

Abstract
Aim
Physiological changes during pregnancy can affect drug disposition. Anticipating these changes will help to maximize drug efficacy and safety in pregnant women. Our objective was to determine if physiologically-based pharmacokinetics (PBPK) can accurately predict changes in the disposition of renally excreted antiretroviral drugs during pregnancy.
Methods
Whole body PBPK models were developed for three renally excreted antiretroviral drugs, tenofovir (TFV), emtricitabine (FTC) and lamivudine (3TC). To assess the impact of pregnancy on PK, time-varying pregnancy-related physiological parameters available within the p-PBPK Simcyp® software package were used. Renal clearance during pregnancy followed glomerular filtration changes with or without alterations in secretion. PK profiles were simulated and compared with observed data, i.e. area under the curves (AUC), peak plasma concentrations (Cmax) and oral clearances (CL/F).
Results
PBPK models successfully predicted TFV, FTC and 3TC disposition for non-pregnant and pregnant populations. Both renal secretion and filtration changed during pregnancy. Changes in renal clearance secretion were related to changes in renal plasma flow. The maximum clearance increases were approximately 30% (TFV 33%, FTC 31%, 3TC 29%).
Conclusions
Pregnancy PBPK models are useful tools to quantify a priori the drug exposure changes during pregnancy for renally excreted drugs. These models can be applied to evaluate alternative dosing regimens to optimize drug therapy during pregnancy.
Keywords: emtricitabine, lamivudine, PBPK, pharmacokinetics, pregnancy, tenofovir
What is Already Known about this Subject
During pregnancy numerous physiological changes can significantly affect drug disposition and dose adjustments may be necessary.
Conducting PK studies in pregnant women is challenging and alternative approaches that can predict changes in drug disposition are needed.
What this Study Adds
This is the first pregnancy PBPK model aimed at predicting maternal drug concentrations of antiretroviral drugs throughout pregnancy.
Changes in renal clearance were simulated and predicted clearances were compared with individual clearances obtained from reported population PK models.
Introduction
In 2012, 35.3 million people were estimated to be living with HIV. Global coverage of antiretroviral (ARV) treatment among HIV-infected pregnant women reached 62% in 2012 1. A fixed dose combination of tenofovir disoproxil fumarate (TDF) + lamivudine (3TC) or emtricitabine (FTC) + efavirenz (EFV) is currently recommended for all pregnant women with HIV by the World Health Organization (WHO) 2.
Physiological changes during pregnancy can be associated with significant variations in pharmacokinetics (PK) 3,4. Ethical, legal and practical considerations often prevent the inclusion of pregnant women in clinical trials. Therefore, off-label drug prescription is not uncommon during pregnancy. Understanding pregnancy-induced changes in PK is essential for designing evidence-based dosing regimens.
Physiologically based pharmacokinetic (PBPK) models are being increasing used to study PK in the pregnant population 5–10. PBPK models are based on actual physiology and pregnant-PBPK models (p-PBPK) incorporate comprehensive information related to anatomical, physiological and metabolic changes that can impact drug disposition. The PK of drugs metabolized by cytochrome (CYP) P450 enzymes can be modified during pregnancy and these metabolic rate changes can be implemented in p-PBPK models 3,6–8,10–12. These models can also account for maternal changes such as increasing body weight, fat mass, cardiac output, plasma volume, glomerular filtration rate (GFR), renal plasma flow and decreasing haematocrit, albumin and α1-acid glycoprotein 3,12.
Our aims were (i) to determine if using the physical and chemical properties of drugs along with the PK parameters in non-pregnant population was able to predict the PK in pregnant population and (ii) to determine the changes in drug exposure of three renally excreted antiretroviral drugs (tenofovir (TFV), FTC and 3TC) in pregnant women.
Methods
Computer software
All simulations were performed using the Simcyp® Simulator (version 13 Release 1). The R software was used for graphics creation (R Core Team (2013), version 3.1.0 URL http://www.R-project.org/).
Development and evaluation of PBPK model
General strategy
The PBPK approach was used to evaluate the effect of pregnancy on the PK of three antiretroviral drugs predominantly eliminated by the kidney. The general workflow is represented in Figure1. PBPK models were firstly developed in non-pregnant populations for intravenous administration (i.v.) and secondly for oral administration. After validation by comparing observed to simulated data we simulated PK profiles throughout pregnancy. We tested different hypotheses about changes in renal clearance and compared simulated clearances throughout pregnancy with clearances obtained by population PK models previously developed.
Figure 1.
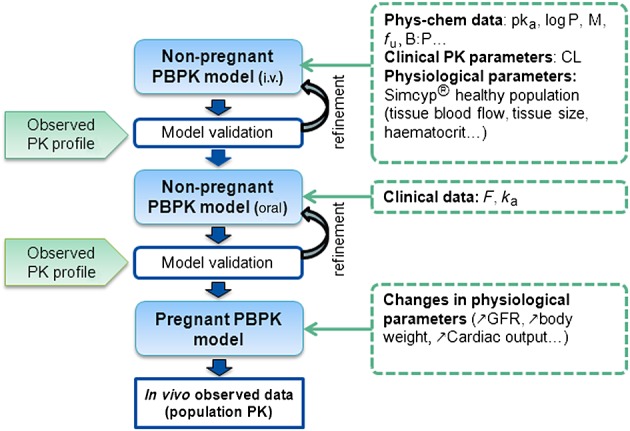
Schematic representation of the workflow of PBPK model development. Hepatic transporter activity for TFV was estimated and there were no i.v. data for FTC. MW molar mass, fu free fraction, B: P blood to plasma ratio, CL clearance, F bioavaibility, ka absorption rate constant, GFR glomerular filtration rate
Non-pregnant population
The full PBPK model comprising 13 tissue/organ compartments was used. Human physiological parameters were obtained using the virtual population implemented in Simcyp®. The physicochemical, biopharmaceutical and PK parameters obtained from literature or in silico prediction tools are summarized in Table1 13–23. Tissue: plasma partition coefficient (Kp) values represent the affinity of a drug for a tissue/organ and are key factors in drugs distribution. They were calculated using Rodgers & Rowland's method 24. Systemic clearance was considered to occur in liver and kidney. For all drugs, absorption was defined by one compartment with first order absorption rate (ka) 23,25,26. TFV is administered as a rapidly hydrolyzed prodrug, TDF. The 300 mg TDF dose was implemented as a 136 mg TFV 27,28. For the three drugs the ka coefficient of variations (CV) were assumed to be 60%. Moreover bioavaibility of TFV is highly variable, then a CV of 60% was assumed 16. All default variabilities were kept for other parameters. Tenofovir PK are described by a bi-compartmental model 30,31. A full PBPK model with blood flow limited distribution did not provide a satisfactory PK profile. Thereafter, a diffusion-limited distribution in hepatic tissue was assumed based on animal data suggesting hepatic accumulation 17,32,33. Moreover TFV was shown to inhibit some hepatic uptake transporters in vitro (OCT1 and ENT1) 34–36. For the liver we considered only uptake transport and passive diffusion. As no value for hepatic transporter activity was available, the parameters were estimated using i.v. data 28, which is a generic refinement strategy especially for drugs in development. Transport parameters were assumed to stay constant whatever the route of administration and throughout pregnancy.
Table 1.
Summary of drug dependent parameters
| Tenofovir [ref.] | Lamivudine [ref.] | Emtricitabine [ref.] | |
|---|---|---|---|
| PBPK model | Full | Full | Full |
| MW (g/mol) | 287 13 | 229 18 | 247 22 |
| pKa | 3.7–6.5 13 | 4.5 18 | 2.65 22 |
| Log P | −2.21 14 | −0.7 20 | −0.43 22 |
| F | 0.28 16 | 0.85 20 | 0.93 22 |
| ka (h−1) | 0.56 26 | 1.04 25 | 0.54 23 |
| fu | 0.993 16 | 0.84* | 0.96 22 |
| Main binding protein | Albumin | Albumin | Albumin |
| B: P ratio | 0.58 17 | 1.5 21 | 1 22 |
| Simcyp predicted Vss (l kg−1) | 0.31* | 0.50* | 0.51* |
| Total CL (l h−1) | 14.2 28 | 23.9 18 | 18 29 |
| CLR (l h−1) | 10.6 28 | 16.8 18 | 13 22,29 |
| CLsecretion: uptake | OAT1 | OCT2 | OCT2 |
| CLint,T | 5.8† | 11.98† | 7.18† |
| efflux | MRP4 | MRP4 | MRP4 |
| CLint,T | 1‡ | 1‡ | 1‡ |
| CLad (l h−1) | 3.6 28 | 7.1 18 | 5 29 |
| Hepatic transport | |||
| CLPD (ml min−1/106 cells) | 4 10−06† | - | - |
| CLint,T (µl min−1/106 cells) | 1.4† | - | - |
*Simcyp prediction toolbox,
†estimated by data fitting,
‡implemented (no impact on plasma PK profile).
MW; molecular weight; F, bioavailability; ka, first order absorption rate; fu, free fraction; B: P ratio, blood to plasma ratio; CLR, renal clearance; CLad, additional systemic clearance; CLPD, passive diffusion clearance; CLint,T, in vitro transporter-mediated intrinsic clearance (µl min−1/106 cells)
The volume of distribution at steady state (Vss) was derived from the dose (D), bioavailability (F), area under the first moment curve (AUMC) and area under the curve (AUC), as follows:
with AUMC and AUC calculated using the trapezoidal rule.
PBPK models evaluation. Firstly, we compared simulated with observed in vivo PK profiles obtained after i.v. administration and oral administration of the standard dosage. Data were digitized from the literature using plot digitilizer 37. Secondly, the evaluation was based on the Cmax and AUC values obtained by non-compartment analyses after administration of the drugs for different dosages and routes of administration. Table2 summarizes input and comparison data used 19,21,28,38–40. PK profiles are mean PK profiles.
Table 2.
Summaries of used studies
| Posology | Observed data | Simulated data | ||||||
|---|---|---|---|---|---|---|---|---|
| Drug | Scheme | Dose | n | Reference | Population | n | Population | Figure |
| TFV | i.v. single dose | 1 mg kg−1 (1 h) | 8 | Deeks et al. 28 | 100 | 2, 3 | ||
| TFV | i.v. single dose | 8 mg kg−1 | 7 | Deeks et al. 28 | 100 | 3 | ||
| TDF | oral single dose | 300 mg | 9 | Wenning et al. 39 | 100 | 2, 3 | ||
| 3TC | i.v. single dose | 0.25 mg kg−1 | 4 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | i.v. single dose | 1 mg kg−1 | 4 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | i.v. single dose | 2 mg kg−1 | 4 | Johnson et al. 21 | HIV infected males | 100 | age: 20–50 years, male | 3 |
| 3TC | i.v. single dose | 8 mg kg−1 | 4 | Johnson et al. 21 | 100 | 2, | ||
| 3TC | oral single dose | 0.25 mg kg−1 | 10 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 0.5 mg kg−1 | 12 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 1 mg kg−1 | 14 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 2 mg kg−1 | 9 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 6 mg kg−1 | 15 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 10 mg kg−1 | 14 | Johnson et al. 21 | 100 | 3 | ||
| 3TC | oral single dose | 150 mg | 24 | Johnson et al. 21 | Healthy volunteers | 100 | 2, 3 | |
| FTC | oral steady-state, twice daily | 25 mg | 8 | Wang et al. 38 | 100 | 3 | ||
| FTC | oral steady-state, twice daily | 100 mg | 8 | Wang et al. 38 | HIV infected males and females | 100 | age: 20–50 years prop.F = 0.5 | 3 |
| FTC | oral steady-state, once daily | 100 mg | 8 | Wang et al. 38 | 100 | 3 | ||
| FTC | oral steady-state, twice daily | 200 mg | 8 | Wang et al. 38 | 100 | 2,3 | ||
| FTC | oral steady-state, once daily | 200 mg | 8 | Wang et al. 38 | 100 | 3 | ||
| TDF | oral steady-state, once daily | 300 mg | 46 | Benaboud et al. 26 | Pregnant women | 100 | Pregnant women 0 < GA < 40 weeks | 4 |
| TDF | oral single dose | 600 mg | 38 | Hirt et al. 23 | Women in labour | 100 | Pregnant women 0 < GA < 40 weeks | 4 |
| 33 < GA < 42 weeks (median = 39 weeks) | age: 20–45 years | |||||||
| TDF | oral steady-state, once daily | 300 mg | 34 | Colbers et al. 42 | Pregnant women | 100 | Pregnant women GA = 33 weeks | 4,5 |
| age: 20–45 years | ||||||||
| 28 < GA < 38 weeks (median = 33 weeks) | ||||||||
| TDF | oral steady-stare, once daily | 300 mg | 19 | Benaboud et al. 26 | Pregnant women | 19 | Pregnant women GA = 33 weeks | 5 |
| 28 < GA < 38 weeks | age: 20–45 years | |||||||
| 3TC | oral steady-state, twice daily | 150 mg | 114 | Benaboud et al. 25 | Pregnant women | 100 | Pregnant women 0 < GA < 40 weeks | 4 |
| 6 < GA < 39 weeks | age: 20–45 years | |||||||
| 3TC | oral steady-state twice daily | 150 mg | 40 | Benaboud et al. 25 | Pregnant women | 40 | Pregnant women GA = 29 weeks | 5 |
| 24 < GA < 34 weeks (median = 29 weeks) | age: 20–45 years | |||||||
| FTC | oral steady-state, twice daily | 200 mg | 83 | Valade et al. 41 | Pregnant women | 100 | Pregnant women 0 < GA < 40 weeks | 4 |
| 5 < GA < 41 weeks (median = 29 weeks) | age: 20–45 years | |||||||
| FTC | oral steady-state, once daily | 200 mg | 27 | Colbers et al. 42 | Pregnant women | 100 | Pregnant women GA = 33 weeks | 4 |
| 28 < GA < 38 weeks (median = 33 weeks) | age: 20–45 years | |||||||
| FTC | oral steady-state, once daily | 200 mg | 26 | Stek et al. 43 | Pregnant women | 100, 26 | Pregnant women GA = 35 weeks | 4, 5 |
| 31 < GA < 38 weeks (median = 35 weeks) | age: 20–45 years | |||||||
GA, gestational age
Pregnant women
Once PBPK models were evaluated on the non-pregnant population, all drugs parameters (e.g. physicochemical properties as molecular weight or log P, Kp values, transport values, etc.) were fixed and only parameters related to physiology (e.g. body weight, blood flow, GFR etc.) were modified in order to mimic the physiology of pregnant women.
Various physiological modifications occurring during pregnancy were taken into account using the Simcyp® module, i.e. weight gain, plasma protein concentration, individual organ/tissue volumes, blood flow and GFRs 12. Studies in non-pregnant and pregnant women for the three drugs showed no significant differences in absorption rate during pregnancy 25,26,41. We then assumed that there were no significant changes in absorption rate during pregnancy 12. The PBPK model was extended by addition of an extra compartment named the foetoplacental unit. This new compartment included the placenta, foetal organs, amniotic fluid, membranes and umbilical cord. Volume and blood flows were lumped and the foetoplacental Kp was assumed to be identical to the muscle Kp (Simcyp® assumption). Using any other tissue Kp value did not result in significant changes in maternal PK profile for these three drugs. The small size of foetal organs does not support an extensive metabolism. Thus, metabolism in foetal organs or placenta was not considered. We focused on the prediction of exposure (area under the curve, AUC) and maximum concentration (Cmax) in pregnant women.
Regarding renal clearance, we assumed that renal filtration follows changes in GFR during pregnancy. Three hypotheses were tested about net renal secretion: (i) no changes occurred during pregnancy, (ii) renal secretion follows GFR changes and (iii) secretion clearance follows renal plasma flow modifications. The clearance components were:
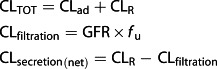 |
The total clearance (CLTOT) was the sum of the renal clearance (CLR) and the additional systemic clearance (CLad). All non-renal clearances were included in CLad. Moreover renal clearance could be split into net secretion (CLsecretion(net)) and filtration (CLfiltration) clearances. Table3 summarizes the Simcyp input used to simulate changes in renal clearance during pregnancy. The equation of Abduljalil et al. describing GFR changes during pregnancy was used to model filtration clearance assuming that only renal filtration changes. The default setting of Simcyp is to scale renal CL by relative changes in GFR in a specific population vs. healthy volunteers. When we implemented only additional systemic clearances and typical renal clearances for healthy volunteers in the simulator, filtration and net secretion were then assumed to follow GFR. We used the mechanistic kidney model to simulate change of net renal secretion clearance following renal plasma flow. Firstly we estimated renal uptake transporter (blood to kidney cells) parameters by using previously simulated PK profiles. The efflux transporter (kidney cells to urine) activity did not impact plasma PK profile and was assumed to be equal to 1 µl min–1/106 cells. The mechanistic kidney model allows, without changing transporter activity, to simulate renal net secretion clearance following renal plasma flow. Amplitudes of GFR and renal plasma flow changes were described according to Abduljalil et al., and the mean increases were 16% and 28%, 37% and 52% and 37% and 32% in the first, second and third trimester for GFR and renal plasma flow 12. Finally, additional changes in renal transporters activity during pregnancy were investigated to explain clearance modifications.
Table 3.
Simcyp input according to renal clearance hypotheses
| Renal clearance changes | ||
|---|---|---|
| CLfiltration | CLsecretion(net) | Simcyp input |
| ∼GFR* | no | • no typical renal clearance, no renal transport |
| • CLad’ = CLad + CLsecretion(net) + CLfiltration | ||
| • CLfiltration ∼ GFR (Abduljalil et al. 12) | ||
| ∼GFR* | ∼GFR* | • no renal transport |
| • Typical renal clearance = CLR | ||
| ∼GFR* | ∼Renal plasma flow* | • no typical renal clearance |
| • renal transport | ||
GFR, glomerular filtration rate; CLad, systemic additional clearance; CLR, renal clerance.
*CLfiltration or CLsecretion(net) follows GFR or renal plasma flow modifications described by Abduljalil et al. 12
Using the p-PBPK model, apparent clearances calculated as the dose-to-AUC ratios were simulated. The apparent clearance ratio between pregnant and non-pregnant women was then calculated throughout pregnancy and compared with data obtained from the literature 23,25,26,41–43. Moreover PK profiles were simulated for the mean observed gestational age (GA) and compared with observed PK data. When data throughout pregnancy were available only data obtained from women with GA similar to the mean observed one (±5 weeks) were kept. Table2 summarizes input and comparison data used 23,25,26,41–43.
Results
Model evaluation for the non-pregnant population
Figure2 shows simulated vs. observed concentration–time courses. The models were roughly satisfactory. However, using a log scale for concentrations showed some differences for the late decay phase where very low concentrations are observed. The predicted Cmax and AUC were all within a two-fold range of observed values and 87.5% were within a 1.25-fold range of observed values (Figure3) 19,21,28,38,39.
Figure 2.
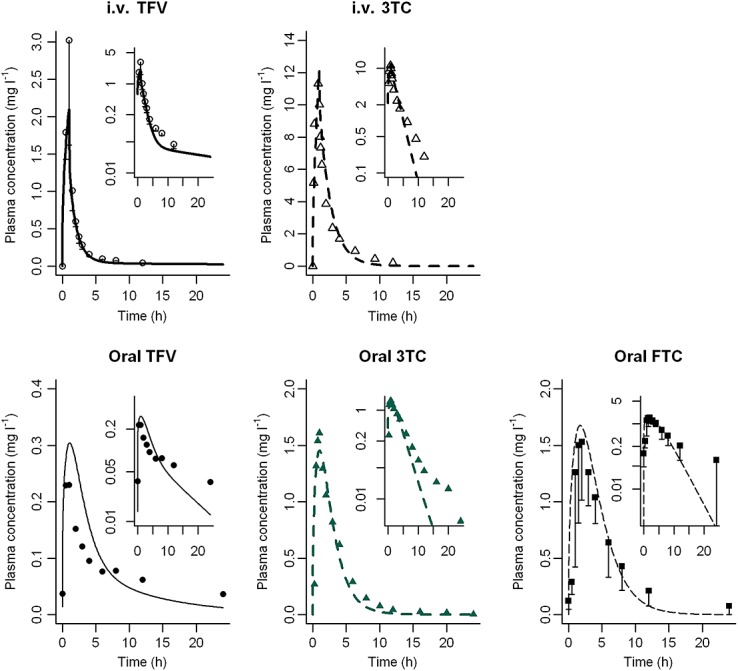
PK profiles in the non-pregnant population. Simulation (lines) of PK profiles for an intravenous administration of 1 mg kg−1 TFV and 8 mg kg−1 3TC and for an oral administration of 300 mg of TDF, 150 mg of 3TC and 200 mg of FTC. We compared these simulations with observed clinical data (TFV circle, 3TC triangle and FTC square).  TFV observed (Deeks et al. 28),
TFV observed (Deeks et al. 28),  3TC observed (Johnson et al. 21),
3TC observed (Johnson et al. 21),  TFV observed (Wenning et al. 39),
TFV observed (Wenning et al. 39),  3TC observed (Wang et al. 38),
3TC observed (Wang et al. 38),  FTC observed,
FTC observed,  TFV predicted,
TFV predicted,  3TC predicted,
3TC predicted,  TFV predicted,
TFV predicted,  3TC predicted,
3TC predicted,  FTC predicted
FTC predicted
Figure 3.
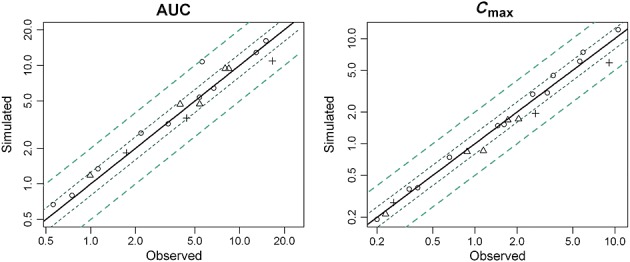
Comparison between simulated and observed PK parameters from several studies in the literature for non-pregnant population 19,21,39. + TFV,  3TC,
3TC,  FTC,
FTC,  Line of unity,
Line of unity,  1.25-fold,
1.25-fold,  2-fold
2-fold
For TFV, i.v. data came from Deeks et al. 28. Eight healthy male volunteers received a 1 h infusion of 1 mg kg–1 day–1 of TFV. The intrinsic clearance for uptake transport in liver was estimated to be 1.4 µl min–1/106 cells and the passive clearance 4.10−6 ml min–1/106 cells. As shown in Figure2, the overall PK profile was well described. Oral data came from the study of Wenning et al. where nine healthy male volunteers received multiple doses of TDF at 300 mg once daily 39. Cmax was then predicted at 0.30 mg l–1, which was close to the observed Cmax of 0.26 mg l–1 (95% confidence interval (CI) 0.22 − 0.31 mg l–1). For the same dose the simulated AUC of 1.84 mg l–1 h was also close to the reported AUC of 1.74 mg l–1 h (95% CI 1.49 − 2.03) 39. TFV volume of distribution (Vd) is 0.76 ± 0.27 l kg–1 after intravenous infusion 28, which is close to the Vss value of 0.74 l kg–1 obtained with our model.
For 3TC, the model evaluation for non-pregnant subjects was achieved by comparing simulated results with the observed data of Van Leeuwen et al. and Moore et al. summarized in the study of Johnson et al. 21. As shown in Figure2, the maximum concentration of 12 mg l–1 predicted by the model for an intravenous administration of 8 mg kg–1 matched the observed Cmax value. Regarding an oral administration of 150 mg the predicted Cmax value was close to the published value: 1.45 vs. 1.54 mg l–1 (range 1.34–1.77 mg l–1), respectively. However we under-predicted the very low concentrations in the late decay phase (Figure2). The AUC reported by Johnson et al. of 5.6 mg l–1 h after oral administration of 4 mg kg–1 of 3TC was not proportional to the dose and was lower than expected (Figure3: circle at (observed: simulated) 5.6 mg l–1 h: 10.73 mg l–1 h). For the same dose regimen Van Leeuwen et al. reported an AUC of 9.2 mg l–1 h which is closer to our model prediction 19. 3TC Vd reported after i.v. infusion was 1.3 ± 0.5 l kg–1, which is consistent with our Vss of 0.83 l kg–1 for the non-pregnant population 19.
To the best of our knowledge no intravenous data for FTC are available. Thus, the model was directly developed for oral administration. Observed data were extracted from the publication of Wang et al. 38. Predicted Cmax obtained after a 200 mg administration was 1.7 mg l–1 (Figure2). The estimated AUC was 9.4 mg l–1 h. These values were close to the values reported by Wang et al., 1.72 ± 0.27 mg l–1 and 10.1 ± 1.8 mg l–1 h for Cmax and AUC, respectively (Figure3). The Vd value was 213 ± 92 l in non-pregnant population which is close to the Vss of 179 l predicted by our model 38.
Extrapolation to pregnant women
Pregnancy-induced changes in renal clearance were assumed to parallel GFR change. Thereby, the predicted clearance values during pregnancy were underestimated (TFV +21%, 3TC +11%, FTC +22%) 23,25,41,42. So according to Xia et al., renal secretion clearance was assumed to increase during pregnancy 44.
For all p-PBPK models, the apparent clearance increased with GA and a small decrease was observed in late pregnancy (Figure4). When renal secretion was assumed to follow GFR, the maximum clearance increase was observed around the 28th week and was approximately 30% (TDF +33%, FTC +31%, 3TC +29%). The Vss also increased during pregnancy (TDF +19%, FTC +24%, 3TC +27%). A Cmax decrease was predicted (TDF −18%, FTC −19%, 3TC −21%). When renal secretion was assumed to follow renal plasma flow, the clearances time-courses were similar but the increases in late pregnancy were less important (Figure4). No significant differences in clearance kinetics throughout pregnancy were observed with our models between these three drugs. As shown in Figure4, almost all clearance observations were within the 90% prediction interval calculated by Simcyp®. These three antiretroviral drugs have wide interindividual variability. Our p-PBPK models were able to describe the observed PK profiles (Figure5). However, few data points in early times were available for TFV and 3TC to really evaluate these parts of the PK profiles.
Figure 4.
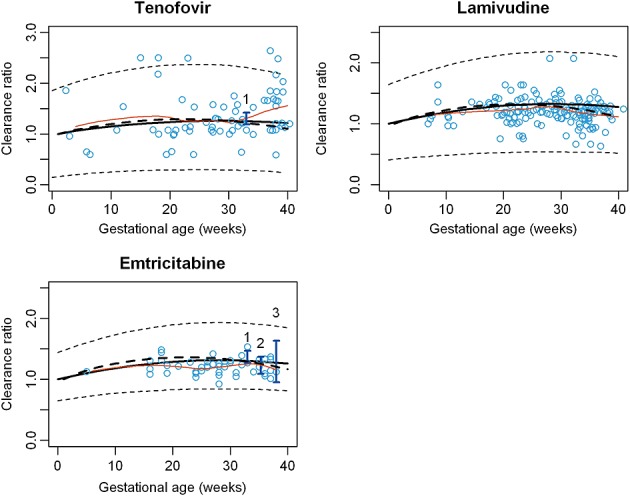
Evolution of clearance throughout pregnancy. Clearance ratio = Cpregnant women: CLnon pregnant women. o Points represent individual clearances obtained by PopPK analyses 25,26,41, I vertical lines represent confidence interval of observed clearances obtained for given mean GA 23,42,43, and the orange line the spline of individual clearance.  mean predicted clearance ratio (GFR),
mean predicted clearance ratio (GFR),  mean predicted clearance ratio (GFR + renal blood flow),
mean predicted clearance ratio (GFR + renal blood flow),  predicted IP,
predicted IP,  spline,
spline,  population PK data, 1 Data form Colbers et al. 42, 2 Data from Stek et al. 43, 3 Data from Hirt et al. 23
population PK data, 1 Data form Colbers et al. 42, 2 Data from Stek et al. 43, 3 Data from Hirt et al. 23
Figure 5.
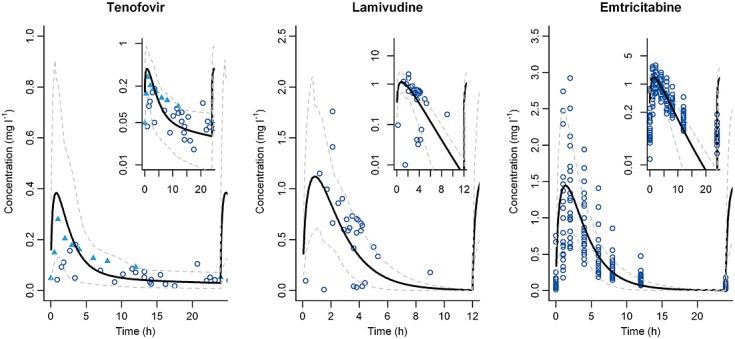
PK profiles in pregnant women 25,26,42,43. Left: Mean simulated tenofovir PK profile (black bold line) and 95% confidence interval (grey dashed line) obtained for a gestational age of 33 weeks (n = 19) compared with observed concentration from Benadoud et al. 26 (blue circle) and Colbers et al. 42 (blue filled triangle). Log scale figure in the top right corner. Middle: Mean simulated lamivudine PK profile (black bold line) and 95% confidence interval (grey dashed line) obtained for a gestational age of 29 weeks (n = 40) compared to observed concentration from Benadoud et al. 25 (blue circle). Log scale figure in the top right corner. Right: Mean simulated emtricitabine PK profile (black bold line) and 95% confidence interval (grey dashed line) obtained for a gestational age of 33 weeks (n = 26) compared with observed concentration from Valade et al. 41 (blue circle). Log scale figure in the top right corner
For 3TC, splitting renal clearances into secretion and filtration and assuming that renal secretion follows renal plasma flow during pregnancy seemed to better fit clearances obtained from population PK (Figure4). This better described clearances in late pregnancy 25 (Figure4).
Oral administration of 150 mg 3TC was simulated for a GA of 29 weeks and compared with data extracted from a population PK study 25 (Figure5). Cmax was predicted to be 1.4 mg l–1.
TFV clearance variation during pregnancy came from a published population PK study 26. These clearances were divided by the mean clearance of non-pregnant women matched by the same age. These ratios were compared with the corresponding clearance ratios simulated by the p-PBPK model (Figure4). Colbers et al. have found an increase of TFV apparent clearance of approximately 30% for 33 weeks of gestation 42. Our model is in agreement with this study and predicts the same increase. Moreover the PK profile obtained after chronic administration of 300 mg TDF was compared with observed data (Figure5): firstly with mean plasma concentration extracted from the publication of Colbers et al. and secondly with population data coming from a therapeutic drug monitoring study 26. Cmax of TFV was close to the 0.28 mg l–1 Cmax reported by Colbers et al. Predicted and observed PK parameters are summarized in Table2.
For FTC, individual clearance ratios throughout pregnancy from a population PK study were compared with our simulations 40 (Figure4). Predicted changes in the apparent clearance for the 33rd and 39th weeks were consistent with observed data 23,42.
The PK profile simulated for a representative pregnant woman was compared with data digitalized from a report 43 (Figure5). For FTC, three studies in the late pregnancy have reported PK alterations. Table4 compares PK parameters obtained by non-compartmental analysis to simulated PK parameters 42. The prediction fold error was less than 1.2 for FTC 43.
Table 4.
| FTC (GA = 35 weeks, n = 26) | TFV (GA = 33 weeks, n = 34) | |||||
|---|---|---|---|---|---|---|
| Stek et al. 42* | Simulated* | Ratio | Colbers et al. 41† | Simulated* | Ratio | |
| AUC (mg l−1 h) | 8 (7.1, 8.9) | 7.3 (5, 10.8) | 0.9 | 2.5 ( 2.2, 2.7) | 1.7 (0.8, 4.3) | 0.7 |
| CL/F (l h−1) | 25 (22.6, 28.3) | 27.2 (18.5, 40.0) | 1.1 | 55 (51, 61) | 78 (32, 176) | 1.4 |
| Cmax (mg l−1) | 1.4 (1.2, 1.6) | 1.4 (0.8, 2.5) | 1 | 0.28 (0.24, 0.31) | 0.27 (0.12, 0.91) | 1 |
| tmax (h) median (range) | 2 (1–4) | 1.8 (0.5 – 2.8) | 0.9 | 1.0 (0.5 – 4.0) | 1.0 (0.4 – 1.6) | 1 |
*Geometric mean (90% confidence interval).
†Geometric mean (95% confidence interval).
Finally, no changes in renal transporter activity were required to describe pregnancy data. Our models suggest that there were no significant changes in renal transporter activity during pregnancy for OCT2, OAT1 and OAT3. Sensitivity analyses were done on transporter activities (Figure6). When the transporter activities were multiplied or divided by 1.5 and 2, the corresponding PK profiles were not significantly different and remained close to the reference PK curves.
Figure 6.
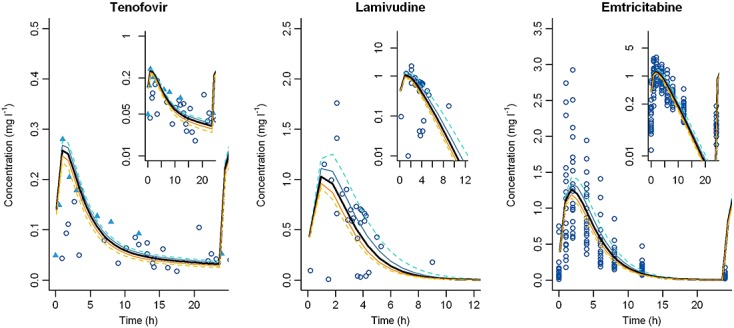
Sensitivity analyses of renal transporter activity. Simulated PK profiles for population representative individuals with transporter activity multiplied (yellow) or divided (blue) by 1.5 and 2 (dashed lines) for the three drugs compared with reference PK profiles (black bold lines)
Discussion
The PBPK models developed were able to predict quantitatively the disposition of three antiretroviral drugs during pregnancy. Drug clearances increased until the 28th week of pregnancy and then slowly decreased. Assuming that renal secretion clearance was related to renal plasma flow during pregnancy allowed a better description of the decrease in 3TC clearance during the late stages of pregnancy. Taking into account the relatively large therapeutic index for these antiretrovirals, the effect of pregnancy on drug disposition appeared not to be significant. The simulated and observed exposures in pregnant women were slightly lower than exposures in non-pregnant women. However, the differences were considered to be negligible and no changes in dose are needed. Moreover, as these three agents have limited plasma protein binding, major changes due to free fraction variations are unlikely to occur and impact clearance 45.
The simulated clearances were compared to clearances obtained by non-compartmental analyses and individual clearances obtained from population PK studies. Significant patient data were used to build these population models and individual clearances throughout pregnancy were estimated 25,26. Our simulations were validated using these in vivo data throughout pregnancy.
A simplified p-PBPK model by Xia et al. for FTC considering only renal elimination and a 40% increase in secretion and GFR predicted a 1.4 fold change in the apparent oral clearance by the 35th week 44. An observed 1.22-fold change 43 was similar to our 1.29-fold predicted change. Xia et al. only included renal excretion and neglected other elimination pathways. This could explain their greater total clearance predicted change, assuming no change in other elimination pathways. Moreover, clearance variations were simulated not only for a given GA but throughout pregnancy.
Few studies have investigated changes in transporter activity during pregnancy, and fewer throughout pregnancy 10,17,44,46,47. Moreover it is difficult to measure the impact of change in transporter activity among other changes. In our models, no changes in transporter activity were required to allow an accurate description of PK during pregnancy. Our results suggest that there were no significant changes in renal transporter activity during pregnancy for OCT2, OAT1 and OAT3. Moreover, sensitivity analyses on transporter activity confirmed the very effect of change in transporter activities (Figure6).
For these three antiretroviral drugs, apart from renal clearance a pathway never accounts for more than 10% of the total clearance. Pregnancy-induced changes in the minor elimination pathways should not result in more than a 5% variation in total apparent clearance, and for this reason they were not included.
The placental–foetal compartment of this model was added as a lumped peripheral compartment and the transplacental transfer was not implemented. This might affect the overall drug disposition in pregnant women. Moreover, the prediction of foetal exposure would be useful to appreciate potentially beneficial or detrimental effects.
In conclusion, these PBPK models successfully predicted the kinetics of TFV, FTC and 3TC with various dosing strategies in non-pregnant and pregnant women. Both renal secretion and filtration varied during pregnancy. Changes in clearance via renal secretion were related to changes in renal plasma flow. Using a pregnancy PBPK modelling approach to predict PK changes can be a powerful tool for drugs with a narrow therapeutic index. The use of PBPK models during drug development could help design clinical trials to study potential exposure changes in pregnant women a priori for compounds excreted by the kidneys.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
We acknowledge the French Agence Nationale de Recherche contre le VIH/SIDA et les hepatites virales (ANRS) for sponsoring this work.
References
- UNAIDS. 2013. UNAIDS 2013 [Internet]. Global report 2013.. Available at http://www.unaids.org/en/resources/documents/2013/ (last accessed 17 December 2013)
- 2013. WHO | Guidelines: HIV [Internet]. WHO.. Available at http://www.who.int/hiv/pub/guidelines/en/ (last accessed 30 July 2014)
- Ke AB, Rostami-Hodjegan A, Zhao P, Unadkat JD. Pharmacometrics in pregnancy: an unmet need. Annu Rev Pharmacol Toxicol. 2014;54:53–69. doi: 10.1146/annurev-pharmtox-011613-140009. [DOI] [PubMed] [Google Scholar]
- Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44:989–1008. doi: 10.2165/00003088-200544100-00001. [DOI] [PubMed] [Google Scholar]
- Luecke RH, Wosilait WD, Pearce BA, Young JF. A computer model and program for xenobiotic disposition during pregnancy. Comput Methods Programs Biomed. 1997;53:201–24. doi: 10.1016/s0169-2607(97)00020-5. [DOI] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. A PBPK Model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the sSite of CYP3A induction. CPT Pharmacomet Syst Pharmacol. 2012;1:e3. doi: 10.1038/psp.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos Biol Fate Chem. 2013;41:801–13. doi: 10.1124/dmd.112.050161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4: PBPK for pregnancy with time-varying physiological parameters. Br J Clin Pharmacol. 2012;74:873–85. doi: 10.1111/j.1365-2125.2012.04363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry PR, Covington TR, Andersen ME, Clewell HJ., III Application of a physiologically based pharmacokinetic model for isopropanol in the derivation of a reference dose and reference concentration. Regul Toxicol Pharmacol. 2002;36:51–68. doi: 10.1006/rtph.2002.1540. [DOI] [PubMed] [Google Scholar]
- Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: how does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos. 2013;41:256–62. doi: 10.1124/dmd.112.050245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19: PBPK prediction of PK changes during pregnancy. Br J Clin Pharmacol. 2014;77:554–70. doi: 10.1111/bcp.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51:365–96. doi: 10.2165/11597440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- Janneh O, Khoo S. Interactions of tenofovir, lamivudine, abacavir and didanosine in primary human cells. Pharmaceutics. 2011;3:326–37. doi: 10.3390/pharmaceutics3020326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuklenyik Z, Martin A, Pau C-P, Garcia-Lerma JG, Heneine W, Pirkle JL, Barr JR. Effect of mobile phase pH and organic content on LC-MS analysis of nucleoside and nucleotide HIV reverse transcriptase inhibitors. J Chromatogr Sci. 2009;47:365–72. doi: 10.1093/chromsci/47.5.365. [DOI] [PubMed] [Google Scholar]
- Gilead Sciences, Inc. Product Information: VIREAD(R) oral tablets, tenofovir disoproxil fumarate oral tablets. 2010.
- Lee WA, He G-X, Eisenberg E, Cihlar T, Swaminathan S, Mulato A, Cundy KC. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother. 2005;49:1898–906. doi: 10.1128/AAC.49.5.1898-1906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ViiV Healthcare Shire Canada. 2011. monographie Heptovir,lamivudine [Internet] [cité 21 janv 2014]. Disponible sur: http://www.gsk.ca/french/docs-pdf/product-monographs/Heptovir.pdf.
- Van Leeuwen R, Lange JM, Hussey EK, Donn KH, Hall ST, Harker AJ, Jonker P, Danner SA. The safety and pharmacokinetics of a reverse transcriptase inhibitor, 3TC, in patients with HIV infection: a phase I study. AIDS Lond Engl. 1992;6:1471–5. doi: 10.1097/00002030-199212000-00008. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. Research Triangle Park. Product Information: Epivir(R), lamivudine.2002.
- Johnson MA, Moore KH, Yuen GJ, Bye A, Pakes GE. Clinical pharmacokinetics of lamivudine. Clin Pharmacokinet. 1999;36:41–66. doi: 10.2165/00003088-199936010-00004. [DOI] [PubMed] [Google Scholar]
- Gilead Sciences, Inc. Product Information: Emtriva(TM), emtricitabine capsules. 2003.
- Hirt D, Urien S, Rey E, Arrive E, Ekouevi DK, Coffie P, Leang SK, Lalsab S, Avit D, Nerrienet E, McIntyre J, Blanche S, Dabis F, Treluyer J-M. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1-infected pregnant women and their neonates. Antimicrob Agents Chemother. 2008;53:1067–73. doi: 10.1128/AAC.00860-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–57. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- Benaboud S, Treluyer JM, Urien S, Blanche S, Bouazza N, Chappuy H, Rey E, Pannier E, Firtion G, Launay O, Hirt D. Pregnancy-related effects on lamivudine pharmacokinetics in a population study with 228 women. Antimicrob Agents Chemother. 2012;56:776–82. doi: 10.1128/AAC.00370-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaboud S, Hirt D, Launay O, Pannier E, Firtion G, Rey E, Bouazza N, Foissac F, Chappuy H, Urien S, Treluyer JM. Pregnancy-related effects on tenofovir pharmacokinetics: a population study with 186 women. Antimicrob Agents Chemother. 2011;56:857–62. doi: 10.1128/AAC.05244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JP, Sueoko CM, Oliyai R, Lee WA, Arimilli MN, Kim CU, Cundy KC. Metabolism and pharmacokinetics of novel oral prodrugs of 9-[(R)-2-(phosphonomethoxy)propyl]adenine (PMPA) in dogs. Pharm Res. 1997;14:1824–9. doi: 10.1023/a:1012108719462. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Barditch-Crovo P, Lietman PS, Hwang F, Cundy KC, Rooney JF, Hellmann NS, Safrin S, Kahn JO. Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiency virus (HIV) therapy, in HIV-\infected adults. Antimicrob Agents Chemother. 1998;42:2380. doi: 10.1128/aac.42.9.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish RG, Leung NWY, Wright TL, Trinh H, Lang W, Kessler HA, Fang L, Wang LH, Delehanty J, Rigney A, Mondou E, Snow A, Rousseau F. Dose range study of pharmacokinetics, safety, and preliminary antiviral activity of emtricitabine in adults with hepatitis B virus infection. Antimicrob Agents Chemother. 2002;46:1734–40. doi: 10.1128/AAC.46.6.1734-1740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnieu M-C, Barkil ME, Livrozet J-M, Cotte L, Miailhes P, Boibieux A, Guitton J, Tod M. Population pharmacokinetics of tenofovir in AIDS patients. J Clin Pharmacol. 2008;48:1282–8. doi: 10.1177/0091270008322908. [DOI] [PubMed] [Google Scholar]
- Jullien V, Tréluyer J-M, Rey E, Jaffray P, Krivine A, Moachon L, Lillo-Le Louet A, Lescoat A, Dupin N, Salmon D, Pons G, Urien S. Population pharmacokinetics of tenofovir in human immunodeficiency virus-infected patients taking highly active antiretroviral therapy. Antimicrob Agents Chemother. 2005;49:3361–6. doi: 10.1128/AAC.49.8.3361-3366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mascio M, Srinivasula S, Bhattacharjee A, Cheng L, Martiniova L, Herscovitch P, Lertora J, Kiesewetter D. Antiretroviral tissue kinetics: in vivo imaging using positron emission tomography. Antimicrob Agents Chemother. 2009;53:4086–95. doi: 10.1128/AAC.00419-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol. 2007;71:619–27. doi: 10.1124/mol.106.028233. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev. 2010;62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud V, Bar-Magen T, Turgeon J, Flockhart D, Desta Z, Wainberg MA. The dual role of pharmacogenetics in HIV treatment: mutations and polymorphisms regulating antiretroviral drug resistance and disposition. Pharmacol Rev. 2012;64:803–33. doi: 10.1124/pr.111.005553. [DOI] [PubMed] [Google Scholar]
- Klein DM, Evans KK, Hardwick RN, Dantzler WH, Wright SH, Cherrington NJ. Basolateral uptake of nucleosides by Sertoli cells is mediated primarily by equilibrative nucleoside transporter 1. J Pharmacol Exp Ther. 2013;346:121–9. doi: 10.1124/jpet.113.203265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huwaldt J. 2010. A plot digitizer [Internet]. Available at http:\\plotdigitizer.sourceforge.net (last accessed 4 April 2014)
- Wang LH, Begley J, St. Claire RL, 3rd, Harris J, Wakeford C, Rousseau FS. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res Hum Retroviruses. 2004;20:1173–82. doi: 10.1089/aid.2004.20.1173. [DOI] [PubMed] [Google Scholar]
- Wenning LA, Friedman EJ, Kost JT, Breidinger SA, Stek JE, Lasseter KC, Gottesdiener KM, Chen J, Teppler H, Wagner JA, Stone JA, Iwamoto M. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2008;52:3253–8. doi: 10.1128/AAC.00005-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KH, Yuen GJ, Hussey EK, Pakes GE, Eron JJ, Jr, Bartlett JA. Population pharmacokinetics of lamivudine in adult human immunodeficiency virus-infected patients enrolled in two phase III clinical trials. Antimicrob Agents Chemother. 1999;43:3025–9. doi: 10.1128/aac.43.12.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valade E, Tréluyer J-M, Dabis F, Arrivé E, Pannier E, Benaboud S, Fauchet F, Bouazza N, Foissac F, Urien S, Hirt D. Modified renal function in pregnancy: impact on emtricitabine pharmacokinetics. Br J Clin Pharmacol. 2014;78:1378–86. doi: 10.1111/bcp.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbers APH, Hawkins DA, Gingelmaier A, Kabeya K, Rockstroh JK, Wyen C, Weizsäcker K, Sadiq ST, Ivanovic J, Giaquinto C, Taylor GP, Moltó J, Burger DM. PANNA network. The pharmacokinetics, safety and efficacy of tenofovir and emtricitabine in HIV-1-infected pregnant women. AIDS Lond Engl. 2013;27:739–48. doi: 10.1097/QAD.0b013e32835c208b. [DOI] [PubMed] [Google Scholar]
- Stek AM, Best BM, Luo W, Capparelli E, Burchett S, Hu C, Li H, Read JS, Jennings A, Barr E, Smith E, Rossi SS, Mirochnick M. Effect of pregnancy on emtricitabine pharmacokinetics. HIV Med avr. 2012;13:226–35. doi: 10.1111/j.1468-1293.2011.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Heimbach T, Gollen R, Nanavati C, He H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pPregnancy. AAPS J. 2013;15:1012–24. doi: 10.1208/s12248-013-9505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71:115–21. doi: 10.1067/mcp.2002.121829. [DOI] [PubMed] [Google Scholar]
- Buist SCN, Cherrington NJ, Choudhuri S, Hartley DP, Klaassen CD. Gender-specific and developmental influences on the expression of rat organic anion transporters. J Pharmacol Exp Ther. 2002;301:145–51. doi: 10.1124/jpet.301.1.145. [DOI] [PubMed] [Google Scholar]
- Yacovino LL, Aleksunes LM. Endocrine and metabolic regulation of renal drug transporters. J Biochem Mol Toxicol. 2012;26:407–21. doi: 10.1002/jbt.21435. [DOI] [PMC free article] [PubMed] [Google Scholar]