to identify the s.c. dose for formal non-inferiority testing in part 2. AUC values were calculated by numerical integration of individual concentration predictions.
rHuPH20 pharmacokinetic analysis
Samples for rHuPH20 pharmacokinetic analyses were taken at day 1 pre-dose, 30 min and 60 min post-dose and days 2, 29 and 57. rHuPH20 concentrations were measured using a validated ELISA. The lower limit of quantification was 500 ng ml–1 14.
Safety assessments
Safety assessments included laboratory assessments, vital signs and electrocardiograms. Patients were assessed for adverse events (AEs) at each visit, including during cycles 1–4 for patients enrolled before cycle 5, and as necessary during the study. AEs were graded according to National Cancer Institute Common Terminology Criteria for AEs (NCI CTC-AE; Version 4.0). AEs occurring during or within 24 h of rituximab administration and considered rituximab-related by the investigator were classified as administration-related reactions (ARRs). Safety data were reviewed regularly by an independent Data Monitoring Committee.
Immunogenicity
Blood samples for detection of human anti-chimeric antibodies (HACAs; anti-rituximab antibodies) and human anti-human antibodies (HAHAs; anti-rHuPH20 antibodies), were taken at cycle 5 and 6 (pre-dose and day 15) for anti-rituximab and at cycle 6 (pre-dose and day 15) for anti-rHuPH20, as well as each follow-up visit until 9 or 24 months after the last rituximab dose for HAHAs or HACAs, respectively. Validated bridging ELISA and electrochemiluminescent assays were used to detect the presence of HACAs in serum and HAHAs in plasma 14.
Patient and nurse preference
After completion of cycle 6, patients and their treating nurses were asked whether they had a preference for s.c. or i.v. rituximab administration.
Study objectives
The primary objective of SAWYER part 1 was to identify a rituximab s.c. dose with Ctrough similar to that obtained with 4 weekly rituximab i.v. 500 mg m–2. This dose will be further investigated in part 2 to establish non-inferiority between rituximab s.c. and i.v. Ctrough. Additional objectives of part 1 were to determine other pharmacokinetic parameters (e.g. AUC) for rituximab s.c., evaluate safety (e.g. ARRs and immunogenicity) and tolerability, and assess patient and nurse administration route preferences.
Statistical analyses and methodology
Safety data were summarized descriptively. Pharmacokinetic data were analyzed according to the treatment received during the cycle in which the sample was collected. Patients served as their own control (i.e. pharmacokinetic values in cycle 5 [i.v.] were compared with those in cycle 6 [s.c.]). Intravenous and s.c. concentration data were combined and analyzed using a population approach informed by post-i.v. administration pharmacokinetic data from NHL and CLL populations and by post-s.c. administration pharmacokinetic data from an NHL population. Individual predicted Ctrough and AUC(0,τ) were derived from the final pharmacokinetic model and summarized using descriptive statistics. An exploratory non-inferiority comparison was conducted to inform dose selection for part 2. Non-inferiority would be inferred if lower boundaries of the 90% confidence intervals (CIs) for the geometric mean ratios of pharmacokinetic parameters of rituximab s.c. vs. i.v. were above 0.8.
Results
Patients
Sixty-four patients were enrolled and 56 received rituximab s.c. in cycle 6. Eight patients did not receive rituximab s.c. five patients discontinued treatment before cycle 5, including one who died shortly after enrolment (27 days after cycle 4, unknown cause of death considered unrelated to study drug) and three patients withdrew after receiving rituximab i.v. in cycle 5. Per-protocol dose adjustments were made based on ongoing pharmacokinetic analyses such that the first 22 patients received rituximab s.c. 1870 mg, 16 received 1400 mg and 17 received 1600 mg. One patient assigned to rituximab s.c. 1870 mg received 1000 mg in error.
Table1 shows baseline patient characteristics. The three dose groups were balanced with respect to age, gender and BSA. As expected for CLL, more men than women were enrolled. The majority of patients had CLL Binet stage B and most had no B symptoms (i.e. fevers, night sweats, weight loss or significant fatigue) at screening.
Table 1.
Baseline patient characteristics
| Baseline characteristic | s.c. 1400 mg (n = 16)* | s.c. 1600 mg (n = 17)† | s.c. 1870 mg (n = 22)‡ |
|---|---|---|---|
| Median age, years (range) | 57.5 (38–77) | 61.0 (45–72) | 58.5 (43–67) |
| Age category, n (%) | |||
| <65 years | 13 (81) | 10 (59) | 20 (91) |
| 65–70 years | 2 (13) | 6 (35) | 2 (9) |
| >70 years | 1 (6) | 1 (6) | – |
| Gender, n (%) | |||
| Male | 10 (63) | 15 (88) | 15 (68) |
| Female | 6 (38) | 2 (12) | 7 (32) |
| Race, n (%) | |||
| White | 16 (100) | 14 (88) | 22 (100) |
| American Indian/Alaska | – | 1 (6) | – |
| Native | – | 1 (6) | – |
| Other | |||
| Hispanic ethnicity, n (%) | 3 (19) | 4 (27) | 1 (5) |
| BSA (m2), median (range) | 1.89 (1.60–2.35) | 1.98 (1.63–2.40) | 1.91 (1.56–2.13) |
| Median time from first CLL diagnosis, months (range) | 8.2 (0.3–95.9) | 10.8 (0.7–141.4) | 20.3 (4.0–101.1) |
| Binet stage | |||
| A | 5 (31) | 4 (24) | 5 (23) |
| B | 10 (63) | 8 (47) | 14 (64) |
| C | 1 (6) | 5 (29) | 3 (14) |
BSA, body surface area; CLL, chronic lymphocytic leukaemia; sc, subcutaneous.
*n = 15 for BSA.
†n = 15 for Hispanic ethnicity and n = 16 for race.
‡n = 21 for Hispanic ethnicity.
Pharmacokinetics
The final dataset contained 859 samples (414 samples for rituximab i.v. and 445 for rituximab s.c.) from 59 patients. Five patients withdrew before cycle 5 and therefore had no pharmacokinetic samples taken. Among these 59 patients, 56 had i.v. and s.c. data and three had i.v. data only. Visual inspection of observed Ctrough concentrations after i.v. or s.c. administration in cycle 5 and 6, respectively, indicated that a rituximab s.c. dose between 1400 and 1650 mg would achieve non-inferior Ctrough and comparable AUC levels relative to those obtained with the standard i.v. dose for CLL.
Table2 presents geometric mean ratios (90% CI) of Ctrough and AUC for incremental s.c. doses (1400–1650 mg) vs. rituximab i.v. 500 mg m–2. For the geometric mean Ctrough ratio of the fixed rituximab s.c. 1600 mg dose at cycle 5, the predicted lower boundary of the 90% CI was 1.01 (5th percentiles 0.82–1.21). For the geometric mean AUC ratio of rituximab s.c. 1600 mg at cycle 5, the predicted lower boundary of the 90% CI was 0.97 (5th percentiles 0.84–1.10). As the lower bound of the lower boundaries of the 90% CIs for both geometric mean ratios were narrowly above 0.8 (0.82 for Ctrough and 0.84 for AUC), a rituximab s.c. dose of 1600 mg was considered the lowest possible dose for formal non-inferiority testing.
Table 2.
Predicted mean Ctrough and mean AUC values for fixed doses of rituximab s.c. (based on 1000 simulated trials)
| Rituximab SC dose | Geometric mean, µg ml–1 (90% CI) | Power†, % | Geometric mean ratio vs. rituximab i.v.* | ||
|---|---|---|---|---|---|
| Ratio | Lower bound of the 90% CI | Upper bound of the 90% CI | |||
| Predicted mean Ctrough values | |||||
| 1400 mg | 65.6 (51.9, 78.9) | 77.2 | 1.05 (0.87, 1.30) | 0.87 (0.71, 1.07) | 1.25 (1.05, 1.60) |
| 1500 mg | 70.3 (56.8, 85.3) | 90.9 | 1.13 (0.93, 1.41) | 0.94 (0.77, 1.16) | 1.35 (1.12, 1.72) |
| 1600 mg | 75.2 (60.1, 90.1) | 96.3 | 1.21 (1.00, 1.48) | 1.01 (0.82, 1.21) | 1.45 (1.18, 1.83) |
| 1650 mg | 77.6 (62.6, 93.1) | 96.9 | 1.24 (1.03, 1.52) | 1.04 (0.84, 1.25) | 1.48 (1.23, 1.89) |
| Predicted mean AUC values | |||||
| 1400 mg | 3260 (2830, 3710) | 71.9 | 0.94 (0.83, 1.07) | 0.84 (0.73, 0.96) | 1.06 (0.93, 1.21) |
| 1500 mg | 3520 (3060, 4030) | 93.1 | 1.01 (0.89, 1.17) | 0.90 (0.79, 1.04) | 1.13 (1.00, 1.31) |
| 1600 mg | 3760 (3250, 4240) | 98.8 | 1.09 (0.95, 1.23) | 0.97 (0.84, 1.10) | 1.22 (1.07, 1.39) |
| 1650 mg | 3870 (3380, 4390) | 99.3 | 1.12 (0.99, 1.27) | 1.00 (0.87, 1.13) | 1.25 (1.11, 1.44) |
AUC, area under the plasma concentration–time curve; CI, confidence interval; Ctrough, trough serum concentration; i.v., intravenous; s.c., subcutaneous.
*Geometric mean Ctrough for rituximab i.v. was 62.5 µg ml–1 (90% CI 49.4, 73.6); Geometric mean AUC for rituximab i.v. was 3470 µg ml–1 (90% CI 3100, 3820).
†Power to achieve non-inferiority.
Figure2 shows model-based simulations of the concentration–time course of fixed dose rituximab s.c. 1600 mg and rituximab i.v. 500 mg m–2 over cycles 5 and 6. The 5th percentile curves for rituximab s.c. and i.v. showed that for rituximab exposure to be matched in patients with higher body weight (i.e. superimposing the two concentration curves), a higher exposure to rituximab s.c. would be experienced in patients with lower body weight (i.e. patients in the 95th percentile).
Figure 2.
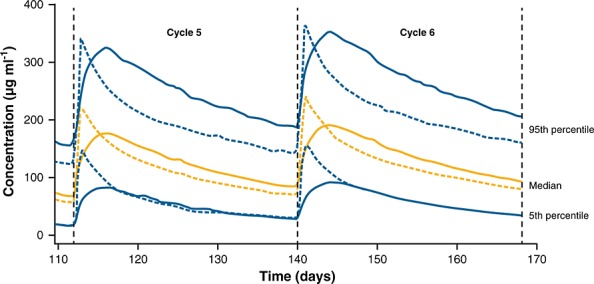
Model-based simulations: concentration-time course for rituximab s.c. 1600 mg and rituximab i.v. 500 mg m−2 during cycles 5 and 6 using 5000 simulations. Dashed lines represent rituximab i.v. and solid lines represent rituximab s.c. i.v., intravenous; s.c., subcutaneous
Parameter estimates for the final model are shown in Supplementary Table S1. All structural parameters were estimated with good precision with the relative standard error (RSE) below 20%. All clearance and volume parameters increased with the body size. These dependencies were described by the power functions of the BSA with power coefficients of 1 and 4/3 for clearance and volume parameters, respectively, mimicking the allometric scaling of weight dependence. The visual predictive check plots of the simulated percentiles overlaid with observed data suggested good agreement between the simulated and observed data, both for i.v. and s.c. doses (Supplementary Figures S1 and S2).
Safety
The safety population included all patients who received a per-protocol rituximab s.c. dose in cycle 6 (n = 55). Comparison of AE profiles for rituximab s.c. and i.v. was restricted to cycles 5 and 6 (Table3) because patients could have enrolled at any time during cycles 1–4. Three patients withdrew from the study after cycle 5, but prior to receiving rituximab s.c., due to AEs (neutropenia [n = 2] and Guillain-Barré syndrome [n = 1]). Safety data for cycles 1–6, and for patients who withdrew after cycle 5 or were incorrectly dosed, are presented in Supplementary Tables S2 and S3.
Table 3.
Overview of safety in cycles 5 and 6
| Patients with AEs | s.c. 1400 mg (n = 16) | s.c. 1600 mg (n = 17) | s.c. 1870 mg (n = 22) |
|---|---|---|---|
| Cycle 5 (rituximab i.v.) | |||
| Any AE, n (%) | 8 (50) | 8 (47) | 13 (59) |
| Total number of events | 11 | 15 | 25 |
| Any grade ≥3 AE, n (%) | 5 (31) | 6 (35) | 5 (23) |
| Total number of events | 5 | 8 | 7 |
| Any ARR, n (%) | – | 2 (12) | − |
| Any serious AE, n (%) | − | 1 (6) | 1 (5) |
| Cycle 6 (rituximab s.c.) | |||
| Any AE, n (%) | 7 (44) | 10 (59) | 18 (82) |
| Total number of events | 12 | 27 | 37 |
| Any grade ≥3 AE, n (%) | 3 (19) | 4 (24) | 3 (14) |
| Total number of events | 4 | 4 | 3 |
| Any ARR, n (%) | 2 (13) | 5 (29) | 5 (23) |
| Any serious AE, n (%) | − | 2 (12) | − |
AE, adverse event; ARR, administration-related reaction; i.v., intravenous; s.c., subcutaneous.
There was a slight increase in the number of AEs reported during cycle 6 (n = 36; 64%) vs. cycle 5 (n = 32; 54%). The majority of AEs were grade 1 or 2 and the most commonly reported in both cycles were neutropenia and leucopenia. Patients who received higher s.c. doses experienced more AEs (n = 7; 44%, n = 10; 59% and n = 18; 82% for 1400 mg, 1600 mg and 1870 mg, respectively). However, a similar pattern was observed in cycle 5 (i.v. administration). More patients experienced at least one grade ≥3 AE during cycle 5 (n = 19; 32%) than during cycle 6 (n = 11; 20%). The most common grade ≥3 AE was neutropenia. The treatment groups (1400 mg, 1600 mg and 1870 mg rituximab s.c.) were well balanced with respect to the incidence of grade ≥3 AEs, both at cycle 5 which was given i.v. (31%, 35% and 23%, respectively) and at cycle 6 (s.c. 19%, 24% and 14%, respectively). Serious AEs (SAEs) were experienced by two patients in cycle 5 (upper respiratory tract infection and febrile neutropenia) and two patients in cycle 6 (diarrhoea and cholecystitis).
More patients experienced ARRs during cycle 6 (n = 12; 21%) compared with cycle 5 (n = 2; 3%). The most frequently occurring ARRs in cycle 6 were pain (n = 4; 7%) and erythema (n = 3; 5%) at the injection site; all were grade 1 or 2.
Immunogenicity
One patient (1.7%) tested positive for HACAs at the 6-month follow-up visit, with no apparent effect on rituximab pharmacokinetics or safety. Six patients (10.7%) were positive for HAHAs, of whom four were positive before rituximab s.c. was administered. No correlation between HAHA-positivity and safety was apparent. The presence of anti-rHuPH20 antibodies prior to the administration of rituximab s.c. is a reflection of the prevalence of such antibodies of unknown etiology in the general population, and has not been associated with any specific AEs or immune phenomena.
Patient and nurse preference
Fifty-six patients (including the patient who received rituximab s.c. 1000 mg) and 56 nurses indicated their preference regarding route of administration. Of these, 52 (92.9%) patients and 53 (94.6%) nurses preferred s.c. administration.
Discussion
Establishing the s.c. route of rituximab administration as an effective and well tolerated alternative to i.v. administration has the potential to not only simplify treatment, but also improve patient convenience and reduce healthcare resource utilization. To enable s.c. administration of clinically effective doses of rituximab, a formulation of 120 mg ml–1 rituximab (12-fold more concentrated than i.v.) with the addition of rHuPH20 (to facilitate s.c. administration), was developed. The clinical development of rituximab s.c. assumes that Ctrough concentrations of rituximab obtained after s.c. administration that are at least as high as those achieved with rituximab i.v. will result in at least the same degree of target site saturation, and therefore achieve the same degree of efficacy.
In SAWYER part 1, pharmacokinetic data collected after administration of one of three s.c. doses were used to refine a pre-existing population pharmacokinetic model that was developed based on clinical data from NHL and CLL populations, including data from the SparkThera trial 14. All structural parameters in the refined model were estimated with good precision with RSE not exceeding 20%. In addition, visual predictive check plots indicated the ability of the model to predict the central tendency and distribution of rituximab concentrations. The final model predicted that a fixed dose of rituximab s.c. 1600 mg would achieve non-inferior Ctrough values compared with 4 weekly rituximab i.v. 500 mg m–2. Given the wide therapeutic window of rituximab 9,10, and its established efficacy and safety 1, a fixed dose approach was considered feasible in adults, provided that the dose selected was sufficient to prevent under-dosing in all patient subgroups. Studies employing high doses of rituximab in CLL did not show any dose limiting toxicity or loss of clinical benefit compared with standard doses 9,10. In the SABRINA and SparkThera (maintenance setting) studies of rituximab s.c., the s.c. route of administration did not have an increased incidence of serious or severe AEs vs. rituximab i.v., over the entire BSA range or in patients with low BSA (BSA ≤1.70 m–2) 14,15. The safety findings were similar for the induction and maintenance settings. Although exposure to rituximab s.c. was reduced with greater body size (BSA), a subset of 15 patients in the SABRINA study with very high BSA (defined as ≥2.1 m–2) had a Ctrough(s.c.): Ctrough(i.v.) geometric mean ratio of 1.327 (90% CI [0.925, 1.905]) and overall response rates were comparable between arms (rituximab s.c. 7/8 patients; rituximab i.v. 6/7 patients). Using modelling and simulation approaches, there appeared to be a minimal risk of underexposure for patients with a BSA <2.7 m2 who are administered rituximab s.c. (F. Hoffmann-La Roche Ltd, data on file). Furthermore, in a study of 12 different monoclonal antibodies, a fixed dosing approach was found to have similar PK and PD outcomes to body size-based dosing 19.
The rituximab s.c. 1600 mg dose selected for further investigation in SAWYER part 2 is higher than the 1400 mg dose that demonstrated non-inferior Ctrough in SparkThera and SABRINA 14,15. This difference reflects the fact that these fixed s.c. doses were bridged to the different weight-based i.v. doses and dosing intervals used for patients with CLL (500 mg m–2 given 4 weekly) vs. FL (375 mg m–2 given 3 weekly, 2 or 3 monthly).
With the limitation of a small sample size and single s.c. administration, the safety profile for s.c. administration appeared similar to that of rituximab i.v. and no new clinically relevant safety signals were identified. The incidence of AEs increased slightly with increasing doses of rituximab s.c.. However, this trend was not observed for grade ≥3 AEs or SAEs. A greater proportion of patients experienced ARRs after treatment with rituximab s.c. than rituximab i.v.. These were primarily transient injection site pain and erythema. This observation was not unexpected and is consistent with results from other rituximab s.c. studies 14,15 and other s.c. monoclonal antibodies 3,20. Interestingly, the increase in ARRs reported with rituximab s.c. did not adversely affect patient preference. More than 90% of patients and treating nurses preferred s.c. to i.v. administration. Preference for s.c. vs. i.v. administration of rituximab will be further assessed in the phase IV PrefMab study (NCT01724021).
The incidence of HACA and HAHA positivity was low and therefore limited conclusions can be drawn regarding their potential effect on the occurrence of AEs or ARRs. However, the available data from part 1 suggest that these antibodies do not adversely affect safety. Further monitoring of immunogenicity and its potential implications will be performed in part 2.
Evaluation of rituximab s.c. efficacy and safety compared with rituximab i.v. is currently ongoing in SAWYER part 2 and, if the results are positive, rituximab s.c. has the potential to replace rituximab i.v. as the standard treatment (plus FC) for CLL. As oral formulations of FC are already available, it is possible that future treatment of patients with CLL could comprise a chemoimmunotherapeutic regimen with minimal i.v. infusions, which would represent an important advance in treatment.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare the following for the previous 3 years: SA reports grants from Roche Canada outside the submitted work, AD reports personal fees from Celgene, Mundipharma and Roche and grants from Chugai and Roche, outside the submitted work, GG reports grants from Roche during the conduct of the study and grants and personal fees from GlaxoSmithKline and Roche and grants from Morphosys, outside the submitted work, XB reports personal fees from Roche and VB reports grants from Roche and personal fees from Janssen-Cilag, outside the submitted work. CM, MB, OC and PS are Roche employees and MB also holds stocks. FHP is a former Roche employee.
This study was sponsored by F. Hoffmann-La Roche Ltd. Support for third-party writing assistance for this manuscript was provided by F. Hoffmann-La Roche Ltd. We also wish to thank all patients and investigators who participated in Part 1 of the SAWYER study.
Contributors
Dr Assouline, lead author, is the Principal Investigator for the SAWYER study.
FHP, CM and PS designed the study, SA, VB, GG, FHP, AD, CM, OC and XB collected the data, MB, FHP, CM, OC and PS performed the data analysis and interpretation. All authors contributed to the writing of the manuscript, provided critical review and approved the final version.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Supporting info item
Table S1 Parameter estimates for the final model
Table S2 Overview of safety during cycles 1 to 6*
Table S3 Adverse events during cycles 5 and 6 for patients who completed cycle 5, but did not receive a per-protocol s.c. dose in cycle 6 (n = 4)
Figure S1 Visual predictive check for final model at cycle 5 (IV administration)
Figure S2 Visual predictive check for final model at cycle 6 (s.c. administration) for a) all doses b) 1870 mg dose c) 1600 mg dose and d) 1400 mg dose
References
- Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von Grünhagen U, Bergmann M, Catalano J, Zinzani PL, Caligaris-Cappio F, Seymour JF, Berrebi A, Jäger U, Cazin B, Trneny M, Westermann A, Wendtner CM, Eichhorst BF, Staib P, Bühler A, Winkler D, Zenz T, Böttcher S, Ritgen M, Mendila M, Kneba M, Döhner H, Stilgenbauer S. International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–74. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- Pivot X, Gligorov J, Müller V, Barrett-Lee P, Verma S, Knoop A, Curigliano G, Semiglazov V, López-Vivanco G, Jenkins V, Scotto N, Osborne S, Fallowfield L. PrefHer Study Group. Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2-positive early breast cancer (PrefHer): an open-label randomised study. Lancet Oncol. 2013;14:962–70. doi: 10.1016/S1470-2045(13)70383-8. [DOI] [PubMed] [Google Scholar]
- Stilgenbauer S, Zenz T, Winkler D, Bühler A, Schlenk RF, Groner S, Busch R, Hensel M, Dührsen U, Finke J, Dreger P, Jäger U, Lengfelder E, Hohloch K, Söling U, Schlag R, Kneba M, Hallek M, Döhner H. German Chronic Lymphocytic Leukemia Study Group. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2009;27:3994–4001. doi: 10.1200/JCO.2008.21.1128. [DOI] [PubMed] [Google Scholar]
- Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. 2007;4:427–40. doi: 10.1517/17425247.4.4.427. [DOI] [PubMed] [Google Scholar]
- Bookbinder LH, Hofer A, Haller MF, Zepeda ML, Keller GA, Lim JE, Edgington TS, Shepard HM, Patton JS, Frost GI. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J Control Release. 2006;114:230–41. doi: 10.1016/j.jconrel.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Thomas JR, Wallace MS, Yocum RC, Vaughn DE, Haller MF, Flament J. The INFUSE-Morphine study: use of recombinant human hyaluronidase (rHuPH20) to enhance the absorption of subcutaneously administered morphine in patients with advanced illness. J Pain Symptom Manage. 2009;38:663–72. doi: 10.1016/j.jpainsymman.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Harb G, Lebel F, Battikha J, Thackara JW. Safety and pharmacokinetics of subcutaneous ceftriaxone administered with or without recombinant human hyaluronidase (rHuPH20) versus intravenous ceftriaxone administration in adult volunteers. Curr Med Res Opin. 2010;26:279–88. doi: 10.1185/03007990903432900. [DOI] [PubMed] [Google Scholar]
- Ismael G, Hegg R, Muehlbauer S, Heinzmann D, Lum B, Kim SB, Pienkowski T, Lichinitser M, Semiglazov V, Melichar B, Jackisch C. Subcutaneous versus intravenous administration of (neo)adjuvant trastuzumab in patients with HER2-positive, clinical stage I-III breast cancer (HannaH study): a phase 3, open-label, multicentre, randomised trial. Lancet Oncol. 2012;13:869–78. doi: 10.1016/S1470-2045(12)70329-7. [DOI] [PubMed] [Google Scholar]
- Keating M, O'Brien S. High-dose rituximab therapy in chronic lymphocytic leukemia. Semin Oncol. 2000;27:86–90. [PubMed] [Google Scholar]
- O'Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J, Lerner S, Keating MJ. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol. 2001;19:2165–70. doi: 10.1200/JCO.2001.19.8.2165. [DOI] [PubMed] [Google Scholar]
- Bittner B, Richter WF, Hourcade-Potelleret F, McIntyre C, Herting F, Zepeda ML, Schmidt J. Development of a subcutaneous formulation for trastuzumab - nonclinical and clinical bridging approach to the approved intravenous dosing regimen. Drug Res. 2012;62:401–9. doi: 10.1055/s-0032-1321831. [DOI] [PubMed] [Google Scholar]
- Bittner B, Richter WF, Hourcade-Potelleret F, McIntyre C, Herting F, Zepeda ML, Schmidt J. Development of a subcutaneous formulation for trastuzumab - nonclinical and clinical bridging approach to the approved intravenous dosing regimen. Drug Res. 2013;63:602. doi: 10.1055/s-0032-1321831. [DOI] [PubMed] [Google Scholar]
- Wynne C, Harvey V, Schwabe C, Waaka D, McIntyre C, Bittner N. Comparison of subcutaneous and intravenous administration of trastuzumab: a phase I/Ib trial in healthy male volunteers and patients with HER2-positive breast cancer. J Clin Pharmacol. 2013;53:192–201. doi: 10.1177/0091270012436560. [DOI] [PubMed] [Google Scholar]
- Salar A, Avivi I, Bittner B, Bouabdallah R, Brewster M, Catalani O, Follows G, Haynes A, Hourcade-Potelleret F, Janikova A, Larouche JF, McIntyre C, Pedersen M, Pereira J, Sayyed P, Shpilberg O, Tumyan G. A comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: results from a two-stage, phase Ib study. J Clin Oncol. 2014;32:1782–91. doi: 10.1200/JCO.2013.52.2631. [DOI] [PubMed] [Google Scholar]
- Davies A, Merli F, Mihaljevic B, Siritanaratkul N, Solal-Céligny P, Barrett M, Berge C, Bittner B, Boehnke A, McIntyre C, Macdonald D. Pharmacokinetics, safety, and efficacy of subcutaneous rituximab in follicular lymphoma (SABRINA): stage 1 analysis of a randomised phase 3 study. Lancet Oncol. 2014;15:343–52. doi: 10.1016/S1470-2045(14)70005-1. [DOI] [PubMed] [Google Scholar]
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhi J, Wenger M, Valente N, Dmoszynska A, Robak T, Mangat R, Joshi A, Visich J. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52:1918–26. doi: 10.1177/0091270011430506. [DOI] [PubMed] [Google Scholar]
- Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User's Guides (1989–2011) Ellicott City: Icon Development Solutions; 2011. [Google Scholar]
- Wang DD, Zhang S, Zhao H, Men AY, Parivar K. Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49:1012–24. doi: 10.1177/0091270009337512. [DOI] [PubMed] [Google Scholar]
- Lundin J, Kimby E, Björkholm M, Broliden PA, Celsing F, Hjalmar V, Möllgård L, Rebello P, Hale G, Waldmann H, Mellstedt H, Osterborg A. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL) Blood. 2002;100:768–73. doi: 10.1182/blood-2002-01-0159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Table S1 Parameter estimates for the final model
Table S2 Overview of safety during cycles 1 to 6*
Table S3 Adverse events during cycles 5 and 6 for patients who completed cycle 5, but did not receive a per-protocol s.c. dose in cycle 6 (n = 4)
Figure S1 Visual predictive check for final model at cycle 5 (IV administration)
Figure S2 Visual predictive check for final model at cycle 6 (s.c. administration) for a) all doses b) 1870 mg dose c) 1600 mg dose and d) 1400 mg dose