

Abstract
Aims
The aim of the present study was to evaluate the pharmacokinetics/pharmacodynamics (PK/PD), safety and tolerability of single intravenous (IV) doses of PF-05231023, a long acting fibroblast growth factor 21 (FGF21) analogue being developed for the treatment of type 2 diabetes mellitus (T2DM).
Methods
T2DM subjects (glycosylated haemoglobin: 7.0–10.5%; on stable metformin therapy and/or diet and exercise) were randomized to receive a single dose of placebo or PF-05231023 (0.5–200 mg). Safety evaluations were performed up to 14 days after dosing. PK and PD endpoints were measured and a PK/PD model was developed for triglyceride – an early marker of drug activity.
Results
No antidrug antibody or serious adverse events (AEs) were observed. The most frequent AEs were gastrointestinal but were generally mild. Plasma PF-05231023 levels peaked immediately post-IV dosing, with mean terminal half-lives of 6.5–7.7 h and 66.5– 96.6 h for intact C- and N-termini, respectively. Intact C-terminus exposures increased proportionally with increasing dose, whereas N-terminus exposures appeared to trend higher than dose-proportionally. Although no apparent effect on plasma glucose was seen, dose-dependent decreases in triglyceride were observed, with a maximum reduction of 48.5 ± 10.0% (mean ± standard deviation) for the 200 mg dose compared with a reduction of 19.1 ± 26.4% for placebo, demonstrating proof of pharmacology. Moreover, a reduction in total cholesterol and low-density lipoprotein cholesterol and an increase in high-density lipoprotein cholesterol were observed in the high-dose groups.
Conclusions
Single IV doses of PF-05231023 up to 200 mg were generally safe and well tolerated by subjects with T2DM. The observed early sign of pharmacology supports further clinical testing of PF-05231023 upon repeated administration.
Keywords: FGF21, pharmacodynamics, pharmacokinetics, T2DM
What is Already Known about this Subject
Nonclinical evidence suggests that PF-05231023 is a functionally active and potent FGF21 agonist with the potential to reduce blood glucose, lipids levels and body weight.
The clinical pharmacology and safety of PF-05231023, a novel FGF21-CovX body conjugate, in humans were unknown prior to the first-in-human single-ascending-dose study.
What this Study Adds
PF-05231023 is safe and well tolerated after a single IV dose in diabetic subjects.
PF-05231023 has a prolonged PK half-life and sustained triglyceride lowering effects after a single dose in humans, and PK/PD modelling supports future clinical trials with a twice-weekly IV dosing regimen.
Introduction
Type 2 diabetes mellitus (T2DM) is one of the largest medical burdens in the world. Several studies have demonstrated that by improving glycaemic control with pharmacological intervention, the risk of T2DM microvascular complications (nephropathy, neuropathy and retinopathy) can be significantly reduced 1–4. Although currently available antidiabetic agents, either singly or in combination, provide effective glycaemic control for some patients, many do not achieve the American Diabetes Association target glycosylated haemoglobin (HbA1c) level, and glucose control tends to deteriorate over time 5–7. Fibroblast growth factor 21 (FGF21) represents a potential new target for the treatment of T2DM and associated comorbidities.
FGF21 stimulates non-insulin-dependent glucose uptake in 3 T3-L1 adipocytes 8. Expression of FGF21 occurs predominantly in the liver and plays an important role in the control of energy balance and glucose metabolism 9. Mice expressing human FGF21 were found to be resistant to weight gain and fat accumulation 8. Daily administration of FGF21 to diabetic rhesus monkeys resulted in a decrease in fasting plasma glucose, fructosamine and triglycerides (TGs), with no evidence of hypoglycaemia 10. The mechanisms by which FGF21 exerts these metabolic effects remain to be fully elucidated. Assessment of FGF21 plasma levels in human subjects has revealed elevated levels in obese individuals, subjects with impaired glucose tolerance and subjects with T2DM, as compared to lean normoglycaemic subjects, and levels appear to correlate with the magnitude of peripheral and hepatic insulin resistance 11. It is possible that this elevation is an adaptation to the insulin resistant state and that further supplementation of FGF21 may help overcome this insulin resistance and lead to improved glycaemic control in subjects with T2DM.
Given the short half-life (t½) of wild-type FGF21 (wtFGF21), 30 min in mice and 2 h in monkeys, several groups are in various stages of pharmaceutical development of long-acting FGF21 analogues and/or antibodies that activate the FGF21 receptor complex, some of which are already being tested in the clinic 12,13. We designed and engineered a long-acting FGF21 mimetic, a CovX body, by covalently linking two recombinant human FGF21 proteins to the Fab of a scaffold antibody, CVX-2000. This molecule, PF-05231023 (also known as CVX-343) demonstrated superior nonclinical pharmacodynamics by extending the serum t½ of FGF21 while preserving full therapeutic functionality 14,15. A single injection demonstrated improved glucose tolerance for 6 days in leptin-deficient obese (ob/ob) mice. In diet-induced obese mice, weekly doses of PF-05231023 reduced body weight, blood glucose and lipid levels. In db/db mice, it increased glucose tolerance, pancreatic β-cell mass and proliferation. The collective nonclinical evidence suggests that PF-05231023 is a functionally active and potent FGF21 agonist, suitable for development as a potential treatment for T2DM. The pleiotropic activities of FGF21 also suggest other potential uses, such as for the treatment of lipid disorders and excess body weight.
This was the first-in-human (FIH) study to assess the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of PF-05231023 after a single intravenous (IV) administration to subjects with T2DM. Both in vitro and in vivo observations in animals suggested differential roles of an intact FGF21 C-terminus vs. an intact FGF21 N-terminus, with the intact FGF21 C-terminus mainly impacting the potency of FGF21 and the intact FGF21 N-terminus being essential for biological signalling 15. Therefore, plasma levels of both intact C-terminus and intact N-terminus of PF-05231023 were assessed in the present study. PK studies were conducted in rats and nonhuman primates in order to predict the PK of PF-05231023 in humans, identify safe exposure margins using the no observed adverse effect level (NOAEL) in toxicology studies, and support dose selections for the FIH study 16.
Materials and methods
Investigational product
PF-05231023 (∼190 kDa) was manufactured at the Sponsor's facility in St Louis, MO, USA. The investigational PF-05231023 substance for injection 30 mg per vial was a lyophilized powder stored at 2–8 °C; it was reconstituted to a liquid formulation at 10 mg ml–1. Each dose of PF-05231023 was diluted to the appropriate concentration and administered intravenously. The placebo was a 0.9% weight/volume (w/v) sodium chloride injection, United States Pharmacopeia (USP).
Subjects
Eighty-four male and female subjects between the ages of 30 and 65 years, inclusive, with a historical diagnosis of T2DM (diagnosed according to the American Diabetes Association guidelines), with body mass index (BMI) of 25–35.5 kg m–2 and a total body weight >50 kg, were planned to be included. Subjects were also required to be on a stable (at least 2 months) treatment of metformin therapy or diet and/or exercise and to have an HbA1c value ≥7% but ≤10.5%. Subjects on background metformin treatment were to remain on the same regimen throughout the duration of the study. Subjects were ineligible if they were current users of antidiabetic agents other than metformin, or had a fasting plasma glucose of ≥270 mg dl–1 or ≤90 mg dl–1. Females were eligible only if previously surgically sterilized or postmenopausal. Subjects were excluded if there was evidence or a history of clinically significant conditions or diseases that posed a risk to the subject's safety if participating in the study, or interfered with study evaluation and procedures, or had a screening 12-lead electrocardiogram (ECG) with a QTc interval >450 ms or a QRS interval >120 ms.
All subjects gave written informed consent, and the protocol was reviewed and approved by the Institutional Review Board(s) (IRB) at each of the investigational centres participating in the study [Elite Research Institute, Miami, FL, USA (approved by the Independent Investigational Review Board); Cetero Research, San Antonio, TX, USA; Profile Institute for Clinical Research, Inc., Chula Vista, CA, USA; Comprehensive Phase One (A Division of Comprehensive NeuroScience Inc.), Miramar, FL, and Ft Meyers, FL, USA (approved by IntegReview Ethical Review Board)]. The trial was conducted in compliance with the ethical principles set forth in the Declaration of Helsinki and its amendments, the International Conference on Harmonization (ICH) E6 Guidance for Good Clinical Practice (CPMP/ICH/135/95) and all other applicable country-specific regulatory requirements and site-specific standard operating procedures (SOPs). The trial was registered at http://clinicaltrials.gov (#NCT01285518).
Study design
This was a randomized, parallel-arm, placebo-controlled, single-ascending-dose study to evaluate the safety, tolerability, PK and PD effects of PF-05231023 after IV administration in subjects with T2DM. The subjects and investigators were blinded to treatment, while Pfizer's clinical research team was unblinded for the purpose of real-time safety monitoring.
The study was conducted in seven cohorts, with 12 subjects per cohort, for a total of 84 volunteers. Within each cohort, subjects were randomized such that 10 received PF-05231023 and two received placebo. The study was conducted at five centres in the United Sates. Two centres participated in each cohort, with each centre recruiting six subjects (five subjects treated with PF-05231023 and one treated with placebo). The cohorts were studied in a sequential dose-escalating fashion, with available PK and safety data from the previous cohort being reviewed prior to initiating dosing of the next cohort.
Screening occurred within 28 days of dosing for each cohort. Subjects were admitted to the clinical research unit (CRU) on Day −1 and remained confined to the CRU until discharge on Day 8, for a total confinement period of 9 days. Follow-up visits occurred on Days 15 and 22 following the single dose of PF-05231023 or placebo.
PF-05231023 was administered to subjects with T2DM as a single dose of 0.5 mg or 1.5 mg IV bolus or 5, 15, 50, 100 or 200 mg 1-h IV infusion on Day 1. The starting dose of 0.5 mg was predicted based on the PK properties in rats and nonhuman primates, and allometric scaling as described previously 16 to generate exposures similar to human endogenous FGF21 plasma levels and thus not expected to elicit pharmacological activity. Using the PK/PD model previously developed for PF-05231023, coupled with projected human PK parameters, IV administration of PF-05231023 to humans in the range 5–10 mg was projected to deliver intact C-terminus and intact N-terminus exposures that elicited glucose lowering as observed in ob/ob mice 14,15. Doses up to 200 mg were tested in this single-dose study in order to adequately explore the safety and tolerability of PF-05231023. This represented a dose that was 20-fold above the projected efficacious dose while maintaining an adequate safety margin compared with the NOAEL in the 4-week monkey safety study.
Safety, immunogenicity and laboratory assessments
Subject safety and tolerability were assessed through physical examinations, laboratory evaluations, vital signs measurements [blood pressure (BP) and pulse rate (PR)], 12-lead ECGs, continuous cardiac monitoring and an ongoing adverse event (AE) assessment. Triplicate supine PR and BP measures were used for vital sign monitoring in Days 1–8. For the purpose of safety monitoring, continuous cardiac monitoring commenced at least 2 h prior to dosing on Day 1 and continued through 8 h postdose. Capillary blood glucose levels were monitored by glucometer before each meal and at bedtime.
Human serum samples were analysed for the presence or absence of anti-PF-05231023 antibodies at QPS, LLC (Newark, DE, USA) using a validated bridging electrochemiluminescent immunoassay (Meso Scale Discovery, Rockville, MD, USA) and following a tiered approach using screening, confirmation and titre/quantitation. In addition, all confirmed positive samples were evaluated for cross-reactivity to FGF21. Samples demonstrating a specific response to PF-05231023 were further characterized for neutralization using a validated cell-based competitive ligand binding assay.
In addition to safety laboratory and immunogenicity assessments, serial blood samples were collected at predose, 0.5 (mid-infusion), 1 (end of infusion), 1.25 (bolus dose only), 1.5, 2.5, 4, 6, 9, 13 and 24 h, and at 2, 3, 4, 5, 7, 14 and 21 days after the start of IV administration for PF-05231023 intact C-terminus and intact N-terminus concentrations. PD measures for fasting plasma glucose (at predose, 1, 2, 3, 4, 5 and 6 days postdose) and insulin (at predose, 1, 2, 4, 6 and 14 days postdose), and for fasting lipid panels including TG, high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and total cholesterol (at predose, 1, 2, 4, 6 and 14 days postdose) were performed.
PK assays
Plasma samples containing aprotinin protease inhibitor were analysed for PF-05231023 concentrations at QPS, LLC, using validated, sensitive and specific enzyme-linked immunosorbent assay (ELISA) methods. It utilized the sponsor's internally generated murine monoclonal anti-FGF21 C-terminus and anti-FGF21 N-terminus capture antibodies for the measurement of intact C-terminus and intact N-terminus of PF-05231023, respectively, and a biotin mouse monoclonal anti-CVX-2000 antibody and streptavidin-conjugated horseradish peroxidase for detection. The lower limit of quantification (LLOQ) for both the PF-05231023 intact C-terminus and intact N-terminus assays was 0.01 µg ml–1. For the C-terminus assay, the between-day assay accuracy, expressed as percentage relative error (%RE), for three quality control (QC) concentration levels, ranged from –0.5% to 3.0%. Assay precision, expressed as the between-day percent coefficient of variation (%CV) of the mean estimated concentrations of QC samples, ranged from 5.2% to 6.4%. For the N-terminus assay, the between-day assay accuracy for the QC samples ranged from –0.7% to 1.5% and the assay precision was 5.1% to 6.7%.
PK analyses
Noncompartmental PK analysis was performed using eNCA (an internally validated system for noncompartmental analysis). Actual sampling times elapsed from dosing time were used for the calculation of PK parameters for individual subjects. These included maximum observed PF-05231023 intact C-terminus and intact N-terminus concentration in plasma (Cmax), time to (Cmax) (tmax), area under the concentration–time curve from time zero to the last observed concentration (AUC0–t) and extrapolated to infinity (AUC0–∞), terminal t½, systemic clearance (CL) and volume of distribution at steady state (Vss). AUC values were calculated using the log-linear trapezoidal rule. AUC0–∞, t½, CL and Vss were reported only where a well-characterized terminal phase was observed, defined as at least 3 data points with an r2 ≥ 0.9 (goodness-of-fit statistic for the log-linear regression) and the percentage of AUC0–∞ due to extrapolation not exceeding 20%. The descriptive statistics of N, mean (median for tmax) and standard deviation (SD) (range for tmax) were calculated for each treatment group.
PD Assays
Insulin was measured using an immunoenzymatic assay (Beckman Coulter, Brea, CA, USA). Glucose was determined using a hexokinase enzymatic method (Roche Diagnostics, Indianapolis, IN, USA). Total cholesterol, TG, direct LDL-C and HDL-C were measured enzymatically using a Roche Modular Analyzer (Roche Diagnostics).
Statistical analysis
This was a Phase 1 clinical trial, with the sample size chosen based on empirical considerations consistent with convention for FIH pharmacological evaluation; therefore, the data have been presented using descriptive statistics only. Placebo subjects from all cohorts were pooled. The descriptive statistics of N, mean and SD for change from baseline (glucose, insulin) or percentage change from baseline (fasting lipids) were calculated for each treatment group. Safety and tolerability data were analysed descriptively by treatment group as well.
PK/PD modelling and simulation
PK and PK/PD modelling was performed using the nonlinear mixed-effect modelling technique and implemented in NONMEM 7.2 (ICON, Dublin, Ireland).
PK/PD modelling was conducted using the observed intact N-terminus PK data as representation of drug exposures in subjects, as suggested by the nonclinical evidence of intact N-terminus’ role in the biological signalling for sustained lipid lowering.
Concentration–response models were fitted to the observed TG data, which employed a sequential PK/PD model. A two-compartment, linear PK model was first developed to describe the intact N-terminus PK profiles after single IV doses of PF-05231023 across the dose range 0.5–200 mg in the current study. The individual predicted intact N-terminus concentrations were assumed to drive the TG response with an indirect response model 17, using a stimulation function on the degradation of TG. The final model included baseline TG as a covariate on Emax as follows:
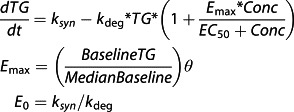 |
where ksyn and kdeg are the rate constants for TG synthesis and degradation processes, Emax is the maximal stimulating effect, EC50 is the PF-05231023 N-terminus plasma concentration to give half of the maximal effect on the degradation of TG and E0 is the TG level at steady state.
Simulations were then performed using this model to assess the PK and PD after 4 weeks of twice-weekly IV administration at 5, 25 or 100 mg PF-05231023.
Results
Subject characteristics
A total of 84 subjects were enrolled and treated across seven cohorts. A total of 70 subjects were randomized and treated with a single dose of PF-05231023 (10 in each of the seven PF-05231023 dose groups), and 14 subjects were treated with a single dose of placebo. All subjects treated completed the study except for one subject in the PF-05231023 100 mg group, who was lost to follow-up after the 7-day inpatient stay. All subjects treated were analysed for AEs, safety evaluations, laboratory data and PD; all subjects who received PF-05231023 were analysed for PK.
The baseline characteristics of the subjects are shown in Table1. The majority of subjects were male (60/84, 71.4%) and white (77/84, 91.7%), and of Hispanic or Latino descent. The mean age ranged from 52.9 to 59.4 years across the treatment groups (overall range 38.0–65.0 years). The mean weight ranged from 81.1 to 95.7 kg (overall range 60.9–124.7 kg) and the mean BMI ranged from 29.7 to 32.1 kg m–2 across the treatment groups (overall range 23.2–35.5 kg m–2). The mean HbA1c at screening ranged from 8.1% to 9.0% across the treatment groups (overall range 7.0–10.5%). The mean baseline fasting glucose and TG ranged from 165 to 192 mg dl–1 (overall range 82–247 mg dl–1) and 148 to 291 mg dl–1 (overall range 57–557 mg dl–1), respectively. Overall, the characteristics of subjects across the treatment groups were similar.
Table 1.
Mean (SD) baseline demographics
| PF-05231023 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | 0.5 mg | 1.5 mg | 5 mg | 15 mg | 50 mg | 100 mg | 200 mg | |||
| n = 14 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | |||
| Age (years) | 52.9 (6.8) | 59.4 (4.1) | 58.4 (3.9) | 53.8 (7.2) | 53.3 (8.2) | 54.5 (8.6) | 55.2 (7.3) | 57.2 (5.7) | ||
| Weight (kg) | 85.1 (13.0) | 81.1 (8.2) | 83.6 (10.7) | 93.6 (13.1) | 87.6 (16.3) | 95.7 (14.7) | 81.2 (12.2) | 86.1 (10.9) | ||
| Height (cm) | 167.6 (9.3) | 164.3 (9.8) | 167.6 (11.0) | 172.7 (9.1) | 170.7 (10.5) | 172.2 (9.8) | 164.7 (8.0) | 169.1 (10.6) | ||
| BMI (kg m–2) | 30.3 (3.4) | 30.1 (2.8) | 29.7 (1.9) | 31.4 (3.5) | 29.9 (3.6) | 32.1 (2.1) | 29.8 (2.9) | 30.1 (2.6) | ||
| Race (% white) | 100 | 90 | 90 | 80 | 90 | 90 | 100 | 90 | ||
| Gender (% male) | 64 | 60 | 70 | 80 | 80 | 70 | 80 | 70 | ||
| HbA1c (%) | 8.6 (1.1) | 9.0 (0.9) | 8.3 (0.6) | 8.9 (0.7) | 8.9 (1.1) | 8.6 (0.7) | 8.2 (1.1) | 8.1 (0.8) | ||
| TG (mg dl–1) | 210 (75) | 148 (65) | 187(74) | 213 (130) | 208 (115) | 291 (177) | 194 (120) | 182 (72) | ||
| FPG (mg dl–1) | 166 (32) | 180 (31) | 168 (38) | 174 (46) | 172 (35) | 192 (37) | 173 (41) | 165 (36) | ||
| Con-Med (% metformin) | 93 | 100 | 100 | 80 | 100 | 80 | 90 | 100 | ||
Abbreviations are as follows: BMI, body mass index (= weight/(height/100)2); Con-Med, concomitant medication; FPG, fasting plasma glucose; HbA1c, glycosylated haemoglobin; n, number of subjects; SD, standard deviation; TG, fasting triglyceride.
Safety, tolerability and immunogenicity
No subject experienced a serious AE (SAE), severe AE, discontinuation due to AE, or dose reduction or temporary discontinuation due to AE. A total of 29/70 (41.4%) subjects who received PF-05231023 experienced AEs (all causalities) and 16/70 (22.9%) experienced treatment-related AEs. The PF-05231023 treatment groups with the highest percentages of subjects experiencing AEs (all causalities and treatment-related) were the 200 mg, 50 mg and 0.5 mg groups. A total of 5/14 (35.7%) placebo-treated subjects experienced AEs (all causalities) and 2/14 (14.3%) experienced treatment-related AEs.
Table2 summarizes the most frequent (occurring in ≥2 subjects overall) AEs of all causality. Diarrhoea was the most frequent AE overall, occurring in a total of 12/70 (17.1%) PF-05231023-treated subjects and no placebo-treated subjects. Nausea occurred in a total of 7/70 (10.0%) PF-05231023-treated subjects and 1/14 (7.1%) placebo-treated subjects. The occurrence of the most frequent AEs among the PF-05231023-treated groups did not appear to be dose dependent.
Table 2.
Treatment-emergent adverse events of all causality occurring in ≥2 subjects
| PF-05231023 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | 0.5 mg | 1.5 mg | 5 mg | 15 mg | 50 mg | 100 mg | 200 mg | |
| System organ class/preferred term | n = 14 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 |
| Gastrointestinal disorders | ||||||||
| Abdominal pain | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Diarrhoea | 0 | 2 | 0 | 1 | 0 | 6 | 0 | 3 |
| Nausea | 1 | 0 | 0 | 2 | 1 | 1 | 1 | 2 |
| Vomiting | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Metabolism and nutritional disorders | ||||||||
| Decreased appetite | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| Nervous system disorders | ||||||||
| Dizziness | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Headache | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 1 |
| Skin and subcutaneous tissue disorders | ||||||||
| Dermatitis contact | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 1 |
None of the most frequent AEs occurring in placebo-treated subjects were considered to be treatment related. In PF-05231023-treated subjects, diarrhoea was the most frequent treatment-related AE (9/70 subjects; 12.9%), followed by nausea (4/70 subjects; 5.7%), abdominal pain and decreased appetite (2/70 subjects; 2.9% for each). The occurrence of the most frequent treatment-related AEs among the PF-05231023-treated groups did not appear to be dose dependent.
All of the AEs in the study were mild in intensity, except for two that were moderate in intensity: an AE of nausea in the PF-05230123 15 mg group and an AE of diarrhoea in the PF-05230123 50 mg group; both of these AEs were considered to be treatment related.
There did not appear to be a dose-dependent effect of PF-05231023 on the occurrences of laboratory abnormalities, and the laboratory abnormalities seen were consistent with those in the diabetic study population. No dose-related trends were observed in ECGs, and findings were consistent with those in the diabetic study population.
In the PF-05231023 200 mg treatment group, mean placebo-adjusted change from baseline values for systolic BP (SBP) and diastolic BP (DBP) trended numerically higher than in the other dose groups. The mean placebo-adjusted increases from baseline in SBP ranged from 4.1 mmHg to 12.1 mmHg on Day 1; and the increases were sustained on Days 2–15, with mean values ranging from 3.5 mmHg to 13.3 mmHg. Consistent mean placebo-adjusted increases from baseline in DBP were also observed in the 200 mg treatment group, ranging from 2.8 mmHg to 7.1 mmHg on Day 1 and 3.1 mmHg to 7.4 mmHg on Days 2–15. No other trends in SBP, DBP or PR were observed across all other PF-05231023-treated groups. Out of the 10 subjects treated with 200 mg PF-05231023, the increases in SBP and DBP were sustained for 24 h after dosing in five and four subjects respectively; in the remaining subjects, the increases were observed intermittently.
No treatment-related antidrug antibodies were detected in subjects after a single IV dose of PF-05231023. A transient pre-existing antidrug antibody response (log2 endpoint titre = 7.01) was detected in one subject prior to receiving PF-05231023 1.5 mg (i.e. baseline predose). This pre-existing antidrug antibody response was further characterized as cross-reactive to FGF21, and negative for neutralization.
PK
Plasma PF-05231023 intact C-terminus and intact N-terminus concentration–time profiles across the studied doses are presented in Figure1. PK parameters from the noncompartmental analysis are summarized in Table3.
Figure 1.
Mean (standard deviation) plasma PF-05231023 intact C-terminus (A) (with insert showing expanded profiles up to 3 days postdose) and intact N-terminus (B) concentration–time profiles (semi-log scale) after a single intravenous bolus (0.5 mg and 1.5 mg) or 1-hour intravenous infusion (5 mg, 15 mg, 50 mg, 100 mg and 200 mg) doses. Lower limit of quantification was 0.01 µg ml–1.  0.5 mg,
0.5 mg,  1.5 mg,
1.5 mg,  5 mg,
5 mg,  15 mg,
15 mg,  50 mg,
50 mg,  100 mg,
100 mg,  200 mg
200 mg
Table 3.
Mean (SD) pharmacokinetic parameters of PF-05231023 intact C- and N-terminus after a single IV bolus or 1-h infusion administration in subjects with type 2 diabetes mellitus
| PF-05231023 | |||||||
|---|---|---|---|---|---|---|---|
| 0.5 mg | 1.5 mg | 5 mg | 15 mg | 50 mg | 100 mg | 200 mg | |
| Parameters | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 | n = 10 |
| Intact C-terminus | |||||||
| C0/Cmax (µg ml–1) | 0.117*(0.048) | 0.363*(0.082) | 1.36 (1.26) | 3.32 (0.66) | 11.7 (4.9) | 28.7 (8.0) | 47.8 (13.8) |
| tmax (h) | NA | NA | 1.5 (1.5, 3.0) | 1.5 (1.0, 2.5) | 1.3 (1.0, 1.5) | 1.5 (1.0, 6.0) | 1.0 (1.0, 2.5) |
| AUC0–t (µg h ml–1) | 0.672 (0.365) | 2.57 (0.48) | 10.2 (4.1) | 29.4 (6.7) | 95.1 (13.7) | 244 (84) | 409 (135) |
| AUC0–∞†(µg h ml–1) | 1.38, 1.58‡ | 2.79 (0.56) | 10.8 (4.0) | 30.1 (7.1) | 95.4 (13.8) | 244 (84) | 410 (135) |
| t½†(h) | 6.19, 6.92‡ | 6.50 (1.08) | 7.17 (1.24) | 7.14 (1.50) | 7.75 (1.13) | 7.50 (1.00) | 7.30 (1.71) |
| CL†(l h–1) | 0.317, 0.36‡ | 0.537 (0.097) | 0.464 (0.157) | 0.497 (0.123) | 0.525 (0.089) | 0.409 (0.120) | 0.488 (0.257) |
| Vss†(l) | 3.14, 3.20‡ | 4.90 (0.93) | 4.60 (1.26) | 5.02 (0.97) | 5.65 (1.92) | 4.08 (1.19) | 4.66 (1.07) |
| Intact N-terminus | |||||||
| C0/Cmax (µg ml–1) | 0.132*(0.048) | 0.415*(0.081) | 1.20 (0.39) | 3.69 (0.71) | 14.2 (3.9) | 28.9 (6.4) | 61.9 (8.1) |
| tmax (h) | NA | NA | 1.5 (1.5, 6.0) | 1.5 (1.0, 2.5) | 1.3 (1.0, 6.0) | 1.5 (1.0, 2.5) | 1.5 (1.0, 2.5) |
| AUC0–t (µg h ml–1) | 2.96 (1.89) | 11.6 (4.4) | 47.7 (10.8) | 169 (50) | 565 (162) | 1210 (390) | 2800 (760) |
| AUC0–∞†(µg h ml–1) | 4.73 (1.59) | 11.7 (4.3) | 49.3 (10.8) | 171 (51) | 581 (169) | 1240 (400) | 2850 (780) |
| t½†(h) | 31.5 (6.6) | 37.6 (9.0) | 66.5 (12.8) | 77.4 (12.0) | 81.5 (12.6) | 87.7 (16.6) | 96.6 (8.1) |
| CL†(l h–1) | 0.106 (0.046) | 0.128 (0.042) | 0.101 (0.020) | 0.0879 (0.0322) | 0.0861 (0.0292) | 0.0805 (0.0256) | 0.0702 (0.0186) |
| Vss†(l) | 4.54 (0.82) | 6.15 (1.18) | 7.17 (1.40) | 7.32 (1.24) | 7.02 (1.47) | 7.00 (1.48) | 7.20 (1.43) |
Data presented are geometric mean (SD), except for t½ [reported as arithmetic mean (SD)] and tmax [reported as median (minimum, maximum)]. All values are reported to three significant figures, except for tmax, for which values are reported to one decimal place. Abbreviations are as follows: AUC0–t, area under the concentration–time curve from time zero to the last observed concentration; AUC0–∞, area under the concentration–time curve from time zero to infinity; CL, clearance; Cmax, maximum observed plasma concentration; C0, back-extrapolated concentration at time 0; IV, intravenous; NA, not applicable; SD, standard deviation; t½: terminal half-life; Vss, volume of distribution at steady state
*C0 is reported for IV bolus dosing at 0.5 mg and 1.5 mg;
†n ≤ 10 for these parameters owing to the fact that the terminal elimination phase cannot be accurately estimated for some subjects in each of the dose groups;
‡n = 2, individual values are listed, terminal elimination phase cannot be accurately estimated for the remaining subjects in the dose group.
Following IV bolus injection of single 0.5 mg or 1.5 mg doses of PF-05231023, peak intact C-terminus concentrations were achieved instantaneously. Following a 1-h IV infusion of single 5 mg to 200 mg doses of PF-05231023, peak intact C-terminus concentrations were generally observed at 1.5 hours postdose (30 min after the end of the infusion). The C-terminus concentrations then declined in a linear manner over time, and the decline was in parallel across the 0.5 mg to 200 mg dose range. Both Cmax and AUC0–∞ increased proportionally with increasing dose. The geometric mean values of PF-05231023 C-terminus CL ranged from 0.41 l h–1 to 0.54 l h–1, and Vss ranged from 4.1 l to 5.7 l across doses from 1.5 mg to 200 mg. Terminal t½ was also consistent across all doses, with arithmetic mean values ranging from 6.5 h to 7.7 h by treatment.
Similar to the intact C-terminus profiles, peak intact N-terminus concentrations were achieved instantaneously following bolus injection and generally at 1.5 h postdose (30 min after the end of the infusion) following IV infusions. The N-terminus concentrations then declined in a biphasic manner over time. Limited by the bioanalytical assay sensitivity (LLOQ of 0.01 µg ml–1), the terminal phase for the 0.5 mg and 1.5 mg dose groups was not fully characterized in the present study. Across the dose range of 5 mg to 200 mg, Cmax of intact N-terminus increased dose-proportionally with increasing dose, and the geometric mean Vss was approximately 7 l. The geometric mean values of the PF-05231023 N-terminus CL, ranging from 0.101 l h–1 to 0.0702 l h–1, trended lower with increasing dose; mean AUC0–∞ trended higher than dose-proportionally; and the arithmetic mean terminal t½, ranging from 66.5 h to 96.6 h, trended longer with increasing dose, across the 5 mg to 200 mg dose range.
PD Effects in T2DM subjects
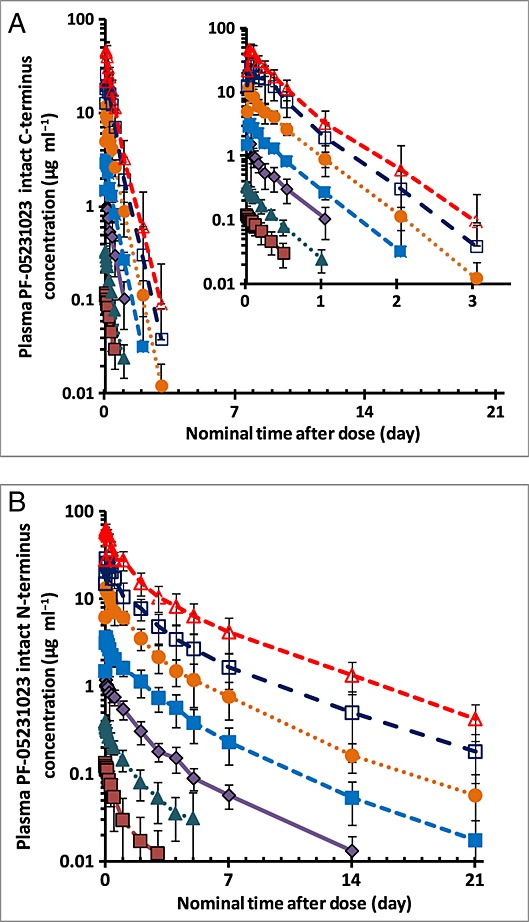
Figure2 shows the effect (change from baseline) –time profiles for PF-05231023 and placebo on fasting plasma glucose and insulin. There was a slight trend of decreases from baseline in fasting glucose up to Day 7 (6 days postdose) for all treatment groups, including placebo, but no dose-dependent pattern among the PF-05231023 groups. Similarly, no discernible changes were observed in mean insulin values up to Day 7 in any of the treatment groups.
Figure 2.
Effects of PF-05231023 on fasting plasma glucose (A) and insulin (B) levels. Time course of mean (standard deviation) change from baseline.  0.5 mg,
0.5 mg,  1.5 mg,
1.5 mg,  5 mg,
5 mg,  15 mg,
15 mg,  50 mg,
50 mg,  100 mg,
100 mg,  200 mg,
200 mg,  Placebo
Placebo
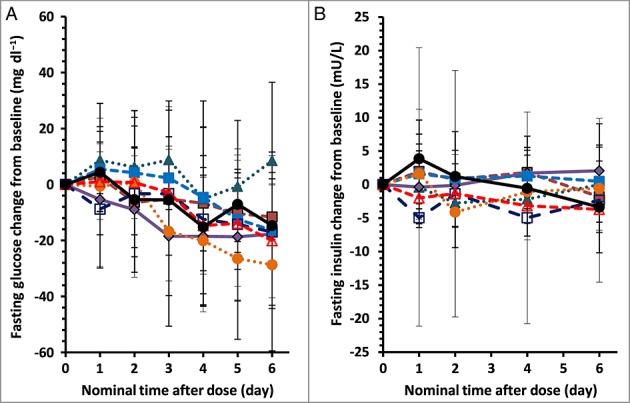
Changes in lipid parameters were observed. Figure3 shows the effect (percentage change from baseline) –time profiles for PF-05231023 and placebo on fasting lipid parameters. A summary of fasting lipid measurements for each of the PF-05231023 treatment groups and placebo at baseline and maximum mean change observed after a single IV administration is provided in Table4. There was a dose-dependent decrease from baseline in mean TGs (Figure3 and Table4). Relative to baseline, apparent mean decreases in fasting TG levels were observed as early as 1 day postdose for all ≥ 5 mg PF-05231023 dosing groups, and maximal TG lowering was observed between Days 4 and 6, with a mean ± SD reduction of 6.8 ± 18.6%, 13.2 ± 26.8%, 14.0 ± 19.8%, 30.2 ± 18.8%, 37.0 ± 13.8%, 36.7 ± 15.9% and 48.5 ± 10.0% observed for the PF-05231023 0.5 mg, 1.5 mg, 5 mg, 15 mg, 50 mg, 100 mg and 200 mg dose groups, respectively, compared with a reduction of 19.1 ± 26.4% for the placebo (Figure3 and Table4). These reductions were maintained up to Day 15 following administration of the PF-05231023 50 mg, 100 mg and 200 mg doses. Small increases from baseline in HDL-C in all treatment groups except for the 0.5 mg group were observed up to Day 7 (Figure3). Only doses ≥ 50 mg showed increases greater than placebo, with the mean ± SD peak increases of 11.7 ± 3.8%, 13.0 ± 10.3% and 17.4 ± 7.6% for the PF-05231023 50 mg, 100 mg and 200 mg groups, respectively, compared with 7.7 ± 31.0% for the placebo group (Table4). With the exception of an initial increase in the 50 mg and 200 mg groups, there was a slight trend of decreases from baseline in LDL-C up to Day 7 across all treatment groups (Figure3); no changes discernible from placebo could be identified at the peak LDL-C reduction (mean peak change ranging from –4.5% to –9.1% for the PF-05231023-treated groups compared with –6.6% for the placebo; Table4). In general, there was a trend of decreases from baseline in total cholesterol up to Day 7, and a dose-dependent response was evident among the PF-05231023 groups across doses from 5 mg to 100 mg, with a mean ± SD peak reduction of 11.4 ± 13.4% for the 100 mg dose; however, changes in the 200 mg dose group (–6.5 ± 18.5%) appeared similar to the changes observed in the placebo group (–4.9 ± 17.5%) (Table4, Figure3).
Figure 3.
Effects of PF-05231023 on fasting lipids: low-density lipoprotein (LDL)-cholesterol (A), high-density lipoprotein (HDL)-cholesterol (B), triglyceride (C) and total cholesterol (D). Time course of mean (standard deviation) percentage change from baseline.  0.5 mg,
0.5 mg,  1.5 mg,
1.5 mg,  5 mg,
5 mg,  15 mg,
15 mg,  50 mg,
50 mg,  100 mg,
100 mg,  200 mg,
200 mg,  Placebo
Placebo
Table 4.
Summary of mean (SD) changes in lipid parameters
| PF-05231023 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | 0.5 mg | 1.5 mg | 5 mg | 15 mg | 50 mg | 100 mg | 200 mg | |
| Lipid panel | n = 13* | n = 10 | n = 10 | n = 10 | n = 10 | n = 5* | n = 10 | n = 10 |
| LDL-C | ||||||||
| Baseline (mg dl–1) | 135.7 (34.7) | 111.1 (41.2) | 127.2 (36.9) | 110.3 (16.2) | 109.2 (24.6) | 132.8 (35.1) | 115.5 (32.6) | 107.1 (39.3) |
| Maximum change from baseline (%) | –6.6 (15.0) | –6.5 (8.2) | –4.5 (12.1) | –7.6 (6.6) | –9.1 (17.7) | –5.3 (18.2) | –7.2 (23.5) | –7.5 (24.3) |
| Time of maximum change (days postdose) | 4 | 1 | 6 | 2 | 6 | 6 | 6 | 6 |
| HDL-C | ||||||||
| Baseline (mg dl–1) | 48.7 (14.0) | 53.3 (12.7) | 40.3 (10.4) | 37.9 (8.9) | 44.8 (12.4) | 43.6 (8.5) | 41.4 (10.0) | 41.8 (10.2) |
| Maximum change frombaseline (%) | 7.7 (31.0) | -3.4 (6.7) | 6.2 (7.8) | 2.8 (7.7) | 5.6 (8.1) | 11.7 (3.8) | 13.0 (10.3) | 17.4 (7.6) |
| Time of maximum change (days postdose) | 1 | 2 | 6 | 4 | 1 | 2 | 6 | 2 |
| Triglyceride | ||||||||
| Baseline (mg dl–1) | 210.2 (74.9) | 148.4 (65.1) | 187.0 (74.4) | 213.0 (129.6) | 207.6 (114.5) | 290.8 (176.8) | 193.8 (120.0) | 182.4 (72.2) |
| Maximum change from baseline (%) | –19.1 (26.4) | –6.8 (18.6) | –13.2 (26.8) | –14.0 (19.8) | –30.2 (18.8) | –37.0 (13.8) | –36.7 (15.9) | –48.5 (10.0) |
| Time of maximum change (days postdose) | 6 | 6 | 6 | 4 | 6 | 6 | 4 | 4 |
| Total cholesterol | ||||||||
| Baseline (mg dl–1) | 208.8 (39.0) | 174.2 (42.7) | 192.7 (47.6) | 178.9 (29.7) | 177.6 (21.8) | 216.8 (43.0) | 186.1 (41.1) | 169.5 (40.7) |
| Maximum change from baseline (%) | –4.9 (17.5) | –0.3 (11.2) | –0.3 (10.4) | –5.3 (10.3) | –8.9 (9.9) | –10.1 (8.2) | –11.4 (13.4) | –6.5 (18.6) |
| Time of maximum change (days postdose) | 6 | 6 | 6 | 4 | 6 | 6 | 6 | 6 |
Data presented as maximum decreases from baseline for LDL-C, triglyceride and total cholesterol, and maximum increases from baseline for HDL-C. Abbreviations are as follows: HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; SD, standard deviation.
*Baseline measurements were missing for one subject in the placebo group and five subjects in the 50 mg group. Data from these subjects are excluded.
PK and PD relationships
A sequential PK/PD model was developed to describe the effects of the PF-05231023 N-terminus concentration–time course and its impact on fasting TGs in subjects with T2DM. The final variables and estimates are shown in Table5. The estimated volume of distribution in the central compartment (Vcentral) of 4.24 l approximated the volume of the vasculature system. CL values were calculated to be 89 ml h–1, faster than a typical monoclonal antibody (average 10 ml h–1). An EC50 of 5.52 µg ml–1 was estimated for TG-lowering effects but with less confidence in the parameter values (%RSE >80%) (Table5). The observed and modelled PK of the N-terminus and its effects on fasting TG are shown in Figure4.
Table 5.
Population pharmacokinetic and pharmacodynamic model variables of N-terminus PF-05231023 and its effects on fasting triglyceride
| Variables | Estimate (%RSE) | Interindividual variability (%) |
|---|---|---|
| Pharmacokinetic variables | ||
| Vcentral (l) | 4.24 (3.7%) | 29.8% |
| k10 (h−1) | 0.021 (2.8%) | 21.6% |
| k12 (h−1) | 0.0116 (7.5%) | 23.8% |
| k21 (h−1) | 0.015 (5.2%) | 11.4% |
| Pharmacodynamic variables | ||
| kdeg (h−1) | 0.00516 (29.8%) | – |
| E0 (mg dl−1) | 180 (5.2%) | 45.3% |
| Emax | 2.68 (50%) | 45.2% |
| EC50 (µg ml−1) | 5.52 (83.5%) | – |
| Baseline power on Emax | 0.674 (34.6%) | – |
Abbreviations are as follows: E0, baseline/steady-state triglyceride level; EC50, half-maximal stimulatory concentration; Emax, maximum stimulatory effect; k10, first-order elimination rate constant from central compartment; k12, first-order distribution rate constant from central to peripheral compartment; k21, first-order distribution rate constant from peripheral to central compartment; kdeg, first-order triglyceride degradation rate constant; Vcentral, volume of central compartment.
Figure 4.
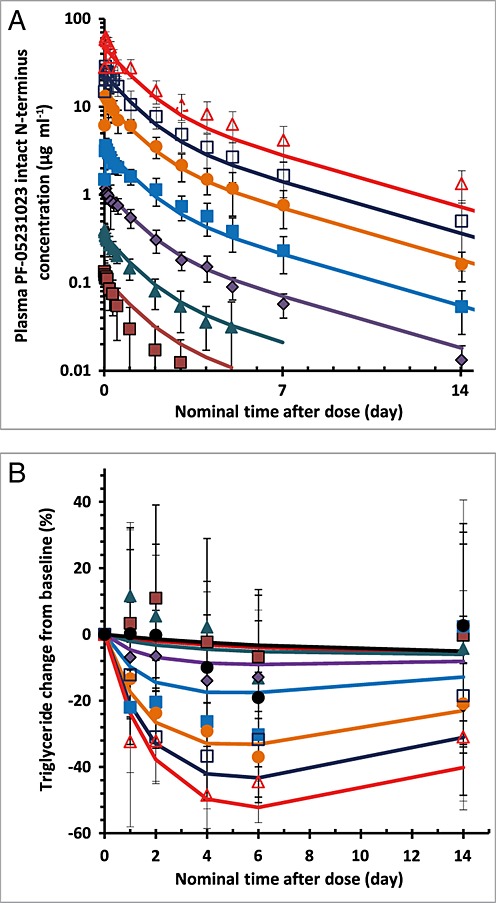
Pharmacokinetic/pharmacodynamic model-predicted time profiles (lines) and the observed time profiles (symbols) of plasma PF-05231023 intact N-terminus concentrations (A) and the percentage change from baseline in fasting triglyceride levels (B) after a single intravenous administration of 0.5, 1.5, 5, 15, 50, 100 or 200 mg PF-05231023. The symbols are the observed mean (standard deviation) values and the lines are model-predicted population mean profiles.  0.5 mg,
0.5 mg,  1.5 mg,
1.5 mg,  5 mg,
5 mg,  15 mg,
15 mg,  50 mg,
50 mg,  100 mg,
100 mg,  200 mg,
200 mg,  0.5 mg-predicted,
0.5 mg-predicted,  , 1.5 mg-predicted,
, 1.5 mg-predicted,  5 mg-predicted,
5 mg-predicted,  15 mg-predicted,
15 mg-predicted,  50 mg-predicted,
50 mg-predicted,  100 mg-predicted,
100 mg-predicted,  200 mg-predicted
200 mg-predicted
Simulated PK and PD profiles after twice-weekly IV administration of PF-05231023 for 4 weeks are presented in Figure5. Steady state for both drug exposure and the effects on TG lowering appeared to have been reached after 3 weeks of dosing. With the twice-weekly IV dosing, the reduction in fasting TG levels can be maintained with relatively small peak-to-trough fluctuation.
Figure 5.
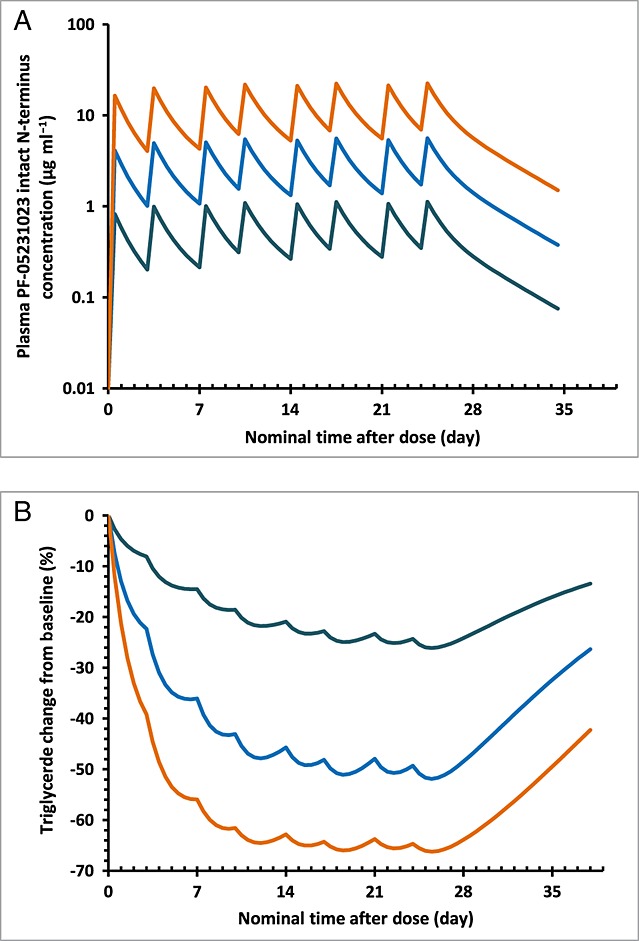
Simulated population mean concentration–time profiles in semi-log scale (A) and the time course of the percentage change from baseline in fasting triglyceride levels (B) after twice-weekly intravenous administration of 5, 25 or 100 mg PF-05231023.  5 mg twice weekly,
5 mg twice weekly,  , 25 mg twice weekly,
, 25 mg twice weekly,  100 mg twice weekly
100 mg twice weekly
Discussion
The effects of FGF21, an important metabolic regulator for glucose and lipid homeostasis, are generally believed to be mediated through the formation of an FGF21/β-klotho/FGFR1C (FGF receptor 1 c isoform) complex, with intact FGF21 C-terminus required for high-affinity binding with β-klotho and the intact FGF21 N-terminus for interaction with FGFR1C. In vivo studies of PF-05231023, a long-acting FGF21 analogue with differential PK characteristics of intact C- and N-termini, in diet-induced obese mice and ob/ob mice coupled with PK/PD modelling indicated that the intact FGF21 N-terminus alone appears to be sufficient to drive glucose lowering as well as body weight loss 15. In the FIH study, a single dose of PF-05231023 was able to elicit robust and sustained changes in fasting lipids, TG in particular, which was also consistent with the role of intact N-terminus in biological signalling, given the apparently longer t½ and therefore more sustained exposures to intact N-terminus than intact C-terminus observed in the T2DM subjects. Consequently, the sequential PK/PD model, developed to characterize the observed reduction in fasting TG levels and later used for simulation of the optimal dosing scheme for the multiple-dose study, used only the exposures of intact N-terminus for PK characterization.
Due to assay limitation, the terminal phase of the intact N-terminus concentration–time profile for the 0.5 mg and 1.5 mg dose groups was not captured in full. Across the dose range of 5 mg to 200 mg PF-05231023, a trend of decreased clearance and increased t½ of intact N-terminus with increasing dose was observed, although the magnitude of the change was small (with a CL of 0.101 l h–1 for the 5 mg dose to 0.0702 l h–1 for the 200 mg dose, and a terminal t½ of 66.5 h for the 5 mg dose to 96.6 h for the 200 mg dose). As target-mediated disposition is not expected for this FGF21 analogue, a simplified PK model assuming linear elimination of PF-5231023 was developed at this stage of clinical development. Similarly, no covariates were explored or included in the PK model. The population PK model of both intact C- and N-termini will be further refined with data from a future multiple-ascending-dose study. The EC50 value for TG lowering was not accurately estimated in the current PK/PD model, warranting the need for more data, preferably in a multiple-dose setting, fully to characterize the dose–response relationship and drug potency. However, the model is sufficient to aid in study design for the subsequent multiple-dose study in terms of optimal PK and PD sampling and predicting doses/regimens with maximal effect on TG lowering. Although designed as a long-acting FGF21 analogue, this CovX body still has a faster CL (89 ml h–1 based on an intact N-terminus) than a typical monoclonal antibody with linear PK (average CL of 10 ml h–1) 18. In order to maintain continuous beneficial effects on the lipid panel as well as potentially elicit other metabolic effects such as glucose lowering and weight loss, twice-weekly IV dosing is preferred and this was confirmed by the simulations using the PK/PD model of TG.
The effect on glucose observed in animals was not translated to humans following a single dose of PF-05231023 up to 200 mg. As this was primarily a FIH safety study, it was conducted with 7 days of in-patient stay, during which time subjects were under a controlled diet. It was therefore not unexpected in the present study to observe a placebo effect on glucose of a similar magnitude to that observed with diet modifications. This effect may have potentially masked any early signal of glycaemic control after single-dose treatment. In the future multiple-dose trial, it will be important to incorporate a diet run-in period to stabilize blood glucose responses prior to initiation of dosing with PF-05231023, with the aim of minimizing placebo effects. It is also worth noting that the majority of the subjects were on stable metformin therapy prior to and throughout the duration of the study. It is possible that FGF21 effects on glycaemic control are blunted when dosed on top of metformin. However, the limited sample size and duration of treatment in the present single-dose study precludes any conclusions regarding FGF21 vs. metformin PD interactions. A direct comparison of combination vs. monotherapy in a multiple-dose setting may be needed to characterize this further. As expected, no effects on body weight were observed in the present single-dose study; this will be further assessed in a multiple-dose setting, with a study duration optimized to monitor changes in body weight. It is interesting to note that another FGF21 analogue tested in the clinic also produced significant improvements in dyslipidaemia, including decreases in LDL-C and TGs and increases in HDL-C 13. Favourable effects on body weight, fasting insulin and adiponectin were also detected after 4 weeks of daily subcutaneous injections, with a statistically nonsignificant trend toward glucose lowering also observed at the end of the treatment 13.
Diarrhoea and nausea were the most frequently observed treatment-related AEs in the PF-05231023-treated subjects. There did not appear to be a dose dependency in the frequency or the intensity of these AEs (Table2). Limited clinical experience with FGF21 has been published in the literature to date; neither diarrhoea nor nausea have been reported previously as frequently observed AEs associated with FGF21 treatment 12. In addition, the gastrointestinal system was not identified as a target organ in a 4-week safety study of PF-05231023 in non-human primates (data on file). A single IV dose of PF-05231023 up to the 200 mg was considered to be generally well tolerated by subjects with T2DM. Whether gastrointestinal system AEs will truly pose a limitation on clinical dosing with PF-05231023 will require further characterization in future trials with multiple-dose administration.
The trend of increased mean SBP and DBP was only observed in the PF-05231023 200 mg treatment group. Scrutiny of individual subject data showed that no more than half of the subjects treated with 200 mg PF-05231023 had consistent increases in BP during the first 24 h postdose. The lack of a consistent increase, the limited sample size and the fact that this increase was limited to a single-treatment group do not permit, at this point, differentiation between a true PD effect versus intracohort variability. Furthermore, no apparent effect on blood pressure had been observed in a 4-week safety study of PF-05231023 in non-human primate (data on file). The effects of PF-05231023 on vital signs should continue to be carefully monitored in future clinical studies.
One subject (in the PF-05231023 1.5 mg group) had a positive anti-PF-05231023 endpoint titre on Day 1 predose, which was further characterized as cross-reactive to FGF21 and negative for neutralization. The observance of pre-existing antibodies is not uncommon in clinical programmes 19,20 and, given this single observation, the overall signal for immunogenicity endpoints was unremarkable.
In summary, PF-05231023 was generally well tolerated and safe following single IV administration to subjects with T2DM. Its PK/PD characteristics support further multiple-dosing assessments in humans, with the potential development of PF-05231023 for the treatment of metabolic disorders such as T2DM, hypertriglyceridaemia, and/or obesity.
Competing Interests
This study was financially supported by Pfizer Inc., New York, NY, USA. All co-authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: support from Pfizer, Inc. for the submitted work; with the exception of Dr Martha Hernandez-Illas, all were employees of Pfizer in the previous 3 years. Ms Rossulek's current affiliation is EMD Serono, Inc., Cambridge, MA, USA; Dr Hudson's current affiliation is GSK, Inc., RTP, NC, USA.
This study was sponsored by Pfizer, Inc.
Contributors
JQD designed the study, interpreted the data and wrote the manuscript. MR designed the study, interpreted the data and contributed to the discussions. VRS designed the study, interpreted the data and contributed to the discussions. DB was responsible for PK and antidrug antibody assay development and sample analysis. YL was responsible for PK data analysis and interpretation. KH contributed to the study design and the discussions. MH was the lead principal investigator of the study and contributed to the study design and safety monitoring. RAC designed the study, interpreted the data and contributed to the discussions.
References
- Stratton IM, Adler AI, Neil HAW, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405–12. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- Gaster B, Hirsch IB. The effects of improved glycemic control on complications in type 2 diabetes. Arch Intern Med. 1998;158:134–40. doi: 10.1001/archinte.158.2.134. [DOI] [PubMed] [Google Scholar]
- Resnick HE, Bardsley J, Foster GL, Ratner RE. Achievement of American Diabetes Association clinical practice recommendations among US adults with diabetes, 1999–2002: the National Health and Nutrition examination survey. Diabetes Care. 2006;29:531–7. doi: 10.2337/diacare.29.03.06.dc05-1254. [DOI] [PubMed] [Google Scholar]
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA. 2004;291:335–42. doi: 10.1001/jama.291.3.335. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–43. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–35. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–40. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, Hansen BC, Shanafelt AB, Etgen GJ. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–81. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32:1542–6. doi: 10.2337/dc09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Moller DE. FGF21-based pharmacotherapy-potential utility for metabolic disorders. Trends Endocrinol Metab. 2014;25:303–11. doi: 10.1016/j.tem.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013;18:333–40. doi: 10.1016/j.cmet.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Huang J, Ishino T, Chen G, Rolzin P, Osothprarop TF, Retting K, Li L, Jin P, Martin MJ, Huyghe B, Talukdar S, Bradshow CW, Palanki M, Violand BN, Woodnutt G, Lappe RW, Ogilvie K, Levin N. Development of a novel long-acting antidiabetic FGF21 mimetic by targeted conjugation to a scaffold antibody. J Pharmacol Exp Ther. 2013;346:270–80. doi: 10.1124/jpet.113.204420. [DOI] [PubMed] [Google Scholar]
- Weng Y, Chabot J, Bernardo B, Yan Q, Zhu Y, Brenner MB, Vage C, Logan A, Calle R, Talukdar S. Pharmacokinetics (PK), pharmacodynamics (PD) and integrated PK/PD modeling of a novel long acting FGF21 clinical candidate PF-05231023 in diet-induced obese and leptin-deficient obese mice. PLoS One. 2015;10 doi: 10.1371/journal.pone.0119104. : e0119104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giragossian C, Vage C, Li J, Pelletier K, Piche-Nicholas N, Rajadhyaksha M, Liras J, Logan A, Calle RA, Weng Y. Mechanistic investigation of the preclinical pharmacokinetics and interspecies scaling of PF-05231023, a fibroblast growth factor 21-antibody protein conjugate. Drug Metab Dispos. 2015;43:803–11. doi: 10.1124/dmd.114.061713. [DOI] [PubMed] [Google Scholar]
- Jusko WJ, Ko HC. Physiologic indirect response models characterize diverse types of pharmacodynamic effects. Clin Pharmacol Ther. 1994;56:406–19. doi: 10.1038/clpt.1994.155. [DOI] [PubMed] [Google Scholar]
- Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first-in-human prediction. Clin Pharmacokinet. 2011;50:131–42. doi: 10.2165/11537430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Xue L, Rup B. Evaluation of pre-existing antibody presence as a risk factor for posttreatment anti-drug antibody induction: analysis of human clinical study data for multiple biotherapeutics. AAPS J. 2013;15:893–6. doi: 10.1208/s12248-013-9497-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Fiscella M, Rajadhyaksha M, Goyal J, Holland C, Gorovits B, Morimoto A. Pre-existing biotherapeutic-reactive antibodies: survey results within the American Association of Pharmaceutical Sciences. AAPS J. 2013;15:852–5. doi: 10.1208/s12248-013-9492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]