

Abstract
Aim
Despite promising preclinical findings regarding clinical utility of farnesyltransferase inhibitors (FTI), such as lonafarnib, success of clinical trials is limited. A multicentre AGO-OVAR-15 phase II trial reported an unfavourable effect of lonafarnib on the outcome of patients with advanced ovarian cancer. This study was performed as a genetic subgroup analysis of the AGO-OVAR-15 trial, and investigated the utility of the promoter polymorphism rs11623866 of the farnesyltransferase ß-subunit gene (FNTB) in predicting the clinical effectiveness of lonafarnib.
Methods
The influence of rs11623866 (c.-609G > C) on FNTB promoter activity was investigated by electrophoretic-mobility-shift assay, luciferase-reporter assay and RT-qPCR. A total of 57 out of 105 patients from the AGO-OVAR-15 trial, treated with carboplatin and paclitaxel ± lonafarnib, was genotyped for rs11623866 by restriction fragment length polymorphism analysis. Genotype-dependent survival analysis was performed by Kaplan–Meier analysis.
Results
The presence of the G allele was associated with increased FNTB promoter activity compared with the C allele. An unfavourable effect of lonafarnib was limited to patients carrying a GG genotype (HRPFS 6.2, 95%CI = 2.01, 19.41, P = 0.002; HROS 9.6, 95%CI = 1.89, 48.54, P = 0.006). Median progression free survival (PFS) for patients with the GG genotype in the lonafarnib treated arm was 10 months, whereas median PFS without FTI-treatment was 40 months. Median overall survival (OS) in the lonafarnib-treated group was 19 months, whereas median OS was not reached in the untreated group.
Conclusions
Discrepancies between preclinical success and clinical failure may be due to the patients' genetic variability of FNTB. Therefore, our results may encourage retrospective evaluation of FNTB polymorphisms in previous FTI studies, especially those reporting positive FTI response.
Keywords: farnesyltransferase inhibitor, lonafarnib, ovarian cancer, rs11623866, single nucleotide polymorphism
What is Already Known about this Subject
Farnesyltransferase inhibitors, such as lonafarnib, have failed to demonstrate clinical benefit in ovarian cancer
Even an adverse effect of this drug was reported in the AGO-OVAR-15 trial.
We investigated the utility of the farnesyltransferase ß-subunit (FNTB) promoter polymorphism rs11623866 in predicting the clinical effect of lonafarnib.
What this Study Adds
Our study contributes to the understanding of lonafarnib and its clinical utility.
It shows that the adverse effect of lonafarnib is limited to patients carrying a rs11623866 GG genotype. No effect was observed for the other genotypes.
Our results encourage retrospective evaluation of FNTB polymorphisms in previous FTI-studies, especially those, reporting a positive FTI response.
Introduction
Ovarian cancer is the leading cause of death among women with gynaecologic malignancies 1. Standard treatment of ovarian cancer constitutes primary radical surgery, aiming at macroscopically complete tumour resection and subsequent platinum- and paclitaxel-based chemotherapy 2. Residual tumour burden after primary surgery is believed to be one of the most relevant prognostic factors for ovarian malignancies 3,4. Advanced ovarian cancer is usually chemotherapy sensitive with an overall clinical response rate of 70–80% 5. However, despite this profound sensitivity to platinum-based chemotherapy and despite continuous attempts to implement maintenance therapies, more than 50% of all patients experience recurrence, resulting in a poor overall prognosis 5,6. Therefore, the development of targeted therapy strategies is highly desirable.
In this context, there are recent advances in the administration of anti-angiogenetic monoclonal antibodies (e.g. bevacizumab) or tyrosine kinase inhibitors (e.g. pazopanib) for targeted ovarian cancer therapy 7,8. However, presently, no predictive biomarkers are available for these kinds of therapies. Apart from this, farnesyltransferase inhibitors (FTI), such as lonafarnib, have also been of significant clinical interest. The FTI lonafarnib abrogates lipid modification of H-Ras and other farnesylation-dependent proteins, such as Rheb, RhoB or centromer-associated motor proteins, thereby interfering with tumourigenic signalling 9,10. Preclinical results showed that lonafarnib, either as single agent or in combination with taxanes, is active not only in a broad spectrum of tumour cell lines in vitro but also in human ovarian cancer and breast cancer animal models 11–13. Due to these encouraging results, a variety of clinical studies investigated the effect of lonafarnib in different cancer entities. However, the majority of trials failed to demonstrate any substantial clinical benefit of lonafarnib. Consequently, the concept of targeting farnesyltransferase activity has not entered clinical practice 14–17. In a randomized clinical trial (AGO-OVAR-15, phase II), we recently analyzed the potential clinical benefit of carboplatin and paclitaxel with or without lonafarnib in first line treatment of epithelial ovarian cancer International Federation of Gynaecology and Obstetrics (FIGO) stages IIB-IV. This clinical trial comprised 105 patients and did not resolve any significant difference in the non-lonafarnib-treated vs. the lonafarnib-treated arm, with regard to progression free survival (PFS) and overall survival (OS). In contrast, lonafarnib treatment appeared to exert a negative effect upon survival, since significant inferiority of the lonafarnib-treated arm was reported in a stratum with residual tumour of more than 1 cm or the presence of distant metastasis (FIGO IV) 18.
So far, farnesyltransferase expression level has been discussed as a potential predictive biomarker for lonafarnib response 19. However, little is known about transcriptional regulation of farnesyltransferases and how this relates to lonafarnib effectiveness 20. We previously demonstrated prognostic relevance of FNTB-promoter core polymorphisms in several cancer entities, such as breast or renal cell cancer 21. Of those, rs11623866 (c.-609G > C) was the only SNP with a sufficient minor allele frequency for analysis in a limited patient cohort and was in linkage disequilibrium to the other FNTB promoter SNPs with lower allele frequency and in silico analysis predicted its potential functionality.
Therefore, we genotyped a subgroup of patients from the AGO-OVAR-15 clinical trial and investigated, in terms of an exploratory genetic study, whether the candidate FNTB promoter polymorphism rs11623866 i) influences farnesyltransferase expression and ii) may be a predictive biomarker for the effect of lonafarnib in ovarian cancer patients.
Methods
Patient characteristics
The current study was based on the recent AGO-OVAR-15, phase II clinical trial (EudraCT number: 2004-004515-26), comprising 105 patients. This trial compared standard chemotherapy (carboplatin and paclitaxel) with or without lonafarnib in primary advanced ovarian cancer. Patients above 18 years with histologically confirmed FIGO stages IIB to IV ovarian cancer were included. They had undergone previous debulking surgery (with the aim of macroscopic complete tumour resection) within 6 weeks before random assignment had been eligible. Lonafarnib was administered at a dose of 100 mg orally twice a day during chemotherapy and was increased thereafter to 200 mg twice a day, up to 6 months as a maintenance therapy. Maintenance therapy was administered for a maximum of 6 months. Patients were stratified according to residual tumour size and FIGO stage 18. Stratum 1 consisted of patients with FIGO IIB to IIIC and a residual tumour up to 1 cm and stratum 2 consisted of patients with FIGO stage IV and/or a residual tumour of more than 1 cm.
In an amendment of the existing approval for the AGO-OVAR-15 trial, we investigated whether rs11623866 could be a predictive biomarker for the effect of lonafarnib. This amendment was approved by the ethics committee, when the AGO-OVAR-15 trial had already started (Ethikkommission der Ärztekammer Nordrhein, Düsseldorf, reference number: 2004-004515-26 / 2005276 / 10-066) and was performed in accordance with good clinical practice guidelines, national laws and the Declaration of Helsinki. To prevent a systematic bias due to selection of long-living patients, we recruited also those patients with available DNA that already died (as suggested by the local ethics committee and in accordance with the declaration of the Central Ethics Committee of Germany 22. In total, DNA samples for genotyping could be obtained from 57 (54.1%) out of 105 participants in the AGO-OVAR-15 (subgroup genetics) trial. Clinicopathological data of this subgroup were comparable with those of the complete study group of the AGO-OVAR-15 trial and are summarized in Table1. Genotype distributions of both treatment arms were not significantly different (P = 0.132).
Table 1.
Baseline characteristics. The table summarizes the patients' clinicopathological data and the rs11623866 genotype distribution in the AGO-OVAR-15 subgroup genetics trial, with regard to the lonafarnib treated arm and the non-treated arm
| All | TC arm | LTC arm | ||||
|---|---|---|---|---|---|---|
| Characteristic | n | % | n | % | n | % |
| Number of patients | 57 | 100 | 27 | 47.4 | 30 | 52.6 |
| Age (years), median (range) | 56 | (21–74) | 56 | (41–74) | 59 | (21–71) |
| Performance status | ||||||
| ECOG 0 | 28 | 49.1 | 13 | 48.1 | 15 | 50.0 |
| ECOG 1 | 27 | 47.4 | 13 | 48.1 | 14 | 46.7 |
| ECOG 2 | 2 | 3.5 | 1 | 3.7 | 1 | 3.3 |
| FIGO stage | ||||||
| IIB-III | 47 | 82.5 | 23 | 85.2 | 24 | 80.0 |
| IV | 10 | 17.5 | 4 | 14.8 | 6 | 20.0 |
| Histology | ||||||
| Serous | 42 | 73.7 | 20 | 74.1 | 22 | 73.3 |
| Endometroid | 4 | 7.0 | 3 | 11.1 | 1 | 3.3 |
| Mucinous | 3 | 5.3 | 2 | 7.4 | 1 | 3.3 |
| Other | 8 | 14.0 | 2 | 7.4 | 6 | 20.0 |
| Primary tumour | ||||||
| Ovary | 52 | 91.2 | 23 | 85.2 | 29 | 96.7 |
| Fallopian tube | 0 | 0 | 0 | |||
| Peritoneum | 5 | 8.8 | 4 | 14.8 | 1 | 3.3 |
| Histologic grading | ||||||
| G1 | 2 | 3.5 | 0 | 2 | 6.7 | |
| G2 | 20 | 35.1 | 12 | 44.4 | 8 | 26.7 |
| G3 | 33 | 57.9 | 14 | 51.9 | 19 | 63.3 |
| Unknown | 2 | 3.5 | 1 | 3.7 | 1 | 3.3 |
| rs11623866 G > C | ||||||
| GG | 20 | 35.1 | 13 | 48.2 | 7 | 23.3 |
| GC | 28 | 49.1 | 10 | 37.0 | 18 | 60.0 |
| CC | 9 | 15.8 | 4 | 14.8 | 5 | 16.7 |
LTC, lonafarnib + TC;
TC, taxane (paclitaxel) + carboplatin.
Polymorphism retrieval and bioinformatic analyses
We retrieved and analyzed data of about 250 putatively functional genetic variations of the FNTB gene region (5' upstream region, 5'UTR, coding region, exon-intron boundaries and 3'UTR) from NCBI dbSNP 23 and the 1000 Genome browser 24. Variations with a minor allele frequency (MAF) of at least 0.01 (1%) in Caucasians were considered to be polymorphisms according to the common definition 25. To locate the core promoter region we performed promoter analysis using PromoterInspector and Gene2Promoter 26. Analysis of putative rs11623866 allele-dependent binding sites of transcription factors was performed with MatInspector 27, Consite 28 and Alibaba2.1 29.
DNA preparation and determination of FNTB rs11623866 genotypes
For rs11623866 genotyping, DNA was extracted from whole blood or routinely processed paraffin-embedded tissue. Dewaxing and isolation of genomic DNA were done following the manufacturer's instructions using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). For polymerase chain reaction (PCR) the Taq DNA Polymerase Master Mix RED (Ampliqon, Herlev, Denmark) was used. Genotypes of the rs11623866 polymorphism were determined by restriction fragment length polymorphism analyses using the ‘slowdown’ PCR 30. The PCR was performed with the following primers: forward: 5'-GCGGACTGACTGTCTATTT-3', reverse: 5'-GACGCCGTCTCAGTATCA -3' resulting in a PCR product of 140 bp. Amplified fragments were digested with restriction enzyme RsaI (New England Biolabs, Beverly, MA, USA) by incubating for 4 h at 37 °C. RsaI specifically cuts PCR products that carry the G allele (101 + 39 bp). Adequate negative and positive controls were routinely used to ascertain correct genotyping. Accuracy of genotyping was additionally validated by direct sequencing of 20 randomly selected samples. This revealed complete concordance with the previous results obtained by PCR product digestion.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts of the SKOV-3 ovarian cancer cell line were prepared using the NuCLEAR Extraction Kit (Sigma, Deisenhofen, Germany), according to the manufacturer's instructions. Sequences of unlabelled and DY-682 fluorescence-labeled oligonucleotides (Eurofins MWG Operon, Ebersberg, Germany) of the double-stranded DNA probes used were as follows: 5'-ATCCTTGTTCCTT(G/C)TACTGCATTTCAG-3' and 5'-CTGAAATGCAGTA(C/G)AAGGAACAAGGAT-3'. EMSAs were performed in triplicate and were carried out with the EMSA buffer kit (Li-COR Bioscience, Lincoln, NE, USA) according to the manufacturer's instructions. Double-stranded DNA probes and SKOV3 nuclear extracts (5 µg) were incubated with 2 µl of 10% binding buffer, 2.5 mm DTT, 0.25% Tween-20, 1 µg of poly (dI–dC) and 0.05% NP-40 in a total volume of 20 µl for 30 min at room temperature and separated by electrophoresis on a 6% polyacrylamide gel in 0.5% Tris-borate-EDTA running buffer. The gels were scanned by direct infrared fluorescence detection on the Odyssey imaging system (Li-COR Bioscience, Lincoln, NE, USA).
Luciferase reporter assay
The promoter region of FNTB, comprising either the G allele or the C allele, was PCR-amplified from genomic DNA and cloned into the pGEM-T Easy vector (Promega, Madison, WI, USA). The amplified promoter region was restricted from the pGEM-T Easy vector and cloned upstream of the firefly luciferase coding region into the pGL4.10 vector (Promega, Madison, WI, USA). SKOV3 and A2780 cells were plated into 96-well plates at a density of 1,5 × 104 cells/well in 100 µl of Dulbecco's modified eagle medium and Roswell Park Memorial Institute medium, respectively, with 10% fetal bovine serum. After 24 h, co-transfections were carried out in 50 µl of medium without serum using 150 ng of the respective pGL4 reporter construct and 50 ng of Renilla luciferase control vector (pGL4.74, Promega, Madison, WI, USA) containing 0.5 µl of Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) per transfection according to the manufacturer's instructions. After 6 h, the transfection mix was removed and cells were incubated with new medium. Twenty-four h after treatment, cells were harvested and assayed for Firefly and Renilla luciferase activities using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) on a Lumat LB 9501 Luminometer (Berthold, Bad Wildbad, Germany). To correct for variable transfection efficiency Firefly luciferase activity was normalized to Renilla luciferase activity.
RT-qPCR
Total RNA was extracted from snap-frozen samples retrieved from the University Hospital of Essen, Germany, utilizing the Qiagen RNeasy kit according to the manufacturer's instructions. One µg of total RNA was applied for cDNA synthesis with oligo dT primers (Roche, Mannheim, Germany) and Superscript II reverse transcriptase (Invitrogen, Karlsruhe, Germany). In order to quantify FNTB mRNA levels, we utilized the QuantiTect SYBR Green PCR kit (Qiagen, Hilden, Germany). Primer sequences for FNTB transcript detection were designed in order to amplify a region near the translation initiation site (160 bp), spanning exon 1 and exon 2 (forward: 5' CTT CTC CGA GTT CTT TCA, reverse: 5' ATC TTT TCT TCT ACT TTT GC). All experimental steps were performed according to the manufacturer's instructions. Quantitative RT-qPCR was performed using the ABI-7500 system (Applied Biosystems, Darmstadt, Germany). The Cq-threshold was adjusted to a fluorescent level above the background signal and within the linear range of each amplification plot. Melting curves were drawn after each PCR run in order to ensure that a single and specific PCR-product was generated. All samples, including non-RT (without reverse transcriptase) and no-template controls were assayed in triplicate. Mean Cq values and deviations between the triplicates were calculated. Samples with a deviation > 0.5 or with any evidence for melting curve abnormality were repeated. In order to allow across sample comparison, FTNB expression was normalized to human beta actin (ACTB) expression as a reference gene 31. Relative expression values were calculated as following: 2 -[Cq(FNTB) - Cq(ACTB)].
Statistical analysis
Clinical variables and genotypes were compared using either Student's t-test, linear anova for continuous variables or Pearson's χ2 test for categorical data. Control for deviation from the Hardy–Weinberg equilibrium was conducted using a publically available Hardy–Weinberg equilibrium calculator 32. Differences with P values <0.05 were regarded as statistically significant. All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, LaJolla, CA, USA) and SPSS software version 21.0 (IBM, Armonk, NY, USA).
Results
Effect of lonafarnib in the studied subpopulation
From 57 out of 105 study patients from the AGO-OVAR-15, we could obtain DNA samples for genotyping. Kaplan–Meier analysis, comparing PFS and OS of the lonafarnib-treated arm vs. the untreated arm in our genetic analysis is shown in Figure1. Curve progression compares with that of the complete study group, as already published 18. The tendency for a negative effect of lonafarnib treatment on PFS is detectable in the subgroup of the current study as well. Furthermore, the lonafarnib-treated arm showed significantly decreased OS (HR 2.178, 95% CI 1.04, 4.46, P = 0.041).
Figure 1.
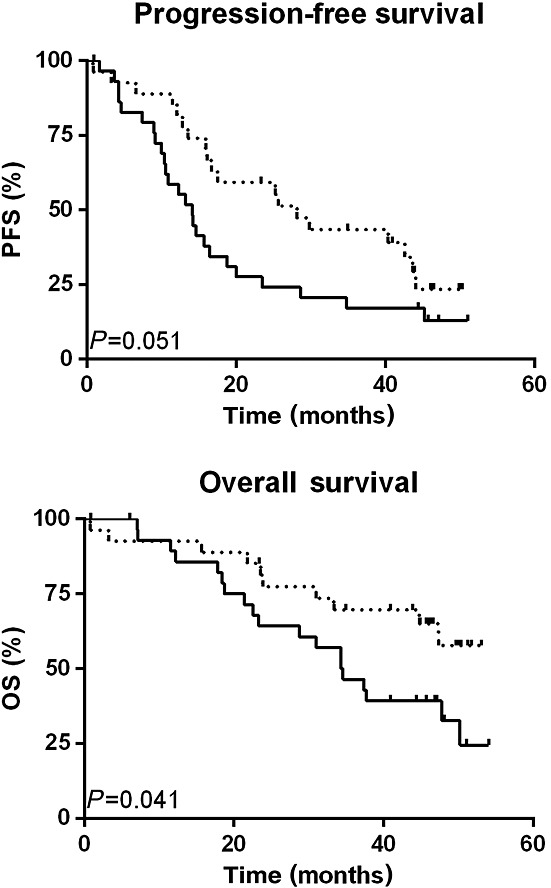
Survival dependent on treatment arm. Kaplan–Meier analysis and the log-rank test were used to calculate survival curves representing progression free and overall survival of 57 patients of the AGO-OVAR-15 study dependent on treatment arm. TC taxane (paclitaxel) + carboplatin (paclitaxel) ( ). LTC lonafarnib + TC (
). LTC lonafarnib + TC ( )
)
Genetic variability of the FNTB gene locus and in silico analysis of rs11623866
Because the FNTB promoter region is not characterized, we used in silico prediction to determine size and localization of the core promoter region. According to this analysis, the core promoter region is located within the first 834 bp 5'upstream of the translation start point. Therefore, we analyzed all genetic variants within 1000 bp 5'upstream of the translation start point as well as all variants located in other putatively functional regions of the FNTB gene, i.e. 5'UTR, exons, exon intron boundaries and 3'UTR. Although databases list about 250 genetic variants within these regions, only eight of them reach a MAF of at least 1% in Caucasians (Figure2A). Three polymorphisms are located within the putative core promoter region. One polymorphism is a synonymous single nucleotide polymorphism in exon 10 and two polymorphisms are part of the 3'UTR of FNTB. Of those, rs11623866 is the only polymorphism with a sufficient minor allele frequency (38%) for analysis in our limited patient cohort. Furthermore, it is in linkage disequilibrium to the other FNTB promoter polymorphisms with lower allele frequencies (data not shown) and in silico analysis predicted its potential functionality due to allele-dependent binding sites of different transcription factors (Figure2B).
Figure 2.
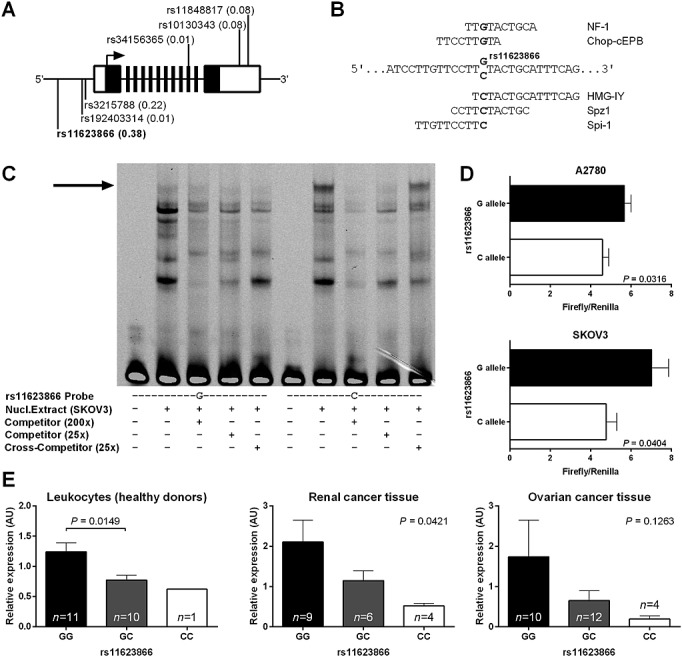
Functional properties of the FNTB G > C polymorphism rs116238662. A) Structure of the FNTB gene locus. Black boxes represent exons, they are not drawn to scale. The 5' and 3'UTR are highlighted in white. Positions of FNTB polymorphisms are indicated together with minor allele frequencies given in brackets. B) In silico transcription factor binding sites, rs11623866 alleles and the surrounding sequence are shown. Transcription factor names and their putative binding sites are shown above and below the corresponding FNTB alleles, respectively. C) Genotype-dependent binding of nuclear proteins to the human FNTB promoter as analyzed by EMSA. Arrow indicates specifically retarded bands observed upon addition of nuclear extracts. A nuclear protein bound the C allele exclusively. D) Genotype-dependent activity of FNTB 5'-regulatory region reporter constructs expressed in A2780 and SKOV3 ovarian carcinoma cells. E) Relative expression of FNTB mRNA in leukocytes of healthy donors, renal cancer and ovarian cancer tissues dependent on rs11623866 genotypes
Genotype-specific binding of SKOV3 nuclear protein to the rs11623866 C allele
We evaluated, whether the rs11623866 polymorphism influences binding of nuclear proteins and may induce differential FNTB promoter activity. An EMSA was performed, using SKOV-3 ovarian cancer cell line nuclear extracts (Figure2C). After incubation with an oligonucleotide probe harbouring the rs11623866 C allele, we observed a distinct band, which was specifically retarded in the presence of a molar excess of a competitor probe but not in the presence of a cross-competitor probe. After incubation with a rs11623866 G allele comprising probe, we did not notice any size corresponding band, independently from the presence of any competitor type. These findings suggest allele-dependent binding of SKOV-3 nuclear protein, preferentially to the C allele.
Presence of the rs11623866 C allele is associated with impaired FNTB promoter activity in vitro
We subsequently investigated, whether the FNTB rs11623866 G > C polymorphism is associated with differential FNTB promoter activity in vitro. Normalized firefly luciferase read-outs of both cell lines revealed reporter activity to be significantly reduced in samples with C allele promoter sequence, compared to G-harbouring promoters (P = 0.0316, P = 0.0404, respectively, Figure2D). This suggests that the C allele is associated with reduced FNTB promoter activity in vitro.
Presence of the rs11623866 C allele parallels impaired FNTB transcription in vivo
Differential promoter activity, associated with rs11623866 G > C polymorphism, may translate into differential rs11623866 G > C dependent FNTB transcript abundance in vivo. To test this assumption, we quantified FNTB expression in leucocytes from healthy subjects and different rs11623866 G > C genotypes (n = 22, Figure2E). The GC genotype was significantly associated with decreased FNTB expression, compared with the GG genotype (P = 0.0149). In snap-frozen specimens from primary human renal cell or ovarian carcinoma (n = 19, n = 26 respectively), we likewise revealed the presence of the GC as well as the CC genotype to parallel decreased FNTB expression, compared with the presence of the GG genotype. However, the difference observed in the ovarian cancer tissues missed statistical significance (P = 0.0421, P = 0.1263, respectively). According to these findings we concluded that differential promoter activity, associated with the rs11623866 G > C polymorphism, is accompanied by impaired FNTB transcription in individuals with a GC/CC genotype in normal and malignant tissue.
Association of the FNTB rs11623866 G > C polymorphism with the clinical effect of lonafarnib
As reported earlier, the whole study population showed a trend for inferiority of the lonafarnib-treated arm. However, for lonafarnib-treated patients with a GG genotype, highly significantly decreased PFS and OS were observed (P < 0.001, P = 0.001, respectively, Figure3A). Concomitantly, hazard ratios were significantly increased for lonafarnib-treated GG genotype carriers (HRPFS 6.2, P = 0.002; HROS 9.6, P = 0.006, Table2). Contrarily, for patients with a GC or CC genotype, no significant difference in the survival curves and hazard ratios could be observed between the two study arms (P = 0.919, P = 0.881, P = 0.625, P = 0.538, Table2, Figure3B + C). To evaluate further the effect of lonafarnib, we analyzed the toxicities by treatment arm for the subgroup genetics and for GG genotype carriers. Distribution and frequency of toxic side effects was comparable with those reported for the complete study population (Supplementary Table 1). Especially for GG genotype carriers, the toxicity profile was comparable between the two study arms (Supplementary Table 2).
Figure 3.
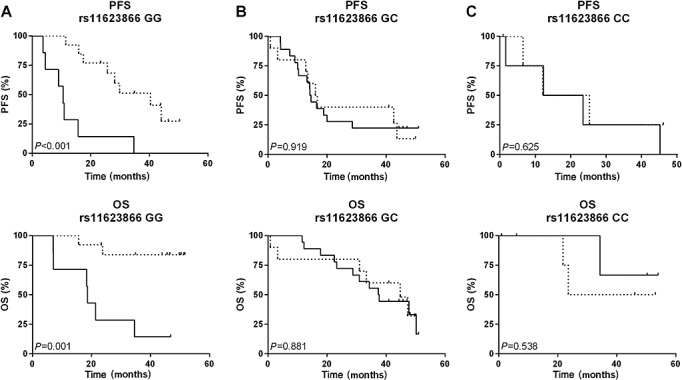
Survival dependent on rs11623866 genotype and treatment arm. Kaplan–Meier analysis and the log-rank test were used to calculate survival curves representing progression free (PFS) and overall survival (OS) of 57 patients of the AGO-OVAR-15 study dependent on rs11623866 genotype and treatment arm. TC taxane (paclitaxel) + carboplatin ( ); LTC lonafarnib + TC (
); LTC lonafarnib + TC ( )
)
Table 2.
Cox regression analyses. The table summarizes results of Cox regression analyses in the AGO-OVAR-15 subgroup genetics trial with regard to rs11623866 genotype, the respective treatment arms and survival (PFS and OS)
| FNTB rs11623866 G > C Genotype | ||||||
|---|---|---|---|---|---|---|
| GG | GC | CC | ||||
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | |
| Progression-free survival | ||||||
| Treatment | ||||||
| TC | 1 | 1 | 1 | |||
| LTC | 6.242 (2.01, 19.41) | 0.002 | 1.046 (0.44, 2.50) | 0.919 | 1.456 (0.32, 6.62) | 0.627 |
| Overall survival | ||||||
| Treatment | ||||||
| TC | 1 | 1 | 1 | |||
| LTC | 9.589 (1.89, 48.54) | 0.006 | 1.080 (0.40, 2.91) | 0.881 | 0.474 (0.04, 5.35) | 0.546 |
CI, confidence interval;
HR, hazard ratio;
LTC, lonafarnib + TC;
TC, taxane (paclitaxel) + carboplatin.
Discussion
The present investigation was performed in association to the AGO-OVAR-15 phase II trial and molecular in vitro and in vivo analysis revealed that the C allele of the FNTB rs11623866 polymorphism was associated with reduced FNTB promoter activity and FNTB mRNA expression compared with the G allele. Moreover, the adverse effect of lonafarnib was restricted to patients carrying the homozygous GG genotype.
On the basis of promising preclinical data, a variety of clinical trials have been performed to investigate the potential clinical utility of lonafarnib as a targeted therapeutic drug, either as a single agent or in combination with chemotherapy. A recent phase II study evaluated the effect of lonafarnib in combination with paclitaxel in a small cohort of patients (n = 33) with resistant non-small cell lung cancer and reported partial response or stable disease in 48% of patients 33. In contrast, the majority of clinical trials failed to unravel any substantial objective response to lonafarnib administration in, for example, haematological malignancies, head and neck, colorectal or urothelial cancers 14–17. Due to these discouraging findings, pharmacological inhibition of farnesyltransferase has not yet been translated into clinical practice.
The present genotyping study was performed in association with the AGO-OVAR-15 phase II trial and evaluated the clinical utility of the FNTB rs11623866 G > C polymorphism as a potential pharmacogenetic predictive biomarker for lonafarnib treatment in ovarian cancer patients. Notably, analysis was conducted at a time when it was yet unknown whether or not the enrolled ovarian cancer patients would benefit from the addition of lonafarnib to standard platinum-based chemotherapy. As its central finding, AGO-OVAR-15 finally revealed no significant difference in the non-lonafarnib-treated vs. lonafarnib-treated arm. Surprisingly, even inferiority in the lonafarnib-treated arm was observed in the stratum with residual tumour of more than 1 cm or the presence of distant metastasis (FIGO IV) 18. A key finding of the present study was that the adverse effect of lonafarnib was restricted to patients carrying the homozygous GG genotype. However, due to the limited number of patients, a further stratification of the subgroup genetics with regard to FIGO stage and residual tumour burden, as conducted in AGO-OVAR-15 18, could not be done. At first glance, these findings may appear counterintuitive. A straightforward hypothesis would imply that increased expression of a molecular target may favour the success of a related targeted therapy. This is demonstrated in, for example, breast cancer patients, in which a correlation between HER2-amplification/overexpression and clinical benefit of trastuzumab (a monoclonal antibody, targeting HER2) is well established 34,35. However, interestingly, our findings are in line with a recent study, correlating farnesyltransferase mRNA expression level with clinical response to a combination therapy of docetaxel and lonafarnib in patients with therapy refractory solid malignancies. In this context, it was reported that patients with lower FNTB baseline expression were more likely to respond to this combination therapy 19. However, the functional background of this effect remains to be addressed in future studies. In this context, we also have to take into consideration that we focused on only one out of a variety of existing SNPs in the FNTB gene. Moreover, control of FNTB expression by its promoter sequence may be dependent on the cellular background as well as on the interaction between transcriptional activators and repressors. Thus, given our data from EMSA analysis, it may be possible that the nuclear protein fraction, specifically binding the C allele, comprises transcriptional repressor protein(s), which may contribute to negative regulation of FNTB transcription. However, in spite of these limitations, the molecular results in our study are indicative that the alleles of the FNTB rs11623866 G > C polymorphism are associated with differential FNTB promoter activity.
Conclusively, given the toxicity of lonafarnib 18 and its limited response among several clinical trials and different cancer entities 14–17, the identification of a predictive biomarker for lonafarnib treatment is highly desirable, since there may still exist a subgroup of patients who benefits from this kind of targeted therapy.
Notably, our genetic substudy was performed in parallel with the AGO-OVAR-15 main trial, i.e. to a time, when it was not clear yet, whether patients will have clinical benefit from lonafarnib, added to platinum-based chemotherapy. Nevertheless, given that rs11623866 enabled a stratification of patients with an adverse effect of lonafarnib, we suppose our study to be a relevant cornerstone, as it implies the general clinical relevance of concomitant and auxiliary SNP analysis among FTI studies as a potential diagnostic tool. However, due to the limited number of patients, the present study should be considered exploratory and our results need to be validated in larger patient cohorts. Nevertheless, our data provide the rationale to evaluate FNTB polymorphisms systematically in other FTI-studies in a retrospective manner, particularly in those trials where at least partial clinical response has been reported. Moreover, apart from the oncological background, it could also be interesting to transfer FNTB polymorphisms analysis, particularly for rs11623866, to other non-oncological diseases, such as chronic hepatitis D or Hutchinson–Gilford progeria syndrome, in which FTIs are candidate drugs of interest 36–38. This may help to dissect the mode of action of FTIs, with regard to FNTB promoter activity and possibly to refine a small proportion of patients, in which the presence of a SNP may be indicative for clinical response to FTIs. Moreover, since lonafarnib confers an adverse outcome in patients with a GG genotype, our study implies a further rationale to investigate whether those patients may possibly benefit from a therapeutic strategy of farnesyltransferase activation.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work.
The authors want to thank the AGO study office staff, especially Mrs Gabriele Elser, for administrating our genetic substudy and for coordinating the obtainment of the patients' consent.
Author Contributions
The principal investigator is Pauline Wimberger, MD, PhD.
HSB, WM, AB, RK, JDK, WS, JS, KW, JH, PH, AB, BS, BA and PW made substantial contributions to the conception and design of the study, to the acquisition data and to analysis/interpretation of the results. HSB, PW, WS and JDK designed and performed in vitro analyses and functional assays. HSB, PW, WS and JDK were involved in drafting the manuscript or revising it. All authors read and approved the manuscript in its final version.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1 Grading of toxicities by treatment arm
Table S2 Grading of toxicities by treatment arm for FNTB rs11623866 GG genotype carriers
References
- Black RJ, Bray F, Ferlay J, Parkin DM. Cancer incidence and mortality in the European Union: cancer registry data and estimates of national incidence for 1990. Eur J Cancer. 1997;33:1075–107. doi: 10.1016/s0959-8049(96)00492-3. [DOI] [PubMed] [Google Scholar]
- du Bois A, Quinn M, Thigpen T, Vermorken J, Avall-Lundqvist E, Bookman M, Bowtell D, Brady M, Casado A, Cervantes A, Eisenhauer E, Friedlaender M, Fujiwara K, Grenman S, Guastalla JP, Harper P, Hogberg T, Kaye S, Kitchener H, Kristensen G, Mannel R, Meier W, Miller B, Neijt JP, Oza A, Ozols R, Parmar M, Pecorelli S, Pfisterer J, Poveda A, Provencher D, Pujade-Lauraine E, Randall M, Rochon J, Rustin G, Sagae S, Stehman F, Stuart G, Trimble E, Vasey P, Vergote I, Verheijen R, Wagner U. 2004 consensus statements on the management of ovarian cancer: final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004) Ann Oncol. 2005;16:viii7–12. doi: 10.1093/annonc/mdi961. [DOI] [PubMed] [Google Scholar]
- du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d'Investigateurs Nationaux Pour les Etudes des Cancers de l'Ovaire (GINECO) Cancer. 2009;115:1234–44. doi: 10.1002/cncr.24149. [DOI] [PubMed] [Google Scholar]
- Wimberger P, Wehling M, Lehmann N, Kimmig R, Schmalfeldt B, Burges A, Harter P, Pfisterer J, du Bois A. Influence of residual tumor on outcome in ovarian cancer patients with FIGO stage IV disease: an exploratory analysis of the AGO-OVAR (Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group) Ann Surg Oncol. 2010;17:1642–8. doi: 10.1245/s10434-010-0964-9. [DOI] [PubMed] [Google Scholar]
- Bookman MA. Extending the platinum-free interval in recurrent ovarian cancer: the role of topotecan in second-line chemotherapy. Oncologist. 1999;4:87–94. [PubMed] [Google Scholar]
- Martin LP, Schilder RJ. Management of recurrent ovarian carcinoma: current status and future directions. Semin Oncol. 2009;36:112–25. doi: 10.1053/j.seminoncol.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P, Cervantes A, Kurzeder C, du Bois A, Sehouli J, Kimmig R, Stahle A, Collinson F, Essapen S, Gourley C, Lortholary A, Selle F, Mirza MR, Leminen A, Plante M, Stark D, Qian W, Parmar MK, Oza AM. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–96. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- Friedlander M, Hancock KC, Rischin D, Messing MJ, Stringer CA, Matthys GM, Ma B, Hodge JP, Lager JJ. A Phase II, open-label study evaluating pazopanib in patients with recurrent ovarian cancer. Gynecol Oncol. 2010;119:32–7. doi: 10.1016/j.ygyno.2010.05.033. [DOI] [PubMed] [Google Scholar]
- Basso AD, Mirza A, Liu G, Long BJ, Bishop WR, Kirschmeier P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J Biol Chem. 2005;280:31101–8. doi: 10.1074/jbc.M503763200. [DOI] [PubMed] [Google Scholar]
- Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- Nielsen LL, Shi B, Hajian G, Yaremko B, Lipari P, Ferrari E, Gurnani M, Malkowski M, Chen J, Bishop WR, Liu M. Combination therapy with the farnesyl protein transferase inhibitor SCH66336 and SCH58500 (p53 adenovirus) in preclinical cancer models. Cancer Res. 1999;59:5896–901. [PubMed] [Google Scholar]
- Niessner H, Beck D, Sinnberg T, Lasithiotakis K, Maczey E, Gogel J, Venturelli S, Berger A, Mauthe M, Toulany M, Flaherty K, Schaller M, Schadendorf D, Proikas-Cezanne T, Schittek B, Garbe C, Kulms D, Meier F. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J Invest Dermatol. 2011;131:468–79. doi: 10.1038/jid.2010.297. [DOI] [PubMed] [Google Scholar]
- Taylor SA, Marrinan CH, Liu G, Nale L, Bishop WR, Kirschmeier P, Liu M, Long BJ. Combining the farnesyltransferase inhibitor lonafarnib with paclitaxel results in enhanced growth inhibitory effects on human ovarian cancer models in vitro and in vivo. Gynecol Oncol. 2008;109:97–106. doi: 10.1016/j.ygyno.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Hanrahan EO, Kies MS, Glisson BS, Khuri FR, Feng L, Tran HT, Ginsberg LE, Truong MT, Hong WK, Kim ES. A phase II study of lonafarnib (SCH66336) in patients with chemorefractory, advanced squamous cell carcinoma of the head and neck. Am J Clin Oncol. 2009;32:274–9. doi: 10.1097/COC.0b013e318187dd57. [DOI] [PubMed] [Google Scholar]
- Ravoet C, Mineur P, Robin V, Debusscher L, Bosly A, Andre M, El Housni H, Soree A, Bron D, Martiat P. Farnesyl transferase inhibitor (lonafarnib) in patients with myelodysplastic syndrome or secondary acute myeloid leukaemia: a phase II study. Ann Hematol. 2008;87:881–5. doi: 10.1007/s00277-008-0536-2. [DOI] [PubMed] [Google Scholar]
- Winquist E, Moore MJ, Chi KN, Ernst DS, Hirte H, North S, Powers J, Walsh W, Boucher T, Patton R, Seymour L. A multinomial phase II study of lonafarnib (SCH 66336) in patients with refractory urothelial cancer. Urol Oncol. 2005;23:143–9. doi: 10.1016/j.urolonc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kemeny N, Kelsen DP, Ilson D, O'Reilly E, Zaknoen S, Baum C, Statkevich P, Hollywood E, Zhu Y, Saltz LB. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann Oncol. 2002;13:1067–71. doi: 10.1093/annonc/mdf173. [DOI] [PubMed] [Google Scholar]
- Meier W, du Bois A, Rau J, Gropp-Meier M, Baumann K, Huober J, Wollschlaeger K, Kreienberg R, Canzler U, Schmalfeldt B, Wimberger P, Richter B, Schroder W, Belau A, Stahle A, Burges A, Sehouli J. Randomized phase II trial of carboplatin and paclitaxel with or without lonafarnib in first-line treatment of epithelial ovarian cancer stage IIB-IV. Gynecol Oncol. 2012;126:236–40. doi: 10.1016/j.ygyno.2012.04.050. [DOI] [PubMed] [Google Scholar]
- Kauh J, Chanel-Vos C, Escuin D, Fanucchi MP, Harvey RD, Saba N, Shin DM, Gal A, Pan L, Kutner M, Ramalingam SS, Bender L, Marcus A, Giannakakou P, Khuri FR. Farnesyl transferase expression determines clinical response to the docetaxel-lonafarnib combination in patients with advanced malignancies. Cancer. 2011;117:4049–59. doi: 10.1002/cncr.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein SA, Hohl RJ. Is there a future for prenyltransferase inhibitors in cancer therapy? Curr Opin Pharmacol. 2012;12:704–9. doi: 10.1016/j.coph.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Bachmann HSLA, Bau M, Schmid KW, Siffert W. FNTB promoter polymorphisms are associated with survival in different cancers. Naunyn-Schmiedeebergs Arch Pharmacol. 2009;379:93–93. , Meeting Abstract: 468. [Google Scholar]
- Zentrale Ethikkommission bei der Bundesärtzekammer. 2003. Die (Weiter-)Verwendung von menschlichen Körpermaterialien von Verstorbenen für Zwecke medizinischer Forschung. Dtsch Arztebl; 100: A 2251.
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes AJ. The essence of SNPs. Gene. 1999;234:177–86. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- Scherf M, Klingenhoff A, Werner T. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: a novel context analysis approach. J Mol Biol. 2000;297:599–606. doi: 10.1006/jmbi.2000.3589. [DOI] [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–84. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–52. doi: 10.1093/nar/gkh372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe N. AliBaba2: context specific identification of transcription factor binding sites. In Silico Biol. 2002;2:S1–15. [PubMed] [Google Scholar]
- Bachmann HS, Siffert W, Frey UH. Successful amplification of extremely GC-rich promoter regions using a novel 'slowdown PCR' technique. Pharmacogenetics. 2003;13:759–66. doi: 10.1097/00008571-200312000-00006. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002 doi: 10.1186/gb-2002-3-7-research0034. ; 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Gaunt TR, Day IN. Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol. 2009;169:505–14. doi: 10.1093/aje/kwn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ES, Kies MS, Fossella FV, Glisson BS, Zaknoen S, Statkevich P, Munden RF, Summey C, Pisters KM, Papadimitrakopoulou V, Tighiouart M, Rogatko A, Khuri FR. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant non-small cell lung carcinoma. Cancer. 2005;104:561–9. doi: 10.1002/cncr.21188. [DOI] [PubMed] [Google Scholar]
- Saracchini S, Foltran L, Tuccia F, Bassini A, Sulfaro S, Micheli E, Del Conte A, Bertola M, Gion M, Lorenzon M, Tumolo S. Phase II study of liposome-encapsulated doxorubicin plus cyclophosphamide, followed by sequential trastuzumab plus docetaxel as primary systemic therapy for breast cancer patients with HER2 overexpression or amplification. Breast. 2013;22:1101–7. doi: 10.1016/j.breast.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Fuchs EM, Kostler WJ, Horvat R, Hudelist G, Kubista E, Attems J, Zielinski CC, Singer CF. High-level ERBB2 gene amplification is associated with a particularly short time-to-metastasis, but results in a high rate of complete response once trastuzumab-based therapy is offered in the metastatic setting. Int J Cancer. 2014 doi: 10.1002/ijc.28660. ; 135: 224-31. DOI: 10.1002/ijc.28660. [DOI] [PubMed] [Google Scholar]
- Gunsar F. Treatment of delta hepatitis. Expert Rev Anti Infect Ther. 2013;11:489–98. doi: 10.1586/eri.13.35. [DOI] [PubMed] [Google Scholar]
- Gordon LB, Massaro J, D'Agostino RB, Sr, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW. Progeria Clinical Trials C. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130:27–34. doi: 10.1161/CIRCULATIONAHA.113.008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima J, Hisama FM, Martin GM. An encouraging progress report on the treatment of progeria and its implications for atherogenesis. Circulation. 2014;130:4–6. doi: 10.1161/CIRCULATIONAHA.114.010648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Grading of toxicities by treatment arm
Table S2 Grading of toxicities by treatment arm for FNTB rs11623866 GG genotype carriers