

Abstract
Aim
Inducers and inhibitors of CYP3A, such as ritonavir and efavirenz, may be used as part of the highly active antiretroviral therapy (HAART) to treat HIV patients. HIV patients with chronic myeloid leukemia or gastrointestinal stromal tumour may need imatinib, a CYP3A4 substrate with known exposure response–relationships. Administration of imatinib to patients on ritonavir or efavirenz may result in altered imatinib exposure leading to increased toxicity or failure of therapy, respectively. We used primary human hepatocyte cultures to evaluate the magnitude of interaction between imatinib and ritonavir/efavirenz.
Methods
Hepatocytes were pre-treated with vehicle, ritonavir, ketoconazole, efavirenz or rifampicin, and the metabolism of imatinib was characterized over time. Concentrations of imatinib and metabolite were quantitated in combined lysate and medium, using LC-MS.
Results
The predicted changes in imatinib CLoral (95% CI) with ketoconazole, ritonavir, rifampicin and efavirenz were 4.0-fold (0, 9.2) lower, 2.8-fold (0.04, 5.5) lower, 2.9-fold (2.2, 3.5) higher and 2.0-fold (0.42, 3.5) higher, respectively. These predictions were in good agreement with clinical single dose drug–drug interaction studies, but not with reports of imatinib interactions at steady-state. Alterations in metabolism were similar after acute or chronic imatinib exposure.
Conclusions
In vitro human hepatocytes predicted increased clearance of imatinib with inducers and decreased clearance with inhibitors of CYP enzymes. The impact of HAART on imatinib may depend on whether it is being initiated or has already been dosed chronically in patients. Therapeutic drug monitoring may have a role in optimizing imatinib therapy in this patient population.
Keywords: drug–drug interactions, HIV drugs, human hepatocytes, imatinib
What is Already Known about this Subject
Imatinib is a CYP3A4 substrate that has been shown to be a victim of 3A4 drug–drug interactions in clinical trials.
Antiretroviral therapy may act as a drug–drug interaction perpetrator in HIV patients treated for a malignancy.
What this Study Adds
Antiretroviral agents were able to change imatinib clearance as predicted by hepatocyte studies and in vitro–in vivo extrapolations.
Our results are validated by accurate predictions of the reported ketoconazole and rifampicin interactions in patients.
Introduction
Highly active antiretroviral therapy (HAART) has significantly reduced the rates of AIDS defining malignancies (Kaposi's sarcoma, non-Hodgkin lymphoma and cervical cancer) 1. As HIV patients live longer with newer and effective antiretroviral therapy, they increasingly experience non-AIDS defining malignancies. Antineoplastic therapy and HAART therapy will need to be applied concomitantly to achieve the simultaneous therapeutic goals of antiretroviral and anticancer activity 2,3.
Imatinib mesylate (Gleevec®, Glivec®) is widely used to treat chronic myeloid leukemia (CML) based on its inhibition of Bcr-Abl and to treat gastrointestinal stromal tumours (GIST) based on its inhibition of c-KIT 4–6. The oral bioavailability of imatinib is reported to vary between 70% and 100% and was recently reported to be dependent on the length of treatment 7,8. The metabolism of imatinib is mainly mediated by CYP3A4 and, to a lesser extent, by CYP2C8 during short term treatment. Ketoconazole (archetype inhibitor of CYP3A) and rifampicin (archetype inducer of CYP3A) have been documented to increase and decrease, respectively, the systemic exposure of imatinib in single dose drug interaction studies 9,10. Upon chronic treatment, CYP2C8 has been reported to play a major role in the metabolism of imatinib due to mechanism based auto-inhibition of CYP3A4 by imatinib 11–14.
Many HAART drugs inhibit and/or induce cytochrome P450, uridine diphosphate glucuronyl transferases (UGTs), ATP binding cassette (ABC) efflux transporters or solute carrier uptake transporters 15,16. Ritonavir, a HIV protease inhibitor, is the most potent inhibitor of CYP3A 17, while efavirenz, a non-nucleoside reverse transcriptase inhibitor (NNRTI), is a mixed inducer and inhibitor of CYP3A 18.
Although prospective, randomized confirmatory studies are lacking, there is ample evidence that imatinib trough concentrations are associated with toxicities on the one hand 19, and with clinical benefit in patients with CML and advanced GIST on the other hand 19–22 suggesting that achieving defined exposures is important.
In this study, we aimed to assess the effect of ketoconazole, ritonavir, rifampicin and efavirenz on the metabolism of imatinib in primary cultures of human hepatocytes. The predicted change in oral clearance of imatinib in the presence of ritonavir/efavirenz was also determined using in vitro–in vivo scale up. Since chronic treatment with imatinib results in auto-inhibition and metabolic switch from CYP3A4 to CYP2C8, we also sought to assess the utility of hepatocytes to assess whether ritonavir would alter the imatinib metabolism after chronic exposure in human hepatocytes.
Methods
Materials
Imatinib, N-desmethyl imatinib (CGP74588) and [D8]-imatinib were obtained from Novartis Pharmaceuticals Co. (East Hanover, NJ, USA). [D8]-N-desmethyl imatinib and ritonavir were procured from Toronto Research Chemicals (Ontario, Canada). Ketoconazole was obtained from Janssen Research Foundation (Titusville, NJ, USA). Hepatocyte maintenance medium was procured from Lonza Inc (Walkersville, MD, USA). Organic solvents used for the extractions and the analytical assays were obtained from Fisher Scientific (Fairlawn, NJ, USA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA).
Primary human hepatocytes cultures
Human liver samples (n = 8) were obtained from Life Technologies (CA, USA) or the Hepatocytes Transplantation Laboratory, University of Pittsburgh or Xenotech, LLC (KS, USA) and processed for hepatocytes as described before 23. These serum free cultures of human hepatocytes are suitable for drug–drug interaction studies as reported before 24. All the drug incubations were carried out 48 h after seeding. The CYP induction potential of the hepatocytes used in all the studies was also evaluated at the end of the study by measuring the activity of CYP3A (formation of testosterone 6β hydroxylation) in separate sets of untreated and rifampicin (10 µm) treated hepatocytes.
Time course of imatinib metabolism
The time course of imatinib metabolism was assessed in the absence and presence of ritonavir or ketoconazole or efavirenz or rifampicin. Hepatocytes were incubated daily with ritonavir (10 μm), ketoconazole positive control (10 μm), efavirenz (10 μm), rifampicin positive control (10 μm) or vehicle control (0.1% dimethyl sulfoxide (DMSO)) for 4 days. On day 5, imatinib (2.5 μm) was incubated along with the above agents for an additional 24 or 48 h. Hepatocyte samples were collected at 0, 4, 8, 24 and 48 h for inhibition studies. The duration of incubation for induction studies was reduced to 24 h and samples were obtained at 0, 2, 4, 8 and 24 h. Drug concentrations were selected based on the steady-state maximum plasma concentration at clinically recommended doses 25. At the end of the study, hepatocytes were scraped from the plates along with culture medium supernatant on ice and the cells were lyzed by sonication. Samples were stored at –80°C until analysis. Additionally, the cytotoxicity assay (MTT assay) was performed to ensure that the selected drug concentrations did not affect the viability of human hepatocytes (data not shown).
Effect of chronic imatinib exposure on metabolism
In an effort to differentiate metabolism of imatinib after chronic exposure relative to acute exposure, 24 h incubations were performed in the absence and presence of ritonavir. To model acute exposure, hepatocytes were incubated daily with ritonavir (10 μm) or vehicle control (0.1% DMSO) for 4 days as described above. On day 5, imatinib was incubated along with the above agents for an additional 24 h. To model chronic exposure, hepatocytes were incubated daily with imatinib and ritonavir (10 μm) or imatinib for 4 days. On day 5, imatinib was incubated along with the above agents for an additional 24 h. These incubations were performed at 2.5 and 10 μm imatinib. Hepatocyte samples were collected on day 5 at 0 and 24 h as a point estimate of alterations of metabolism.
LC-MS quantitation of imatinib and desmethyl imatinib
Imatinib and active desmethyl-metabolite were quantitated with an Agilent 1100 Autosampler and Binary pump (Agilent, Delaware, USA) hyphenated to a Thermo Electron MSQ detector (Thermo Fisher Scientific, MA, USA). Analytes were chromatographically separated on a Phenomenex Luna C18(2) column (5 µm, 2 × 150 mm) kept at ambient temperature. The gradient mobile phase system was comprised of solvent A (methanol 0.1% formic acid) and solvent B (water 0.1% formic acid). The initial mobile phase was 30% A and 70% B at a flow rate of 0.2 ml min–1 held for 4 min. Subsequently, solvent A was increased to 60% over 6 min, where it was held until 13 min. Next, A was increased to 80% over 0.5 min with a flow rate of 0.3 ml min–1 and held until 16 min. Finally, A was lowered back to initial conditions over 0.5 min, where it was held until 24 min, followed by injection of the next sample. Mass spectrometer settings were probe voltage 3 kV, cone voltage 40 V and probe temperature 500°C. The SIM monitored were m/z 493.9, 501.9, 479.9 and 487.9 for imatinib, [D8]-imatinib, desmethyl imatinib, and [D8]-desmethyl imatinib, respectively. Aliquots of 200 µl of hepatocyte medium were extracted with 1000 µl acetonitrile and 10 µl of internal standard mix (2 µg ml–1 of [D8]-imatinib (internal standard for imatinib) and [D8]-desmethyl imatinib (internal standard for desmethyl imatinib) in methanol : water (50: 50; v/v)). After vortexing and centrifugation, the supernatant was transferred to a glass tube and then evaporated to dryness under nitrogen. Subsequently, the dried residue was reconstituted with 100 µl of methanol : H2O 30 :70, (v/v) mobile phase and then 2 µl of the sample was injected into the LC-MS system. The ion chromatograms were integrated and quantified using Thermo Electron Excalibur 1.4 (Thermo Fisher Scientific, MA, USA). The assay was linear between 10 and 1000 ng ml–1 with acceptable QC accuracy (97.5–106% and 98.8–106%) and precision (<7.9 CV% and <7.6 CV%) for imatinib and desmethyl imatinib, respectively, as determined from independent QC samples (n = 6). Samples with concentrations above the upper limit of the calibration range were diluted to within the calibration range with control media.
Data analysis
Each study was performed in at least three different cultures of human hepatocytes in duplicate. The half-life (t1/2) and area under the concentration–time curve from time 0 to 48 h (AUC(0,48 h) for imatinib in primary cultures of human hepatocytes were calculated non-compartmentally with Phoenix WinNonlin (Pharsight Corp, Cary, North Carolina, USA).
In the acute and chronic exposure experiment, each study was performed once in duplicate.
Calculation of apparent intrinsic clearance
The apparent intrinsic clearance (CLint, app) of imatinib was calculated from the half-life of imatinib in the absence and presence of ritonavir, ketoconazole, efavirenz or rifampicin in primary cultures of human hepatocytes using equation 1 26,
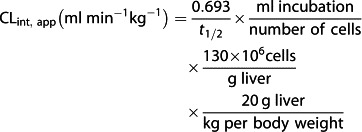 |
1 |
where incubation volume is 1 ml and the number of cells is 1.5 × 106.
Prediction of hepatic clearance
The hepatic clearance (CLh) of imatinib was predicted from CLint, app using the well-stirred liver model as follows 27–29,
| 2 |
Hepatic blood flow (Qh) = 21 ml min–1 kg–1 and hepatic availability (Fh), which was calculated using equations 3 and 4,
| 3 |
| 4 |
where Kp = partition ratio = 4.38 30, Vc = adherent cell volume = 0.0051 ml 31 and Vm = volume of culture medium = 1 ml. Equation 3 was chosen 27–29 as in our experience it was found to predict best for low extraction ratio drugs.
Prediction of oral clearance
Imatinib has been reported to be well-absorbed from the gastro-intestinal tract 8. Consequently, the intestinal clearance is assumed to be negligible for imatinib, particularly in the presence of ritonavir or ketoconazole. Therefore, the fraction absorbed (fa) and fraction of dose escaping gut metabolism (Fg) are assumed to be 1 for the prediction of oral clearance using equation 5 32.
| 5 |
The bias of CLoral prediction was estimated from the geometric mean ratio of the predicted and observed values, and calculated as average fold-error (AFE) from equation 6 whereas the precision of the prediction was assessed from root mean squared prediction error (RMSE) using equations 7 and 8, respectively 33.
| 6 |
| 7 |
| 8 |
Statistical analysis
All the results are expressed as mean ± 95% CI. The statistical differences among the different treatment groups were analyzed by non-parametric Friedman analysis of variance (anova) followed by Dunn's multiple comparison post-hoc test. P < 0.05 was considered as statistically significant. The data from the chronic exposure experiment were handled descriptively.
Results
The time course of metabolism of imatinib in the absence and in the presence of ritonavir or efavirenz was assessed in primary cultures of human hepatocytes and compared with that of the corresponding positive controls, ketoconazole and rifampicin. The demographics of human liver donors are shown in Table1. The CYP3A4/5 activities in control hepatocytes ranged from 51 to 265 pmol min–1 per million cells (based on formation of 6-β-hydroxy testosterone from testosterone) at the end of the study period. All the batches of human hepatocytes used in this study retained CYP induction potential as confirmed by the greater than 2.5-fold increase in CYP3A activity upon treatment with rifampicin.
Table 1.
Demographic profiles of human liver donors
| Liver ID | Age (years) | Gender | Race | Diagnosis |
|---|---|---|---|---|
| Hu 1595 | 31 | Female | Caucasian | Liver mass – Focal nodular hyperplasia |
| Hu 1600 | 40 | Male | Caucasian | Metastatic rectal adenocarcinoma |
| H 1165 | 13 | Male | Caucasian | Anoxia |
| H 1174 | 21 | Female | Caucasian | Cardiovascular arrest |
| HH 2018 | 68 | Male | Caucasian | Cholangiocarcinoma |
| HH 2019 | 68 | Male | Caucasian | Metastatic colon cancer |
| Hu 1488 | 31 | Male | Caucasian | Colorectal cancer |
| HH 2020* | 62 | Female | Caucasian | Metastatic colon cancer |
*Used in chronic exposure experiment.
The average concentration–time profile for imatinib and the formation of desmethyl imatinib in primary cultures of human hepatocytes treated with vehicle, ritonavir and positive control (ketoconazole) are shown in Figure1A and B. The individual and mean values of AUC(0,48 h) for the time course of metabolism of imatinib and the formation of desmethyl imatinib in the absence and presence of ritonavir or ketoconazole in human hepatocytes are shown in Table2. Treatment with ritonavir resulted in a 1.6-fold increase in the exposure (AUC(0,48 h)) of imatinib with a 4.4-fold reduction in the exposure to desmethyl imatinib as compared with vehicle treatment. Ketoconazole treatment increased the exposure of imatinib 1.8-fold and reduced the exposure of desmethyl imatinib 3.2-fold. Treatment with ritonavir significantly increased the half-life of imatinib 2.8-fold. Additionally, metabolite to parent imatinib (M : P) AUC(0,48 h) ratio was decreased 7.4-fold, respectively, by ritonavir. Ketoconazole treatment resulted in a 4.0-fold increase in half-life and 6.3-fold reduction in M : P AUC(0,48 h) ratio as compared with vehicle treatment.
Figure 1.
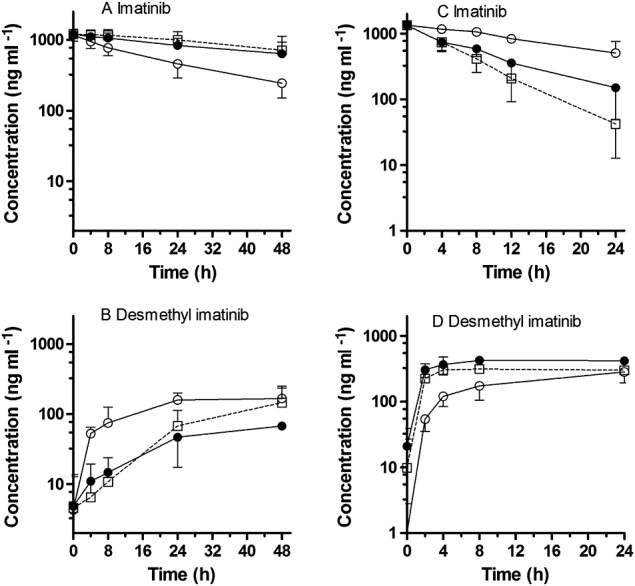
Time course of imatinib depletion and formation of desmethyl imatinib in primary cultures of human hepatocytes treated with either DMSO (○), ritonavir 10 μm (●) and ketoconazole 10 μm (□) [A, B] or DMSO (○), efavirenz 10 μm (●) and rifampicin 10 μm (□) [C, D]. Data are representative of three individual experiments each performed in duplicate. Error bars represent standard deviation
Table 2.
Parameters of imatinib metabolism and desmethyl imatinib metabolite formation in primary cultures of human hepatocytes treated with vehicle (DMSO), ritonavir or ketoconazole and incubated with imatinib (2.5 μm).
| Imatinib | Metabolite | Imatinib | Imatinib | Imatinib | Imatinib | ||
|---|---|---|---|---|---|---|---|
| Treatment liver ID | AUC(0,48 h) (µg ml–1 h) | AUC(0,48 h) (µg ml–1 h) | AUC(0,48 h) M : P ratio | % metabolized at 48 h | t½ (h) | CLint, app (ml min–1 kg–1) | CLoral,pred (mL/min/kg) |
| DMSO | |||||||
| Hu 1595 | 16.7 | 8.4 | 0.51 | 86 | 17 | 1.17 | 1.15 |
| Hu 1600 | 25.3 | 5.8 | 0.23 | 73 | 26 | 0.78 | 0.76 |
| H 1165 | 33.9 | 4.4 | 0.13 | 77 | 24 | 0.84 | 0.82 |
| H 1174 | 28.0 | 5.9 | 0.21 | 82 | 19 | 1.03 | 1.00 |
| Mean (95% CI) | 26.0 (14.6, 37.4) | 6.1 (3.5, 8.8) | 0.27 (0, 0.53) | 80 (71, 89) | 22 (15, 28) | 0.96 (0.67, 1.24) | 0.93 (0.65-1.21) |
| Ritonavir | |||||||
| Hu 1595 | 26.2 | 4.3 | 0.16 | 77 | 23 | 0.88 | 0.86 |
| Hu 1600 | 43.4 | 0.94 | 0.022 | 40 | 64 | 0.32 | 0.31 |
| H 1165 | 46.6 | 1.7 | 0.036 | 51 | 49 | 0.41 | 0.40 |
| H 1174 | 51.1 | 0.90 | 0.018 | 29 | 104 | 0.19 | 0.19 |
| Mean (95% CI) | 41.8 (24.5, 59.1) | 2.0 (0, 4.5) | 0.059 (0, 0.17) | 49 (17, 82) | 60 (6, 114) | 0.45 (0, 0.93) | 0.44 (0-0.96 |
| Ratio to DMSO | 1.62 (1.31, 1.93) | 0.30 (0.02, 0.58) | 0.19 (0.003, 0.38)* | 0.62 (0.26, 0.98) | 2.8 (0, 5.6)* | 0.46 (0.09, 0.83)* | 0.46 (0.09-0.83)* |
| Ketoconazole | |||||||
| Hu 1595 | 26.7 | 5.3 | 0.20 | 77 | 23 | 0.89 | 0.87 |
| Hu 1600 | 58.2 | 1.7 | 0.028 | 10 | 210 | 0.10 | 0.093 |
| H 1165 | 48.5 | 5.1 | 0.11 | 61 | 32 | 0.62 | 0.61 |
| H 1174 | 55.1 | 0.88 | 0.016 | 25 | 100 | 0.20 | 0.20 |
| Mean (95% CI) | 47.1 (24.5, 69.7) | 3.2 (0, 6.9) | 0.088 (–0, 0.22) | 43 (0, 9.3) | 91 (0, 229) | 0.45 (0, 1.0) | 0.44 (0-1.0) |
| Ratio to DMSO | 1.82 (1.21, 2.44)* | 0.56 (0, 1.3) | 0.36 (0, 0.92) | 0.53 (0, 1.1) | 4.0 (0, 11) | 0.46 (0, 1.0) | 0.46 (0-1.0)** |
M : P ratio, the ratio of desmethyl imatinib to imatinib.
*P < 0.05 vs. DMSO, non-parametric Friedman anova followed by Dunn's multiple comparison test.
**The individual ratios to DMSO were 0.76, 0.12, 0.74 and 0.20 for a mean of 0.46. This corresponds to a fold decrease in clearance (inverse of the ratio) of 1.3, 8.2, 1.3 and 5.0 for a mean of 4.0.
The average concentration–time profile for imatinib and the formation of desmethyl imatinib in primary cultures of human hepatocytes treated with vehicle control (DMSO), efavirenz and rifampicin (inducer positive control) are shown in Figure1C and D. The individual and mean values of AUC(0,24 h) for the time course of metabolism of imatinib and the formation of desmethyl imatinib in the presence and absence of efavirenz or rifampicin are presented in Table3. Efavirenz treatment decreased the exposure (AUC(0,24 h)) of imatinib 2.3-fold and increased the exposure of desmethyl imatinib by 3.1-fold as compared with treatment with vehicle. Treatment with rifampicin resulted in a 3.0-fold decrease in the imatinib exposure and a 2.1-fold increase in the exposure of desmethyl imatinib. Efavirenz treatment resulted in an almost 2-fold decrease in half-life of imatinib as compared with vehicle control. The M : P AUC(0,24 h) ratio was increased 7.8-fold, by efavirenz. Rifampicin treatment resulted in a 2.9-fold reduction in the half-life, and a 6.1-fold increase in M : P AUC(0,24 h) ratio, compared with vehicle control.
Table 3.
Parameters of imatinib metabolism and desmethyl imatinib metabolite formation in primary cultures of human hepatocytes treated with vehicle (DMSO), efavirenz or rifampicin and incubated with imatinib (2.5 μm)
| Imatinib | Metabolite | Imatinib | Imatinib | Imatinib | Imatinib | ||
|---|---|---|---|---|---|---|---|
| Treatment liver ID | AUC(0,24 h) (µg ml–1 h) | AUC(0,24 h) (µg ml–1 h) | AUC(0,24 h) M : P ratio | % metabolized at 24 h | t½ (h) | CLint, app (ml min–1 kg–1) | CLoral,pred (ml min–1 kg–1) |
| DMSO | |||||||
| HH 2018 | 17.6 | 5.3 | 0.30 | 64 | 17 | 1.22 | 1.19 |
| HH 2019 | 23.5 | 2.7 | 0.11 | 43 | 16 | 1.24 | 1.21 |
| Hu 1488 | 16.9 | 2.7 | 0.16 | 79 | 11 | 1.84 | 1.80 |
| Mean (95% CI) | 19.3 (13.6, 25.1) | 3.6 (1.2, 6.0) | 0.19 (0.0, 0.35) | 62 (33, 91) | 15 (10, 20) | 1.43 (0.87, 1.99) | 1.40 (0.85, 1.95) |
| Efavirenz | |||||||
| HH 2018 | 10.7 | 9.3 | 0.87 | 82 | 11 | 1.86 | 1.82 |
| HH 2019 | 6.05 | 10.4 | 1.7 | 97 | 5.3 | 3.77 | 3.69 |
| Hu 1488 | 11.9 | 10.2 | 0.86 | 88 | 8.6 | 2.34 | 2.29 |
| Mean (95% CI) | 9.55 (4.6, 14.5) | 10.0 (9.1, 10.9) | 1.1 (0.38, 1.9) | 89 (77, 101) | 8.2 (3.8, 12.6) | 2.66 (1.08, 4.24) | 2.60 (1.05, 4.14) |
| Ratio to DMSO | 0.52 (0.15, 0.90) | 3.1 (1.2, 5.0)* | 7.9 (0, 18) | 1.6 (0.57, 2.5) | 0.59 (0.21, 0.97) | 1.95 (0.42, 3.47) | 1.95 (0.42, 3.48) |
| Rifampicin | |||||||
| HH 2018 | 5.2 | 6.8 | 1.30 | 98 | 5.0 | 4.04 | 3.95 |
| HH 2019 | 8.9 | 6.7 | 0.75 | 94 | 6.0 | 3.32 | 3.25 |
| Hu 1488 | 5.5 | 6.6 | 1.19 | 99 | 4.2 | 4.79 | 4.69 |
| Mean (95% CI) | 6.5 (3.3, 9.8) | 6.7 (6.5, 6.9) | 1.1 (0.62, 1.5) | 97 (93, 101) | 5.1 (3.6, 6.5) | 4.05 (2.88, 5.22) | 3.96 (2.82, 5.11) |
| Ratio to DMSO | 0.33 (0.27, 0.40) | 2.1 (1.0, 3.2) | 6.2 (3.6, 8.8) | 1.7 (0.89, 2.4) | 0.35 (0.28, 0.42) | 2.86 (2.24, 3.48) | 2.87 (2.25, 3.49) |
M : P ratio, the ratio of desmethyl imatinib to imatinib.
*P < 0.05 vs. DMSO, non-parametric Friedman anova followed by Dunn's multiple comparison test.
The individual and mean apparent intrinsic clearance (CLint, app) values of imatinib in primary cultures of human hepatocytes treated with either vehicle (DMSO), ritonavir or ketoconazole are shown in Table2 and these values upon treatment with vehicle, efavirenz or rifampicin are presented in Table3. The CLint, app of imatinib was decreased 2.8-fold and 4.0-fold (range 1.3–8.0 fold) respectively, by ritonavir and ketoconazole. In case of efavirenz or rifampicin treatment, the CLint, app of imatinib was increased 2.0-fold and 2.9-fold, respectively. The imatinib human CLhep values were predicted from CLint, app using the well-stirred liver model and are presented in Figure2. Under the given experimental conditions and assumptions for the predictions, the values of both CLhep and CLoral (both expressed as ml min–1 kg–1) are similar and, therefore, they are presented in the same figure (Figure2). The parameters used for the prediction of hepatic clearance and the comparisons of observed and predicted CLoral values for imatinib with prediction errors are shown in Table4.
Figure 2.
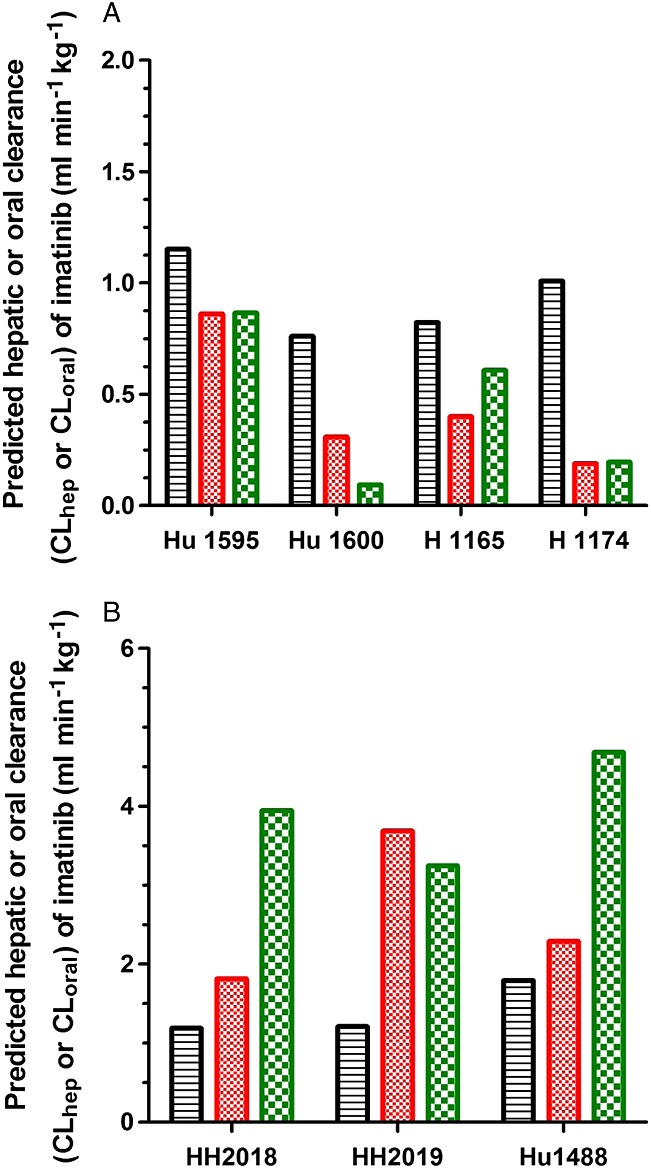
Predicted human hepatic or oral clearance (CLhep or CLoral) of imatinib from primary cultures of human hepatocytes treated with either DMSO ( ), ritonavir 10 μm (
), ritonavir 10 μm ( ) and ketoconazole (
) and ketoconazole ( ) 10 μm (A) or DMSO (
) 10 μm (A) or DMSO ( ), efavirenz 10 μm (
), efavirenz 10 μm ( ) and rifampicin 10 μm (
) and rifampicin 10 μm ( ) (B)
) (B)
Table 4.
The parameters used for the prediction of hepatic clearance, the comparisons of observed and predicted CLoral values for imatinib with prediction errors, and predictions of change in CLoral and literature values
| Parameters | Values | Reference | Comment |
|---|---|---|---|
| Kp | 4.38 | 30 | |
| fu, inc | 0.98 | ||
| Imatinib | |||
| Mean observed CLoral (l h–1) | 12.3 | ||
| Mean predicted CLoral (l h–1) | 4.8 | ||
| Bias of prediction (AFE) | 0.434 | ||
| Precision of prediction (RMSE) | 7.7 | ||
| Ketoconazole | |||
| Predicted change in CLoral* | 1.3–8.0-fold reduction | ||
| Reported change in CLoral* | 1.4-fold reduction | 9 | Single imatinib dose |
| Ritonavir | |||
| Predicted change in CLoral* | 2.8-fold reduction | ||
| Reported change in CLoral* | No effect | 14 | Ritonavir added to SS-imatinib |
| Rifampicin | |||
| Predicted change in CLoral* | 2.9-fold increase | ||
| Reported change in CLoral* | 3.9-fold increase | 10 | Single imatinib dose |
| Efavirenz | |||
| Predicted change in CLoral* | 2.0-fold increase | ||
| Reported change in CLoral* | 1.5-fold increase | 49 | Single imatinib dose added to SS-efavirenz |
| Reported change in CLoral* | 1.2-fold increase | 49 | SS imatinib added to SS-efavirenz |
Kp = Partition ratio; fu, inc = fraction unbound in hepatocytes; CLoral = Oral clearance; AFE = average fold error; RMSE = root mean square error; SS = steady-state; Observed CLoral values (13.3, 8.1, 10, 14.3, 19.2, 9.2, 12 l h–1) were obtained from the literature (n = 7) 25,57–62; Predicted CLoral values (4.84, 3.19, 3.45, 4.24, 5.0, 5.09, 7.55 l h–1) were obtained from six different vehicle treated human hepatocytes and converting CLoral from ml min–1 kg–1 to l h–1 assuming a 70 kg person.
*relative to imatinib alone
Acute exposure to imatinib over 24 h resulted in imatinib depletion and desmethyl imatinib generation (Figure3), as expected. Addition of ritonavir reduced imatinib depletion and abolished desmethyl imatinib generation. After chronic (4 day) exposure, hepatocytes had accumulated imatinib and desmethyl imatinib to some degree. Imatinib depletion and desmethyl imatinib generation patterns were similar after acute and chronic imatinib exposure, respectively.
Figure 3.
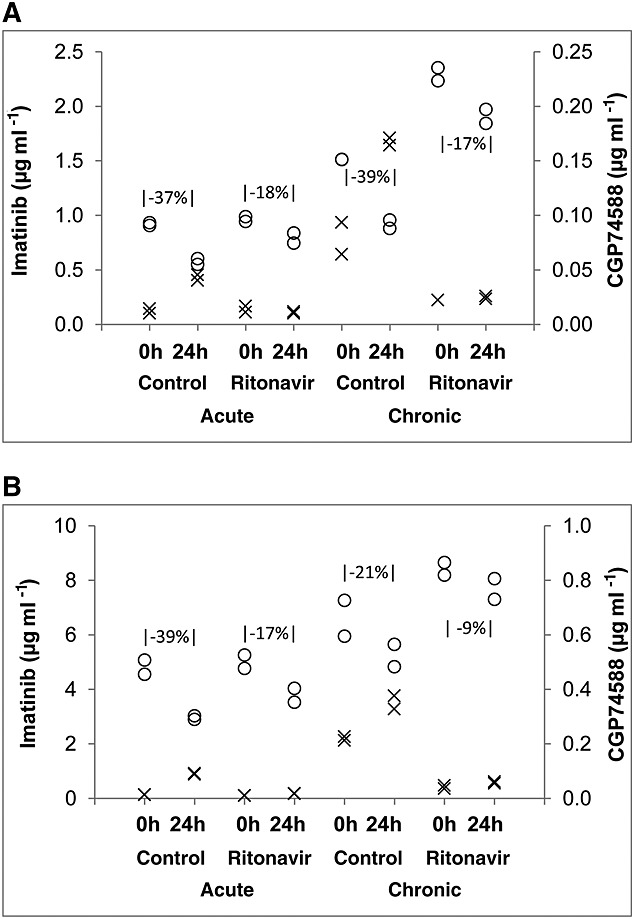
Imatinib depletion and formation of desmethyl imatinib (CGP74588) in primary cultures of human hepatocytes treated with either DMSO or ritonavir simulating acute and chronic imatinib exposure at 2.5 μm [A] or 10 μm [B]. Decrease in imatinib between 0 and 24 h is indicated in the graphs. The open circle (○) indicates imatinib depletion and the cross (x) represents the formation of desmethyl imatinib
Discussion
In vitro–in vivo extrapolation (IVIVE) is useful to predict human pharmacokinetic and potential drug interactions 34. The average predicted human CLoral of imatinib from vehicle treated human hepatocytes cultures was approximately 2.6-fold lower than that of the clinically observed average CLoral of imatinib, which was comparable with reported predictions 28,32.
Imatinib exhibits wide inter-patient variability in exposure, which is associated with toxicity and clinical benefit in patients with CML and advanced GIST 20–22,35–37. It is therefore essential to identify the sources of variability and optimize the likelihood that patients achieve exposures associated with response, e.g. through therapeutic drug monitoring 19,38. Imatinib is mainly metabolized by CYP3A4 and CYP2C8 and is expected to have major interactions with enzyme inducers and inhibitors. Many HAART drugs modulate metabolizing enzymes and transporters, resulting in complex interactions. Previously, ketoconazole and rifampicin were reported to change imatinib clearance significantly in patients 9,10. The present study evaluated the interaction of ketoconazole, rifampicin, and the HAART drugs, ritonavir and efavirenz, on imatinib using primary cultures of human hepatocytes.
Our data suggest that ketoconazole would decrease single dose imatinib clearance 1.3 to 8.0-fold, which may be explained by inhibition of both CYP3A and CYP2C8 39,40. Ketoconazole showed variable extent of inhibition with two of the experiments showing 1.3-fold inhibition, and two showing 5.2- and 8.2-fold inhibition. This likely reflects the biological variability for the susceptibility to azole drug–drug interactions, by virtue of CYP3A5 expressers (CYP3A5*1 carriers) being less susceptible to e.g. fluconazole mediated inhibition 41. The predicted ketoconazole effect is in agreement with clinical observations of approximately 1.4-fold decreased imatinib clearance 9. Ritonavir moderately inhibited imatinib metabolism and strongly reduced desmethyl imatinib exposure, in line with the inhibitory effects of ritonavir on CYP3A (strong) and 2C8 (moderate) 11–13,42–44. Of note, ritonavir inhibits CYP3A and is also reported to induce CYP3A. Results from our laboratory and other studies with ritonavir in vivo suggest that ritonavir mediated CYP3A inhibitory effects prevail over induction effects 12,45. The change in imatinib exposure may not be mirrored by an inverse similar sized effect on metabolite exposure because not only the formation, but also the clearance, of the metabolite is CYP catalyzed and would be modulated by ritonavir. Our data suggest ritonavir decreases imatinib clearance 2.8-fold. Though imatinib is a P-gp substrate, we do not think the inhibitory effect of ritonavir on P-gp would increase imatinib bioavailability because imatinib is nearly completely bioavailable 8.
Rifampicin, which induces CYP3A and CYP2C8, decreased imatinib exposure 3.0-fold and more than doubled desmethyl imatinib exposure 46. This agrees with clinical observations of a 3.9-fold increased imatinib clearance 10. Efavirenz is a mixed type inducer/inhibitor of CYP3A and has potent inhibitory effects on CYP2C8 and CYP2B6 47,48. In human hepatocytes, efavirenz decreased imatinib exposure 2.3-fold and more than tripled desmethyl imatinib, indicating that efavirenz mediated CYP3A induction prevails over its CYP3A inhibition 18. This resulted in a predicted 2.0-fold increase in imatinib clearance.
The interactions predicted and interactions observed in healthy volunteer studies after single imatinib doses were in agreement for ketoconazole and rifampicin 9,10, which supports the predictions for ritonavir and efavirenz. Interestingly, there are clinical data available on the combination of these agents with imatinib in patients. Ritonavir was added to chronic imatinib therapy in 11 cancer patients and did not change imatinib AUC (subjects serving as their own control) 14. Studies in expressed enzymes and human liver microsomes (HLM) suggested that ritonavir completely inhibited CYP3A4 catalyzed imatinib metabolism, but only partly (50–80%) inhibited imatinib metabolism in HLM, implicating an important role for other CYP enzyme(s) in the metabolic fate of imatinib 14. Although this diminishes the role for CYP3A4 in imatinib metabolism, the complete absence of a clinical interaction is still surprising and unexpected. Administration of imatinib to HIV patients on chronic ritonavir or efavirenz resulted in interaction trends, but sample numbers were too small to be conclusive 49.
There have been recent reports on a metabolic shift of imatinib over time, possibly contributing to the differences in the results of single dose and chronic dosing of imatinib. Imatinib metabolism is predicted to be mainly mediated by CYP3A4 (60%) and to a lesser extent by CYP2C8 (40%) during short term treatment. However, during long term treatment, the contribution of CYP2C8 (65–75%) is larger than that of CYP3A4 (25-35%) due to mechanism based auto-inhibition of CYP3A4 by imatinib 12,13. Simulations have shown that this change in the primary enzyme involved causes the 40% increase of single dose imatinib AUC in presence of itraconazole that decreases to about 20% increase in imatinib AUC after multiple imatinib doses 13. Although the authors considered this result to be in line with the absence of a ritonavir interaction on chronic imatinib as reported by van Erp et al. 14, our data point to the stronger inhibition of imatinib metabolism by ritonavir relative to ketoconazole and would suggest that an interaction might still have been seen. Many of the studies of the imatinib metabolic switch have been performed in HLM, which severely limits the experimental duration. In addition, solvent concentrations of up to 1% were used in incubations, which are known to affect metabolism 50,51. It has been reported that it may take several days of in vivo treatment to reach the maximal inhibitory effect of a mechanism-based inhibitor 52–54. Therefore, we performed an additional experiment with 4 days of imatinib pre-incubation allowing for the mechanism based inhibition to take place. Ritonavir abolished desmethyl imatinib generation in hepatocytes, which is in contrast to a previous report on HLM experiments where ritonavir could not completely abolish imatinib demethylation 14. Imatinib depletion and desmethyl imatinib generation patterns were similar after acute and chronic imatinib exposure, respectively, which is not consistent with the hypothesis of time-dependent susceptibility of imatinib to ritonavir mediated metabolic inhibition in hepatocytes. Susceptibility of imatinib metabolism to ritonavir was also not different after acute or chronic exposure. The complete absence of an effect of ritonavir on imatinib exposure in subjects in the van Erp et al. study 14 may be explained by a complete mechanism based inhibition of CYP3A by imatinib after 2 months of imatinib treatment.
Our results underline both the complexity of imatinib metabolism and its drug–drug interactions and the potential limitations of HLM and hepatocyte studies in predicting drug–drug interactions, in particular with regards to limited duration experiments of certain drugs that undergo a shift in metabolic route over time, as exemplified by imatinib and its auto-inhibition of CYP3A4, and changing oral bioavailability 7,13. In addition, this also implies that the metabolic drug–drug interaction studies of imatinib which were limited to single dose administrations of imatinib, such as that of St John's Wort and imatinib 55,56, may not be reflective of the interaction at steady-state use of imatinib.
Based on our results and the existing literature, suggested imatinib dose adjustments in the context of antiretroviral therapy may be dependent on whether imatinib or HAART is the pre-existing therapy. Patients who are on imatinib therapy for over 3 months, and require addition of ritonavir containing antiretroviral therapy may not require any imatinib dose adjustments, as imatinib clearance is no longer additionally susceptible to ritonavir. However, for patients who are on ritonavir containing antiretroviral therapy and are initiating imatinib therapy, a lower dose of imatinib (200 mg) may be warranted to minimize potential side effects. After chronic treatment, the metabolic fate of imatinib will have achieved steady-state, and the imatinib dose of 400 mg or higher may be tolerated 14,49. Clinical confirmation of our hypotheses is desired. The effect of efavirenz on imatinib exposure in patients on long term imatinib treatment will need to be studied in a well-controlled trial comparing within-subject changes, before more specific recommendations may be formulated.
Given the extensive literature on imatinib exposure–response relationships with a target plasma imatinib concentration of 1000 ng ml–1, the wide inter-patient variability in imatinib clearance, and the limited availability of clinical data on the range of possible imatinib drug interactions, therapeutic drug monitoring may be especially critical in the setting of antiretroviral co-medication 38.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare JHB, MAR, and RV had support from the NIH for the submitted work and JHB had research support from Novartis and Bristol-Myers Squibb in the previous 3 years. There are no other relationships or activities that could appear to have influenced the submitted work.
This work is funded by contract N02-CM-62212 and supported by grant P30-CA-047904 (Cancer Pharmacokinetics and Pharmacodynamics Facility at University of Pittsburgh Cancer Institute) and P30 CA006973 (Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) from the National Cancer Institute.
References
- Shiels MS, Pfeiffer RM, Gail MH, Hall HI, Li J, Chaturvedi AK, Bhatia K, Uldrick TS, Yarchoan R, Goedert JJ, Engels EA. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103:753–62. doi: 10.1093/jnci/djr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudek MA, Flexner C, Ambinder RF. Use of antineoplastic agents in patients with cancer who have HIV/AIDS. Lancet Oncol. 2011;12:905–12. doi: 10.1016/S1470-2045(11)70056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfitano A, Barbaro G, Perretti A, Barbarini G. Human immunodeficiency virus-associated malignancies: a therapeutic update. Curr HIV Res. 2012;10:123–32. doi: 10.2174/157016212799937227. [DOI] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CDM, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–37. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- Eechoute K, Fransson MN, Reyners AK, de Jong FA, Sparreboom A, van der Graaf WT, Friberg LE, Schiavon G, Wiemer EA, Verweij J, Loos WJ, Mathijssen RH, De Giorgi U. A long-term prospective population pharmacokinetic study on imatinib plasma concentrations in GIST patients. Clin Cancer Res. 2012;18:5780–7. doi: 10.1158/1078-0432.CCR-12-0490. [DOI] [PubMed] [Google Scholar]
- Peng B, Dutreix C, Mehring G, Hayes MJ, Ben Am M, Seiberling M, Pokorny R, Capdeville R, Lloyd P. Absolute bioavailability of imatinib (Glivec®) orally versus intravenous infusion. J Clin Pharmacol. 2004;44:158–62. doi: 10.1177/0091270003262101. [DOI] [PubMed] [Google Scholar]
- Dutreix C, Peng B, Mehring G, Hayes M, Capdeville R, Pokorny R, Seiberling M. Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother Pharmacol. 2004;54:290–94. doi: 10.1007/s00280-004-0832-z. [DOI] [PubMed] [Google Scholar]
- Bolton AE, Peng B, Hubert M, Krebs-Brown A, Capdeville R, Keller U, Seiberling M. Effect of rifampicin on the pharmacokinetics of imatinib mesylate (Gleevec, STI571) in healthy subjects. Cancer Chemother Pharmacol. 2004;53:102–06. doi: 10.1007/s00280-003-0722-9. [DOI] [PubMed] [Google Scholar]
- Gschwind HP, Pfaar U, Waldmeier F, Zollinger M, Sayer C, Zbinden P, Hayes M, Pokorny R, Seiberling M, Ben Am M, Peng B, Gross G. Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos. 2005;33:1503–12. doi: 10.1124/dmd.105.004283. [DOI] [PubMed] [Google Scholar]
- Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165:2787–98. doi: 10.1111/j.1476-5381.2011.01732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filppula AM, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response. Drug Metab Dispos. 2013;41:50–9. doi: 10.1124/dmd.112.048017. [DOI] [PubMed] [Google Scholar]
- van Erp NP, Gelderblom H, Karlsson MO, Li J, Zhao M, Ouwerkerk J, Nortier JW, Guchelaar HJ, Baker SD, Sparreboom A. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin Cancer Res. 2007;13:7394–400. doi: 10.1158/1078-0432.CCR-07-0346. [DOI] [PubMed] [Google Scholar]
- Piscitelli SC, Gallicano KD. Interactions among drugs for HIV and opportunistic infections. N Engl J Med. 2001;344:984–96. doi: 10.1056/NEJM200103293441307. [DOI] [PubMed] [Google Scholar]
- Boffito M, Acosta E, Burger D, Fletcher CV, Flexner C, Garaffo R, Gatti G, Kurowski M, Perno CF, Peytavin G, Regazzi M, Back D. Therapeutic drug monitoring and drug-drug interactions involving antiretroviral drugs. Antivir Ther. 2005;10:469–77. [PubMed] [Google Scholar]
- Sevrioukova IF, Poulos TL. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc Natl Acad Sci U S A. 2010;107:18422–7. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PF, DiCenzo R, Morse GD. Clinical pharmacokinetics of non-nucleoside reverse transcriptase inhibitors. Clin Pharmacokinet. 2001;40:893–905. doi: 10.2165/00003088-200140120-00002. [DOI] [PubMed] [Google Scholar]
- Gotta V, Buclin T, Csajka C, Widmer N. Systematic review of population pharmacokinetic analyses of imatinib and relationships with treatment outcomes. Ther Drug Monit. 2013;35:150–67. doi: 10.1097/FTD.0b013e318284ef11. [DOI] [PubMed] [Google Scholar]
- Larson RA, Druker BJ, Guilhot F, O'Brien SG, Riviere GJ, Krahnke T, Gathmann I, Wang Y. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111:4022–28. doi: 10.1182/blood-2007-10-116475. [DOI] [PubMed] [Google Scholar]
- Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard MA, Lassalle R, Marit G, Reiffers J, Begaud B, Moore N, Molimard M, Mahon FX. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109:3496–99. doi: 10.1182/blood-2006-07-036012. [DOI] [PubMed] [Google Scholar]
- Demetri GD, Wang Y, Wehrle E, Racine A, Nikolova Z, Blanke CD, Joensuu H, von Mehren M. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141–47. doi: 10.1200/JCO.2008.20.4818. [DOI] [PubMed] [Google Scholar]
- Pillai VC, Venkataramanan R, Parise RA, Christner SM, Gramignoli R, Strom SC, Rudek MA, Beumer JH. Ritonavir and efavirenz significantly alter the metabolism of erlotinib--an observation in primary cultures of human hepatocytes that is relevant to HIV patients with cancer. Drug Metab Dispos. 2013;41:1843–51. doi: 10.1124/dmd.113.052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoroski BJ, Zhang S, Cai H, Hutzler JM, Frye R, Tracy TS, Strom SC, Lehmann T, Ang CY, Cui YY, Venkataramanan R. Induction and inhibition of cytochromes P450 by the St. John's wort constituent hyperforin in human hepatocyte cultures. Drug Metab Dispos. 2004;32:512–8. doi: 10.1124/dmd.32.5.512. [DOI] [PubMed] [Google Scholar]
- Peng B, Hayes M, Resta D, Racine-Poon A, Druker BJ, Talpaz M, Sawyers CL, Rosamilia M, Ford J, Lloyd P, Capdeville R. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22:935–42. doi: 10.1200/JCO.2004.03.050. [DOI] [PubMed] [Google Scholar]
- Lu C, Li P, Gallegos R, Uttamsingh V, Xia CQ, Miwa GT, Balani SK, Gan LS. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34:1600–5. doi: 10.1124/dmd.106.010793. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Itoga H, Kohno Y, Nagata K, Yamazoe Y. Prediction of oral clearance from in vitro metabolic data using recombinant CYPs: comparison among well-stirred, parallel-tube, distributed and dispersion models. Xenobiotica. 2005;35:627–46. doi: 10.1080/00498250500159371. [DOI] [PubMed] [Google Scholar]
- Riley RJ, McGinnity DF, Austin RP. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos. 2005;33:1304–11. doi: 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- Witherow LE, Houston JB. Sigmoidal kinetics of CYP3A substrates: an approach for scaling dextromethorphan metabolism in hepatic microsomes and isolated hepatocytes to predict in vivo clearance in rat. J Pharmacol Exp Ther. 1999;290:58–65. [PubMed] [Google Scholar]
- Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–9. doi: 10.1158/1535-7163.MCT-10-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky S, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. 5th edn. New York: W.H. Freeman and Company; 2000. [Google Scholar]
- Chiba M, Ishii Y, Sugiyama Y. Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009;11:262–76. doi: 10.1208/s12248-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP-glucuronosyltransferase cofactors in alamethicin-activated human liver microsomes. Drug Metab Dispos. 2009;37:82–9. doi: 10.1124/dmd.108.023853. [DOI] [PubMed] [Google Scholar]
- Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, Yates JW, Ku MS, Gibson CR, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Bjornsson T, Lave T, Poulin P. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: Comparative assessement of prediction methods of human clearance. J Pharm Sci. 2011;100:4090–110. doi: 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
- Guilhot F, Hughes TP, Cortes J, Druker BJ, Baccarani M, Gathmann I, Hayes M, Granvil C, Wang Y. Plasma exposure of imatinib and its correlation with clinical response in the Tyrosine Kinase Inhibitor Optimization and Selectivity Trial. Haematologica. 2012;97:731–8. doi: 10.3324/haematol.2011.045666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Kumar L, Meena R, Velpandian T. Drug monitoring of imatinib levels in patients undergoing therapy for chronic myeloid leukaemia: comparing plasma levels of responders and non-responders. Eur J Clin Pharmacol. 2009;65:545–9. doi: 10.1007/s00228-009-0621-z. [DOI] [PubMed] [Google Scholar]
- Gao B, Yeap S, Clements A, Balakrishnar B, Wong M, Gurney H. Evidence for therapeutic drug monitoring of targeted anticancer therapies. J Clin Oncol. 2012;30:4017–25. doi: 10.1200/JCO.2012.43.5362. [DOI] [PubMed] [Google Scholar]
- Beumer JH. Without therapeutic drug monitoring, there is no personalized cancer care. Clin Pharmacol Ther. 2013;93:228–30. doi: 10.1038/clpt.2012.243. [DOI] [PubMed] [Google Scholar]
- Lu C, Hatsis P, Berg C, Lee FW, Balani SK. Prediction of pharmacokinetic drug-drug interactions using human hepatocyte suspension in plasma and cytochrome P450 phenotypic data. II. In vitro-in vivo correlation with ketoconazole. Drug Metab Dispos: the biological fate of chemicals. 2008;36:1255–60. doi: 10.1124/dmd.107.018796. [DOI] [PubMed] [Google Scholar]
- Park JY, Kim KA, Shin JG, Lee KY. Effect of ketoconazole on the pharmacokinetics of rosiglitazone in healthy subjects. Br J Clin Pharmacol. 2004;58:397–402. doi: 10.1111/j.1365-2125.2004.02161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuypers DR, de Jonge H, Naesens M, Vanrenterghem Y. Effects of CYP3A5 and MDR1 single nucleotide polymorphisms on drug interactions between tacrolimus and fluconazole in renal allograft recipients. Pharmacogenet Genomics. 2008;18:861–8. doi: 10.1097/FPC.0b013e328307c26e. [DOI] [PubMed] [Google Scholar]
- Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin Pharmacol Ther. 2011;90:666–73. doi: 10.1038/clpt.2011.164. [DOI] [PubMed] [Google Scholar]
- Walsky RL, Gaman EA, Obach RS. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J Clin Pharmacol. 2005;45:68–78. doi: 10.1177/0091270004270642. [DOI] [PubMed] [Google Scholar]
- O'Brien SG, Meinhardt P, Bond E, Beck J, Peng B, Dutreix C, Mehring G, Milosavljev S, Huber C, Capdeville R, Fischer T. Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome P450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer. 2003;89:1855–9. doi: 10.1038/sj.bjc.6601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt DJ, von Moltke LL, Daily JP, Harmatz JS, Shader RI. Extensive impairment of triazolam and alprazolam clearance by short-term low-dose ritonavir: the clinical dilemma of concurrent inhibition and induction. J Clin Psychopharmacol. 1999;19:293–6. doi: 10.1097/00004714-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Madan A, Graham RA, Carroll KM, Mudra DR, Burton LA, Krueger LA, Downey AD, Czerwinski M, Forster J, Ribadeneira MD, Gan LS, LeCluyse EL, Zech K, Robertson P, Jr, Koch P, Antonian L, Wagner G, Yu L, Parkinson A. Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab Dispos. 2003;31:421–31. doi: 10.1124/dmd.31.4.421. [DOI] [PubMed] [Google Scholar]
- Xu C, Desta Z. In vitro analysis and quantitative prediction of efavirenz inhibition of eight cytochrome P450 (CYP) enzymes: major effects on CYPs 2B6, 2C8, 2C9 and 2C19. Drug Metab Pharmacokinet. 2013;28:362–71. doi: 10.2133/dmpk.dmpk-12-rg-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 2007;82:197–203. doi: 10.1038/sj.clpt.6100122. [DOI] [PubMed] [Google Scholar]
- Koon HB, Krown SE, Lee JY, Honda K, Rapisuwon S, Wang Z, Aboulafia D, Reid EG, Rudek MA, Dezube BJ, Noy A. Phase II trial of imatinib in AIDS-associated Kaposi's sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol. 2014;32:402–8. doi: 10.1200/JCO.2012.48.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauret N, Gauthier A, Nicoll-Griffith DA. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab Dispos. 1998;26:1–4. [PubMed] [Google Scholar]
- Hickman D, Wang JP, Wang Y, Unadkat JD. Evaluation of the selectivity of in vitro probes and suitability of organic solvents for the measurement of human cytochrome P450 monooxygenase activities. Drug Metab Dispos. 1998;26:207–15. [PubMed] [Google Scholar]
- Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet. 1998;35:361–90. doi: 10.2165/00003088-199835050-00003. [DOI] [PubMed] [Google Scholar]
- Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29:687–710. doi: 10.1097/FTD.0b013e31815c16f5. [DOI] [PubMed] [Google Scholar]
- Grime KH, Bird J, Ferguson D, Riley RJ. Mechanism-based inhibition of cytochrome P450 enzymes: an evaluation of early decision making in vitro approaches and drug–drug interaction prediction methods. Eur J Pharm Sci. 2009;36:175–91. doi: 10.1016/j.ejps.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Frye RF, Fitzgerald SM, Lagattuta TF, Hruska MW, Egorin MJ. Effect of St John's wort on imatinib mesylate pharmacokinetics. Clin Pharmacol Ther. 2004;76:323–9. doi: 10.1016/j.clpt.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Smith P, Bullock JM, Booker BM, Haas CE, Berenson CS, Jusko WJ. The influence of St John's wort on the pharmacokinetics and protein binding of imatinib mesylate. Pharmacotherapy. 2004;24:1508–14. doi: 10.1592/phco.24.16.1508.50958. [DOI] [PubMed] [Google Scholar]
- Delbaldo C, Chatelut E, Re M, Deroussent A, Seronie-Vivien S, Jambu A, Berthaud P, Le Cesne A, Blay JY, Vassal G. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12:6073–78. doi: 10.1158/1078-0432.CCR-05-2596. [DOI] [PubMed] [Google Scholar]
- Schmidli H, Peng B, Riviere GJ, Capdeville R, Hensley M, Gathmann I, Bolton AE, Racine-Poon A. Population pharmacokinetics of imatinib mesylate in patients with chronic phase chronic myeloid leukaemia: results of a phase III study. Br J Clin Pharmacol. 2005;60:35–44. doi: 10.1111/j.1365-2125.2005.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A, Rochat B, Eap CB, Henry H, Biollaz J, Buclin T. Population pharmacokinetics of imatinib and the role of α1-acid glycoprotein. Br J Clin Pharmacol. 2006;62:97–112. doi: 10.1111/j.1365-2125.2006.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber G, Wex T, Schleyer E, Troeger U, Hosius C, Malfertheiner P. Imatinib for hepatocellular cancer - focus on pharmacokinetic/pharmacodynamic modelling and liver function. Cancer Lett. 2008;260:146–54. doi: 10.1016/j.canlet.2007.10.041. [DOI] [PubMed] [Google Scholar]
- van Erp N, Gelderblom H, van Glabbeke M, Van Oosterom A, Verweij J, Guchelaar HJ, Debiec-Rychter M, Peng B, Blay JY, Judson I. Effect of cigarette smoking on imatinib in patients in the soft tissue and bone sarcoma group of the EORTC. Clin Cancer Res. 2008;14:8308–13. doi: 10.1158/1078-0432.CCR-08-1303. [DOI] [PubMed] [Google Scholar]
- Judson I, Ma P, Peng B, Verweij J, Racine A, di Paola ED, Van Glabbeke M, Dimitrijevic S, Scurr M, Dumez H, van Oosterom A. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma Group. Cancer Chemother Pharmacol. 2005;55:379–86. doi: 10.1007/s00280-004-0876-0. [DOI] [PubMed] [Google Scholar]