

Abstract
Epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNAs, have been implicated in a number of complex diseases. Schizophrenia and other major psychiatric and neurodevelopmental disorders are associated with abnormalities in multiple epigenetic mechanisms, resulting in altered gene expression during development and adulthood. Polymorphisms and copy number variants in schizophrenia risk genes contribute to the high heritability of the disease, but environmental factors that lead to epigenetic modifications may either reduce or exacerbate the expression of molecular and behavioral phenotypes associated with schizophrenia and related disorders. In the present paper, we will review the current understanding of molecular dysregulation in schizophrenia, including disruption of the dopamine, NMDA, and GABA signaling pathways, and discuss the role of epigenetic factors underlying disease pathology.
Keywords: Schizophrenia, epigenetics, neurodevelopment, DNA methylation, histones, microRNA
1. Introduction
In recent years, many diseases, particularly neuropsychiatric disorders, have been found to be the result of complex interactions between genetic susceptibility and environmental insults (Gavin and Akbarian, 2012; Robertson, 2005; Robertson and Wolffe, 2000; Schanen, 2006; Tsankova et al., 2007). Schizophrenia is a complex and disabling disorder defined by psychotic, affective, and cognitive symptoms. Positive symptoms include hallucinations, psychosis, and mania, while negative symptoms include anhedonia and social withdrawal. Although cognitive functioning is generally intact, working memory may be severely impaired. The onset of the disease typically occurs during late adolescence or early adulthood, a critical period in neurodevelopment that is characterized by activity-dependent synaptic pruning and final maturation of the prefrontal cortex (PFC), the region of the brain that regulates higher cognitive functions such as working memory and emotional control. The positive symptoms in schizophrenia are primarily associated with upregulation of dopamine (DA) signaling throughout the brain, while the negative and cognitive symptoms are caused both by abnormal dopamine signaling and a complicated interaction between GABAergic signaling and hypoactive NMDA function in the PFC and hippocampus. At a neuromorphological level, brains from schizophrenic patients exhibit a reduction in the number and complexity of neuronal connections in the cortex, suggesting aberrant synaptic pruning during the late stages of neurodevelopment. Reduced dendritic complexity in the PFC correlates with the cognitive deficits common to schizophrenia.
Despite the global impairment and severity of the disease, there are few effective therapies available, many of which have severe side effects. Antipsychotic drugs primarily target dopamine receptor signaling at the dopamine D2 receptor. In animal models, antipsychotics that target D2 receptors induce dopamine neuron inactivation or block depolarization; treatment with amphetamine increases hyperactivity and other psychosis-relevant behaviors, and that treatment with the antipsychotic clozapine reverses these effects (Herrera et al., 2013; Valenti et al., 2011). Recently it has been suggested that hyperactivity of the DA signaling system may be related to reduced inhibitory neuron function in the hippocampus and ventral tegmentum, as selective reduction of parvalbumin interneurons in the hippocampus results in an increase in downstream dopamine neuron activity (Boley et al., 2014; Gilani et al., 2014).
The importance of dopamine signaling in schizophrenia is based on 3 lines of evidence: first, the most effective drugs for schizophrenia are the D2R-targeting antipsychotics, with up to 90% D2 receptor occupancy; second, most patients with schizophrenia are highly sensitive to dopamine agonists, showing an immediate re-emergence of psychosis and other positive symptoms; and third, functional imaging studies have provided overwhelming evidence for dopamine hyperactivity in the brain, even in asymptomatic or drug naive patients (Salavati et al., 2014; Seeman, 2010). Functional imaging has overwhelmingly shown an upregulation of presynaptic striatal dopamine activity, variations in dopaminergic signaling correlate with executive functioning in the prefrontal cortex, and manipulation of dopamine signaling results in network disruption in numerous brain regions including the prefrontal cortex, hippocampus, striatum, basal ganglia, and putamen (Cole et al., 2013; Tan et al., 2007).
Although there is a strong functional association between DA signaling and schizophrenia, genome-wide association studies (GWAS) have not identified statistically significant loci containing dopamine-related genes. Quantitative trait locus and candidate gene mapping have been somewhat more successful, and several particularly strong candidates have been found, including DISC1, NRG1, and a cluster of GABA-A receptor subunits on chromosome 5 (Gogos and Gerber, 2006). However, many of these studies have failed to replicate, possibly due to population variability, very small effect sizes of risk genes, epistasis, and environmental factors. Because multiple large genome-wide studies have identified a strong overlap between schizophrenia and other major psychiatric disorders, there is now an effort to define studies based on endophenotypes, rather than diagnosis (Consortium, 2013; Craddock and Sklar, 2013). One DA-related representative of the endophenotypic approach is catechol-O-methyltransferase (COMT), which assists in clearing DA from synapses. The COMT Val158Met polymorphism has been well-studied in humans: in control subjects and unaffected siblings of schizophrenic patients, the Val allele is associated with reduced prefrontal DA levels and impaired working memory, particularly during more difficult tasks, but function can be rescued with amphetamine treatment (Tan et al., 2007). In both control and schizophrenic patients, the Val allele negatively impacts both working memory and negative symptoms (Ceaser et al., 2013). Interestingly, a number of studies have shown that COMT interacts with polymorphisms in other schizophrenia risk genes, including AKT, MTHFR, DRD2, DTNBP1 (Tan et al., 2012).
While DA antagonists are largely effective at reducing or eliminating psychotic and hallucinatory symptoms in schizophrenic patients, antipsychotics have little effect on the affective and cognitive symptoms in schizophrenia. Recent research suggests that the interaction of NMDA and GABA signaling in the PFC may also be important to the pathogenesis of the disease. NMDA signaling is required for proper synapse formation and maintenance, particularly during adolescence, but is reduced in schizophrenia, a phenotype known as hypofrontality (Bhatt et al., 2009). In animal models, genetic or pharmacological induction of NMDA hypofunction is known to recapitulate both positive and negative behavioral endophenotypes similar to those observed in schizophrenia. NMDA antagonists can produce a wide array of relevant positive, negative, and cognitive behaviors such as hyperactivity, anhedonia, and social withdrawal in both acute and chronic paradigms, along with including dysregulation in dopamine signaling, interruption of normal inhibitory-excitatory circuitry in the prefrontal cortex, and cognitive defects induced by aberrant prefrontal cortex-hippocampus connectivity (Blot et al., 2013; Neill et al., 2014; Svensson, 2000). Mice engineered to express just 5% of the obligatory NMDAR subunit NR1 display schizophrenia-like behavior, including increased locomotion and stereotypies (Mohn et al., 1999).
NMDA function is significantly impaired in patients with schizophrenia. NMDA, D-cycloserine, and other endogenous NMDAR agonists are reduced in post-mortem prefrontal and striatal tissue from schizophrenia patients, as are the NMDAR subunit NR2 and the downstream NMDA signaling components NRG1 and ERBB4 (Benneyworth et al., 2011; Geddes et al., 2011). NMDA antagonists induce psychosis and cognitive impairment in normal subjects, and exacerbate symptoms in psychiatric patients (Enomoto et al., 2007; Malhotra et al., 1997). Most recently, copy number variant analysis and exome sequencing of patients with schizophrenia show a significant over-representation of mutations in brain development-related genes, particularly genes associated with the NMDA signaling pathway (Gilman et al., 2012; Hall et al., 2014).
The activity of prefrontal glutamatergic neurons is tightly regulated by GABAergic interneurons, and, like NMDA signaling, GABAergic signaling is dysregulated in schizophrenia. Levels of GAD1, the enzyme responsible for synthesizing GABA, are reduced in post-mortem schizophrenic brains, as are a number of genes associated with GABA synthesis and signaling (Hashimoto et al., 2008). Polymorphisms in two GABA-expressed genes downstream from NMDA, NRG1 and ERBB4 are associated with aberrant GABAergic interneuron development and migration, and elimination of the NR1 subunit of the NMDA receptor in a subset of cortical GABAergic interneurons during postnatal development in mice results in a number of behavioral and molecular phenotypes associated with schizophrenia (Belforte et al., 2010; Flames et al., 2004). The most-replicated change is the decrease in the number of parvalbumin (PVALB)-positive interneurons and reduced expression of PVALB mRNA (Lewis et al., 2012; Uchida et al., 2014). Parvalbumin neurons are considered crucial to the pathogenesis of schizophrenia, as they are strongly associated with the regulation of NMDA signaling.
A final important aspect in the pathogenesis of schizophrenia is the neurodevelopmental timecourse. Although some symptoms, such as reduced sociability, may be observed in childhood, the onset of schizophrenia generally does not occur until late adolescence or early adulthood. While neurogenesis and massive dendritic connectivity characterize the early postnatal period, the adolescent period is characterized by NMDA-dependent synaptic pruning and the final maturation of the GABA-glutamate circuitry in the prefrontal cortex (Bale et al., 2010). Schizophrenia is believed to be 70–80% heritable, but the risk for monozygotic twins is only 50%, suggesting that environmental factors may be as important as genetic risk factors (McGuffin and Gottesman, 1999). In the following review, we suggest that the role of environment on the development and course of schizophrenia are mediated by epigenetic factors including DNA promoter methylation/hydroxymethylation, histone expression and post-translational modifications, and the interaction between these factors and other environmentally responsive molecules such as microRNAs (miRNAs) and other non-coding RNAs.
2. DNA Methylation
Alterations in DNA methylation have been detected in many neuropsychiatric disorders, including autism, bipolar disorder, borderline personality disorder, and schizophrenia. DNA methyltransferases (DNMTs) catalyze the transfer of methyl groups to DNA, resulting in 5-methylcytosine (5-mC) modification of CpG islands in or near gene promoter regions. This modification generally represses transcription. In contrast, TET enzymes can catalyze the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC), resulting in DNA demethylation and subsequent transcriptional de-repression (Figure 1A) (Dong et al., 2012; Grayson and Guidotti, 2013; Guo et al., 2011; Kato and Iwamoto, 2014).
Figure 1. Common epigenetic modifications.
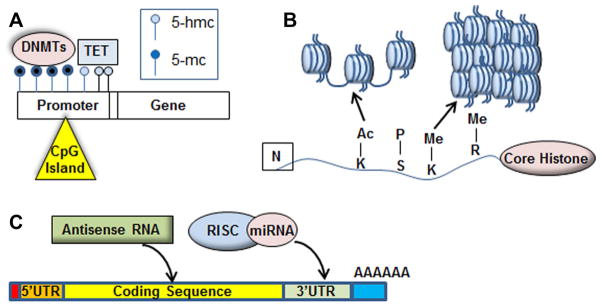
(A) DNA methyltransferases (DNMTs) modify CpG islands, which are found primarily in the promoter region of genes, with 5-methylcytosine (5-mC). An increase in promoter methylation (dark blue circles) results in reduced gene expression, while TET enzyme modification of 5-mC to 5-hmC results in de-repression of gene expression. (B) The four core histones undergo amino acid-specific acetylation, methylation, and phosphorylation. The post-translational modifications result in either the relaxation of the nucleosome structure (euchromatin), which allows transcription factor access to genes, or causes nucleosomes to tighten into heterochromatin, blocking transcription at specific loci. (C) Post-transcription factors including microRNAs, antisense RNA, and long non-coding RNA act at promoter regions, coding regions, and 3′UTR seed sequences to regulate the expression of target mRNAs.
A recent DNA methylome study identified numerous changes in DNA methylation at differentially methylated regions (DMRs) in schizophrenia and bipolar disorder, and a study of monozygotic twins discordant for psychosis found that DMRs involved in known pathways for psychiatric disorders and brain development were over-represented (Dempster et al., 2011; Xiao et al., 2014). Expression of several DNMTs are upregulated in brains from schizophrenia patients, resulting in the hypermethylation and downregulation of schizophrenia-associated genes, including brain-derived neurotrophic factor (BDNF), the glucocorticoid receptor (NR3C1), GAD1, and reelin (RELN) (Grayson et al., 2005, Roth, 2009 #604; Wong et al., 2010). Interestingly, RELN promoter methylation in temporal-cortical tissue from normal subjects increases 25-fold during adolescence, suggesting that altered epigenetic regulation of RELN may play a role in neurodevelopmental changes associated with schizophrenia (Lintas and Persico, 2010).
COMT promoter methylation is also disrupted in schizophrenia, although the methylation is variable and can be affected by antipsychotic therapy, environmental factors, and genotype, including the COMT Val158Met polymorphism (Lott et al., 2013). In control subjects, those homozygous for the COMT Val allele show RELN promoter hypermethylation and decreased RELN expression (Abdolmaleky et al., 2008; Abdolmaleky et al., 2006). Other downstream effects of aberrant COMT methylation include up- or down-regulation of dopamine receptor activity, reduced GAD1 expression, and disrupted prefrontal NMDA signaling (David et al., 2005; Kalkman and Loetscher, 2003).
GADD45, which recruits deaminases and glycosylases to promoter regions, is also a regulatory factor in DNA methylation (Cortellino et al., 2011; Rai et al., 2008). GADD45b binding at the BNDF promoter is significantly decreased in major psychosis, and is associated with BDNF promoter hypermethylation and reduced expression (Gavin and Akbarian, 2012). This same region is also associated with repressive histone interactions. Another member of the GADD45 family, GADD45a, has been shown to bind acetylated histones, suggesting that this family of proteins may be good targets for therapeutic intervention (Carrier et al., 1999).
Although many studies have focused on promoter hypermethylation in schizophrenia, recent studies have shown that TET1 and 5-hmC DNA modifications are also elevated in cortical tissue from schizophrenic patients. Expression of both DNMTs and TET enzymes are increased in corticolimbic tissue from post-mortem SCZ brains, and global analysis has shown that methylation is significantly reduced in schizophrenic patients compared to their un-affected twins (Bonsch et al., 2012; Dong et al., 2012; Guidotti et al., 2014). A similar discordant twin analysis found that the most significant difference between affected and unaffected twins was a 25% increase in hypomethylation at the promoter for ST6GALNAC1, a gene involved in stress-activated kinase signaling (Dempster et al., 2011).
Finally, there is strong evidence that pre- and postnatal environmental factors can affect promoter methylation of genes associated with schizophrenia and other major psychiatric disorders. Reduced postnatal maternal care can alter genome-wide and specific gene methylation, including at the GAD1 and glucocorticoid receptor promoters. Many of these early-life DNA modifications are stable and modify adult behaviors and neurobiology, such as regulation of anxiety by GABAergic signaling and propensity of suicidality (Bagot et al., 2012; McGowan et al., 2009; Zhang et al., 2010). DNA methylation may also interact with genomic factors such as parental imprinting, as in hypomethylation of the paternal copy of the schizophrenia risk gene LRRTM1 (Brucato et al., 2014).
3. Histone Modifications and Chromatin Structure
Chromatin is the DNA-protein complex that regulates the availability of DNA to transcription factors. Chromatin is found in two forms, euchromatin and heterochromatin. Euchromatin is less tightly wound and is associated with active transcription, while heterochromatin is more tightly wound, blocking transcription factor access to gene promoter regions. Histone transition between the euchromatin and heterochromatin state are mediated by post-translational modifications, including acetylation and methylation, of the four major histone proteins that comprise the nucleosome (Figure 1B). Histone modifications occur at specific amino acids in the protein: acetyl groups are attached by histone acetyltransferases (HATs) and can be removed by histone deacetylases (HDACs), while histone methylation is performed by histone methyltransferases (HMTs).
The use of valproic acid (VPA), an HDAC inhibitor, in the treatment of schizophrenia and bipolar disorder suggests that histone regulation may play an important role in disease pathogenesis. In rats, HDAC inhibitors can reverse the effect of postnatal stress on RELN and glucocorticoid levels by increasing acetylation of at the amino terminal of H3 (Weaver et al., 2006). In patients with schizophrenia, both TET1 and HDAC1 are elevated in postmortem hippocampal and PFC tissue, particularly in tissue from female patients (Benes et al., 2007; Dong et al., 2012). Overexpression of HDAC1 may result in chromatin deactylation, causing transcriptional repression of a number of genes. HDAC inhibitors are therefore considered to be good targets for new therapeutic drugs that are more effective and have fewer off-target effects than VPA.
Histone 3 (H3) seems to be particularly dysregulated in schizophrenia. Specifically, di- and tri-methylation of H3K9 and H3R27, which can regulate GAD1 expression, is elevated in cortical neurons and adjacent non-neuronal cells in post-mortem tissue from schizophrenic patients, reducing expression of genes typically associated with neuronal metabolism (Akbarian, 2010). Lymphocyte studies in schizophrenia patients also indicate in an abnormal increase in heterochromatin, likely due to increased H3K9 methylation and reduced H3K9 acetylation, both modifications associated with an increase in heterochromatin (Chase et al., 2013; Gavin et al., 2009; Kosower et al., 1995). Three HMTs, G9a, GLP, and SETDB1, which are responsible for the majority of H3K9me modifications across the genome, are increased in lymphocytes from SCZ patients; notably, SETDB1 is the only HMT that specifically functions to specifically di- and tri- methylate H3K9 (Wang et al., 2003; Zee et al., 2010). Overall, heterochromatin associated with histone methylation is increased in cortical tissue from schizophrenia patients: levels of methylated H3K4 are reduced at nearly 600 loci, including near multiple NMDAR subunits and genes involved in neurodevelopment (Sharma et al., 2008).
4. Association between epigenetic factors and microRNAs
MicroRNAs (miRNAs) assist in the regulation of many mechanisms in the nervous system, including neuronal migration, neuronal differentiation, synaptic plasticity, and adult neurogenesis, by binding to target sites in the 3′ UTR of mRNAs and either preventing translation or targeting the mRNA for degradation (Figure 1C) (Cheng et al., 2009; Kosik, 2006). A number of studies have associated altered miRNA expression or activity with major psychiatric disorders (Miller and Wahlestedt, 2010). DICER1, which plays a major role in miRNA processing, is affected in schizophrenia cases that are associated with copy number variation, as are several miRNAs located nearby, while another enzyme important for miRNA processing, DGCR8, is deleted in DiGeorge Syndrome, a neurodevelopmental disorder that includes schizophrenia-like symptoms. A mouse model of DiGeorge Syndrome exhibits schizophrenia-like behavioral phenotypes, along upregulation of pri-miRNAs and downregulation of mature miRNAs in the brain. These characteristics are also observed when only Dcgr8 is deleted (Stark et al., 2008).
Several specific miRNAs, including miR-132, miR-137, and miR-181b, have been implicated in schizophrenia and other psychiatric disorders. MiR-132 is of particular interest, as it is developmentally regulated during adolescence by NMDA signaling, and targets both MECP2, a chromatin-modifying protein that causes the neurodevelopmental disorder Rett Syndrome, and DNMT3A, an activity-dependent DNA methyltransferase that is elevated in cortical tissue from adult schizophrenic patients (Kundakovic et al., 2009). More than 10% of genes that are over-expressed in PFC tissue from schizophrenic patients are predicted miR-132 targets, consistent with the observed downregulation of miR-132 (Miller et al., 2012).
Polymorphisms near the miR-137 locus have been identified in several large genome-wide association studies, and follow-up work has shown that genes predicted to be miR-137 targets, including TCF4, CACNA1C, CSMD1, and WBP1L, are altered in schizophrenia (Consortium, 2013; Consortium, 2011; Hill et al., 2014; Ripke et al., 2011). MiR-137 is expressed in both the developing and adult mouse brain and plays a role in regulating cell proliferation and differentiation. In mice, overexpression of miR-137 in the embryonic brain leads to a reduction in neuronal proliferation and induction of premature differentiation of neural stem cells, but has the opposite effect in adult brains (Hill et al., 2014). In humans, unaffected carriers of the risk allele of the miR-137 risk allele (rs1625579; TT genotype) have working and episodic memory deficits, and schizophrenic patients with the TT genotype exhibit reduced PFC activation during working memory tasks (Cummings et al., 2013; van Erp et al., 2014).
5. Crosstalk between DNA Methylation, Histone Modifications, and miRNAs
Multiple epigenetic factors may act on single genes or gene networks. For example, promoter methylation and SNP polymorphisms in schizophrenia risk genes can interact, as in COMT (Sections 1 and 2), or allele-specific promoter hypomethylation of a single nucleotide polymorphism in the serotonin 2A receptor (HTR2A). HTR2A is a secondary target of a number of antipsychotic medications, and its promoter is hypomethylated in an allele-specific manner in both schizophrenia patients and their unaffected relatives, indicating that this finding is unaffected by antipsychotic treatment (Ghadirivasfi et al., 2011).
We have identified co-regulation of miR-132 and miR-137, both strongly associated with schizophrenia, and BDNF and MECP2, also associated with neurodevelopment and major psychiatric disorders (Figure 2). In this pathway, transcription of miR-132 and BDNF is induced by activity-dependent NMDA signaling via the CREB signaling pathway. MiR-132 then targets the 3′ UTR of MECP2, which targets both BDNF and miR-137 (Ausio et al., 2014). Finally, miR-132 appears to modulate NMDA receptor signaling in a feedback loop that would regulate its own transcription.
Figure 2. CREB signaling co-regulates epigenetic mechanisms and schizophrenia-associated miRNAs.

Activity-dependent NMDA signaling induces the CREB signaling pathway, which upregulates the expression of miR-132 and BDNF. Both NMDA signaling and miR-132 expression are significantly reduced in schizophrenia, and BDNF expression is dysregulated. MiR-132 inhibits multiple genes including DNMT3A, which causes hypermethylation and reduced gene expression of target genes, and MECP2, which regulates both DNA methyltransferase and histone modification activity. MECP2 also inhibits expression of the schizophrenia-associated miRNA miR-137, and inhibits BDNF expression via interaction with the transcriptional repressor REST.
All members of the miR-132/MECP2/miR-137/BDNF network are subject to epigenetic regulation at multiple levels. MECP2 regulates expression of genes such as BDNF through both protein interactions and histone modification: it inhibits BDNF expression by disinhibition of the transcriptional repressor REST, and binds to heterochromatin marked by H3K9me and H3K27me, altering developmental and neurodevelopmental dominant splice variant expression of BDNF by controlling transcription factor access to the multiple BDNF promoters (Mitchelmore and Gede, 2014). Finally, MECP2 inhibits miR-137 expression through an unknown mechanism (Szulwach et al., 2010); Thambirajah, 2012 #903}.
6. Overlap between Schizophrenia and Other Neurodevelopmental Disorders
The master epigenetic regulator MECP2 can be linked to many other neurodevelopmental disorders. One common neurodevelopmental disorder, Rett Syndrome, is associated with multiple polymorphisms in MECP2 (Amir et al., 1999). These mutations, which may occur at multiple different nucleotides in the gene, prevent MECP2 from binding to methylated DNA (Goffin et al., 2012). MECP2 is also implicated in both Prader-Willi Syndrome (PWS) and Angelmann Syndrome (AS), two different neurodevelopmental disorders caused by differential parental contributions at a single locus, the Prader-Willi Syndrome/Angelmann Syndrome Imprinting Center (PWS/AS-IC: 15q11.2-q13.1). Abnormal maternal imprinting causes Angelmann Syndrome, which is characterized by severe cognitive and neurological impairments, while lack of a paternal copy of the locus causes Prader-Willi Syndrome, which is associated with obsessive-compulsive behavior and endocrine abnormalities (Johnstone et al., 2006). MECP2 exerts its effects across multiple neurodevelopmental disorders due to its ability to regulate by promoter methylation and histone activity, thereby exerting a transcriptome-wide effect (Monteggia and Kavalali, 2009).
The PWS/AS-IC contains the gene for the E3 ubiquitin ligase UBE3A; deletion of this gene causes PWS, while abnormal maternal imprinting prevents MECP2 from binding to the methylated promoter, disrupting UBE3A expression and resulting in Angelmann Syndrome (Yasui et al., 2011). Interestingly, another E3 ubiquitin ligase, MIB1, is targeted by the schizophrenia-associated miR-137. A second gene near the PWS/AS Imprinting Center, the GABA-B receptor subunit GABRB3, has been shown to be both polymorphic and hypermethylated in some cases of autism (Buxbaum et al., 2002). Finally, having two maternal copies of a fragment of the 15q11.2-q13.1 locus is a risk factor for schizophrenia, psychosis, and autism (Ingason et al., 2011).
7. Conclusion
Schizophrenia and other neuropsychiatric disorders are highly polygenic and heritable, but it has been difficult to identify individual causative genes. Phenotypic expression of the disorders is due to complex interactions between risk alleles and environmental risk factors, including prenatal and postnatal stressors, paternal age, and cannabis use (Abdolmaleky et al., 2004; Lewis and Levitt, 2002; Matrisciano et al., 2013; Rapoport et al., 2012). Epigenetic factors, including DNA methylation, histone post-transcriptional modification and subsequent regulation of chromatin structure, and microRNA regulation of signaling pathways--including those involved in DNA methylation and histone activity--play important roles in neurodevelopment and are all capable of regulating large numbers of genes at once. Therefore, it is likely that epigenetic mechanisms in schizophrenia and other psychiatric disorders mediate the interaction between genetic risk alleles and environmental factors by dynamic modification of the genome in response to positive or negative environmental stimuli.
Acknowledgments
Funding: This work was funded by NIH grant R00MH92321 (B.H.M)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdolmaleky HM, Ahmadkhaniha HR, Nohesara S, Smith CL. Epigenetic and genetic aberrations of the brain dopaminergic system in schizophrenia and bipolar disorder: achievements and prospective. Iranian Journal of Psychiatry and Behavioral Sciences. 2008;2:4–13. [Google Scholar]
- Abdolmaleky HM, Cheng KH, Faraone SV, Wilcox M, Glatt SJ, Gao F, Smith CL, Shafa R, Aeali B, Carnevale J, Pan H, Papageorgis P, Ponte JF, Sivaraman V, Tsuang MT, Thiagalingam S. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15:3132–45. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, Tsuang MT. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. American Journal of Medical Genetics Part B (Neuropsychiatric Genetics) 2004;127B:51–59. doi: 10.1002/ajmg.b.20142. [DOI] [PubMed] [Google Scholar]
- Akbarian S. The molecular pathology of schizophrenia--focus on histone and DNA modifications. Brain Res Bull. 2010;83:103–7. doi: 10.1016/j.brainresbull.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ausio J, de Paz AM, Esteller M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends in Molecular Medicine. 2014:1–12. doi: 10.1016/j.molmed.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Bagot RC, Zhang TY, Wen X, Nguyen TT, Nguyen HB, Diorio J, Wong TP, Meaney MJ. Variations in postnatal maternal care and the epigenetic regulation of metabotropic glutamate receptor 1 expression and hippocampal function in the rat. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17200–7. doi: 10.1073/pnas.1204599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–9. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–9. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneyworth MA, Roseman AS, Basu AC, Coyle JT. Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett. 2011;488:267–71. doi: 10.1016/j.neulet.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–82. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Blot K, Bai J, Otani S. The effect of non-competitive NMDA receptor antagonist MK-801 on neuronal activity in rodent prefrontal cortex: an animal model for cognitive symptoms of schizophrenia. J Physiol Paris. 2013;107:448–51. doi: 10.1016/j.jphysparis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Boley AM, Perez SM, Lodge DJ. A fundamental role for hippocampal parvalbumin in the dopamine hyperfunction associated with schizophrenia. Schizophr Res. 2014;157:238–43. doi: 10.1016/j.schres.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsch D, Wunschel M, Lenz B, Janssen G, Weisbrod M, Sauer H. Methylation matters? Decreased methylation status of genomic DNA in the blood of schizophrenic twins. Psychiatry Res. 2012;198:533–7. doi: 10.1016/j.psychres.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Brucato N, DeLisi LE, Fisher SE, Francks C. Hypomethylation of the paternally inherited LRRTM1 promoter linked to schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2014;165:555–63. doi: 10.1002/ajmg.b.32258. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. 2002;7:311–6. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- Carrier F, Georgel PT, Pourquier P, Blake M, Kontny HU, Antinore MJ, Gariboldi M, Myers TG, Weinstein JN, Pommier Y, Fornace AJ. Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol Cell Biol. 1999;19:1673–85. doi: 10.1128/mcb.19.3.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceaser A, Csernansky JG, Barch DM. COMT influences on prefrontal and striatal blood oxygenation level-dependent responses during working memory among individuals with schizophrenia, their siblings, and healthy controls. Cogn Neuropsychiatry. 2013;18:257–83. doi: 10.1080/13546805.2012.698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase KA, Gavin DP, Guidotti A, Sharma RP. Histone methylation at H3K9: evidence for a restrictive epigenome in schizophrenia. Schizophr Res. 2013;149:15–20. doi: 10.1016/j.schres.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 2009 doi: 10.1038/nn.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DM, Oei NY, Soeter RP, Both S, van Gerven JM, Rombouts SA, Beckmann CF. Dopamine-dependent architecture of cortico-subcortical network connectivity. Cereb Cortex. 2013;23:1509–16. doi: 10.1093/cercor/bhs136. [DOI] [PubMed] [Google Scholar]
- Consortium C-DGotPG. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–9. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium SPG-WAS. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381:1654–62. doi: 10.1016/S0140-6736(13)60855-7. [DOI] [PubMed] [Google Scholar]
- Cummings E, Donohoe G, Hargreaves A, Moore S, Fahey C, Dinan TG, McDonald C, O’Callaghan E, O’Neill FA, Waddington JL, Murphy KC, Morris DW, Gill M, Corvin A. Mood congruent psychotic symptoms and specific cognitive deficits in carriers of the novel schizophrenia risk variant at MIR-137. Neurosci Lett. 2013;532:33–8. doi: 10.1016/j.neulet.2012.08.065. [DOI] [PubMed] [Google Scholar]
- David HN, Ansseau M, Abraini JH. Dopamine-glutamate reciprocal modulation of release and motor responses in the rat caudate-putamen and nucleus accumbens of “intact” animals. Brain Res Brain Res Rev. 2005;50:336–60. doi: 10.1016/j.brainresrev.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, Kalidindi S, Picchioni M, Kravariti E, Toulopoulou T, Murray RM, Mill J. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–96. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry. 2012;2:e159. doi: 10.1038/tp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T, Noda Y, Nabeshima T. Phencyclidine and genetic animal models of schizophrenia developed in relation to the glutamate hypothesis. Methods Find Exp Clin Pharmacol. 2007;29:291–301. doi: 10.1358/mf.2007.29.4.1075358. [DOI] [PubMed] [Google Scholar]
- Flames N, Long JE, Garratt AN, Fischer TM, Gassmann M, Birchmeier C, Lai C, Rubenstein JL, Marín O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004;44:251–61. doi: 10.1016/j.neuron.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Gavin DP, Akbarian S. Epigenetic and post-transcriptional dysregulation of gene expression in schizophrenia and related disease. Neurobiol Dis. 2012;46:255–62. doi: 10.1016/j.nbd.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Gavin DP, Rosen C, Chase K, Grayson DR, Tun N, Sharma RP. Dimethylated lysine 9 of histone 3 is elevated in schizophrenia and exhibits a divergent response to histone deacetylase inhibitors in lymphocyte cultures. J Psychiatry Neurosci. 2009;34:232–7. [PMC free article] [PubMed] [Google Scholar]
- Geddes AE, Huang XF, Newell KA. Reciprocal signalling between NR2 subunits of the NMDA receptor and neuregulin1 and their role in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:896–904. doi: 10.1016/j.pnpbp.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Ghadirivasfi M, Nohesara S, Ahmadkhaniha HR, Eskandari MR, Mostafavi S, Thiagalingam S, Abdolmaleky HM. Hypomethylation of the serotonin receptor type-2A Gene (HTR2A) at T102C polymorphic site in DNA derived from the saliva of patients with schizophrenia and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:536–45. doi: 10.1002/ajmg.b.31192. [DOI] [PubMed] [Google Scholar]
- Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S, Chuhma N, Glickstein S, Merker RJ, Xu Q, Small SA, Anderson SA, Ross ME, Moore H. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A. 2014;111:7450–5. doi: 10.1073/pnas.1316488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Chang J, Xu B, Bawa TS, Gogos JA, Karayiorgou M, Vitkup D. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat Neurosci. 2012;15:1723–8. doi: 10.1038/nn.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Allen M, Zhang L, Amorim M, Wang I-TJ, Reyes A-RS, Mercado-Berton A, Ong C, Cohen S, Hu L, Blendy JA, Carlson GC, Siegel SJ, Greenberg ME, Zhou Z. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nature Neuroscience. 2012;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogos JA, Gerber DJ. Schizophrenia susceptibility genes: emergence of positional candidates and future directions. Trends Pharmacol Sci. 2006;27:226–33. doi: 10.1016/j.tips.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138–66. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Dong E, Gavin DP, Grayson DR, Sharma RP, Smith RC, Tueting P, Zhubi A. Toward the identification of peripheral epigenetic biomarkers of schizophrenia. J Neurogenet. 2014;28:41–52. doi: 10.3109/01677063.2014.892485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Trent S, Thomas KL, O’Donovan MC, Owen MJ. Genetic Risk for Schizophrenia: Convergence on Synaptic Pathways Involved in Plasticity. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13:147–61. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera AS, Casanova JP, Gatica RI, Escobar F, Fuentealba JA. Clozapine pre-treatment has a protracted hypolocomotor effect on the induction and expression of amphetamine sensitization. Prog Neuropsychopharmacol Biol Psychiatry. 2013;47:1–6. doi: 10.1016/j.pnpbp.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Hill MJ, Donocik JG, Nuamah RA, Mein CA, Sainz-Fuertes R, Bray NJ. Transcriptional consequences of schizophrenia candidate miR-137 manipulation in human neural progenitor cells. Schizophr Res. 2014;153:225–30. doi: 10.1016/j.schres.2014.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingason A, Kirov G, Giegling I, Hansen T, Isles AR, Jakobsen KD, Kristinsson KT, le Roux L, Gustafsson O, Craddock N, Möller HJ, McQuillin A, Muglia P, Cichon S, Rietschel M, Ophoff RA, Djurovic S, Andreassen OA, Pietiläinen OP, Peltonen L, Dempster E, Collier DA, St Clair D, Rasmussen HB, Glenthøj BY, Kiemeney LA, Franke B, Tosato S, Bonetto C, Saemundsen E, Hreidarsson SJ, Nöthen MM, Gurling H, O’Donovan MC, Owen MJ, Sigurdsson E, Petursson H, Stefansson H, Rujescu D, Stefansson K, Werge T, Investigators G. Maternally derived microduplications at 15q11-q13: implication of imprinted genes in psychotic illness. Am J Psychiatry. 2011;168:408–17. doi: 10.1176/appi.ajp.2010.09111660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone KA, DuBose AJ, Futtner CR, Elmore MD, Brannan CI, Resnick JL. A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum Mol Genet. 2006;15:393–404. doi: 10.1093/hmg/ddi456. [DOI] [PubMed] [Google Scholar]
- Kalkman HO, Loetscher E. GAD(67): the link between the GABA-deficit hypothesis and the dopaminergic- and glutamatergic theories of psychosis. J Neural Transm. 2003;110:803–12. doi: 10.1007/s00702-003-0826-8. [DOI] [PubMed] [Google Scholar]
- Kato T, Iwamoto K. Comprehensive DNA methylation and hydroxymethylation analysis in the human brain and its implication in mental disorders. Neuropharmacology. 2014;80:133–9. doi: 10.1016/j.neuropharm.2013.12.019. [DOI] [PubMed] [Google Scholar]
- Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7:911–20. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- Kosower NS, Gerad L, Goldstein M, Parasol N, Zipser Y, Ragolsky M, Rozencwaig S, Elkabetz E, Abramovitch Y, Lerer B. Constitutive heterochromatin of chromosome 1 and Duffy blood group alleles in schizophrenia. Am J Med Genet. 1995;60:133–8. doi: 10.1002/ajmg.1320600209. [DOI] [PubMed] [Google Scholar]
- Kundakovic M, Chen Y, Guidotti A, Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol. 2009;75:342–54. doi: 10.1124/mol.108.051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–32. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lintas C, Persico AM. Neocortical RELN promoter methylation increases significantly after puberty. Neuroreport. 2010;21:114–8. doi: 10.1097/WNR.0b013e328334b343. [DOI] [PubMed] [Google Scholar]
- Lott SA, Burghardt PR, Burghardt KJ, Bly MJ, Grove TB, Ellingrod VL. The influence of metabolic syndrome, physical activity and genotype on catechol-O-methyl transferase promoter-region methylation in schizophrenia. Pharmacogenomics J. 2013;13:264–71. doi: 10.1038/tpj.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–50. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, Nicoletti F, Guidotti A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–94. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–8. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin P, Gottesman Risk factors for schizophrenia. N Engl J Med. 1999;341:370–1. author reply 372. [PubMed] [Google Scholar]
- Miller BH, Wahlestedt C. MicroRNA dysregulation in psychiatric disease. Brain Research. 2010:89–99. doi: 10.1016/j.brainres.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, Willoughby D, Kenny PJ, Elsworth JD, Lawrence MS, Roth RH, Edbauer D, Kleiman RJ, Wahlestedt C. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci U S A. 2012;109:3125–30. doi: 10.1073/pnas.1113793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelmore C, Gede L. Brain Derived Neurotrophic Factor: epigenetic regulation in psychiatric disorders. Brain Res. 2014;1586:162–72. doi: 10.1016/j.brainres.2014.06.037. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–36. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Kavalali ET. Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission. Biol Psychiatry. 2009;65:204–10. doi: 10.1016/j.biopsych.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill JC, Harte MK, Haddad PM, Lydall ES, Dwyer DM. Acute and chronic effects of NMDA receptor antagonists in rodents, relevance to negative symptoms of schizophrenia: a translational link to humans. Eur Neuropsychopharmacol. 2014;24:822–35. doi: 10.1016/j.euroneuro.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry. 2012;17:1228–38. doi: 10.1038/mp.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, Scolnick E, Cichon S, St Clair D, Corvin A, Gurling H, Werge T, Rujescu D, Blackwood DH, Pato CN, Malhotra AK, Purcell S, Dudbridge F, Neale BM, Rossin L, Visscher PM, Posthuma D, Ruderfer DM, Fanous A, Stefansson H, Steinberg S, Mowry BJ, Golimbet V, De Hert M, Jonsson EG, Bitter I, Pietilainen OP, Collier DA, Tosato S, Agartz I, Albus M, Alexander M, Amdur RL, Amin F, Bass N, Bergen SE, Black DW, Borglum AD, Brown MA, Bruggeman R, Buccola NG, Byerley WF, Cahn W, Cantor RM, Carr VJ, Catts SV, Choudhury K, Cloninger CR, Cormican P, Craddock N, Danoy PA, Datta S, de Haan L, Demontis D, Dikeos D, Djurovic S, Donnelly P, Donohoe G, Duong L, Dwyer S, Fink-Jensen A, Freedman R, Freimer NB, Friedl M, Georgieva L, Giegling I, Gill M, Glenthoj B, Godard S, Hamshere M, Hansen M, Hansen T, Hartmann AM, Henskens FA, Hougaard DM, Hultman CM, Ingason A, Jablensky AV, Jakobsen KD, Jay M, Jurgens G, Kahn RS, Keller MC, Kenis G, Kenny E, Kim Y, Kirov GK, Konnerth H, Konte B, Krabbendam L, Krasucki R, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nature Reviews Genetics. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–9. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- Salavati B, Rajji TK, Price R, Sun Y, Graff-Guerrero A, Daskalakis ZJ. Imaging-Based Neurochemistry in Schizophrenia: A Systematic Review and Implications for Dysfunctional Long-Term Potentiation. Schizophr Bull. 2014 doi: 10.1093/schbul/sbu132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(Spec No 2):R138–50. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- Seeman P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin Schizophr Relat Psychoses. 2010;4:56–73. doi: 10.3371/CSRP.4.1.5. [DOI] [PubMed] [Google Scholar]
- Sharma RP, Grayson DR, Gavin DP. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res. 2008;98:111–7. doi: 10.1016/j.schres.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–60. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- Svensson TH. Dysfunctional brain dopamine systems induced by psychotomimetic NMDA-receptor antagonists and the effects of antipsychotic drugs. Brain Res Brain Res Rev. 2000;31:320–9. doi: 10.1016/s0165-0173(99)00048-x. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, Santistevan NJ, Li W, Zhao X, Jin P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–41. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HY, Callicott JH, Weinberger DR. Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia. Cereb Cortex. 2007;17(Suppl 1):i171–81. doi: 10.1093/cercor/bhm069. [DOI] [PubMed] [Google Scholar]
- Tan HY, Chen AG, Chen Q, Browne LB, Verchinski B, Kolachana B, Zhang F, Apud J, Callicott JH, Mattay VS, Weinberger DR. Epistatic interactions of AKT1 on human medial temporal lobe biology and pharmacogenetic implications. Mol Psychiatry. 2012;17:1007–16. doi: 10.1038/mp.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–67. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Uchida T, Furukawa T, Iwata S, Yanagawa Y, Fukuda A. Selective loss of parvalbumin-positive GABAergic interneurons in the cerebral cortex of maternally stressed Gad1-heterozygous mouse offspring. Transl Psychiatry. 2014;4:e371. doi: 10.1038/tp.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Cifelli P, Gill KM, Grace AA. Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci. 2011;31:12330–8. doi: 10.1523/JNEUROSCI.2808-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Erp TG, Guella I, Vawter MP, Turner J, Brown GG, McCarthy G, Greve DN, Glover GH, Calhoun VD, Lim KO, Bustillo JR, Belger A, Ford JM, Mathalon DH, Diaz M, Preda A, Nguyen D, Macciardi F, Potkin SG. Schizophrenia miR-137 locus risk genotype is associated with dorsolateral prefrontal cortex hyperactivation. Biol Psychiatry. 2014;75:398–405. doi: 10.1016/j.biopsych.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell. 2003;12:475–87. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–5. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Hyde TM, Cassano HL, Deep-Soboslay A, Kleinman JE, Weickert CS. Promoter Specific Alterations of Brain-Derived Neurotrophic Factor Mrna in Schizophrenia. Neuroscience. 2010;169:1071–1084. doi: 10.1016/j.neuroscience.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Camarillo C, Ping Y, Arana TB, Zhao H, Thompson PM, Xu C, Su BB, Fan H, Ordonez J, Wang L, Mao C, Zhang Y, Cruz D, Escamilla MA, Li X. The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS One. 2014;9:e95875. doi: 10.1371/journal.pone.0095875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui DH, Scoles HA, Horike S, Meguro-Horike M, Dunaway KW, Schroeder DI, Lasalle JM. 15q11.2-13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum Mol Genet. 2011;20:4311–23. doi: 10.1093/hmg/ddr357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA. In vivo residue-specific histone methylation dynamics. J Biol Chem. 2010;285:3341–50. doi: 10.1074/jbc.M109.063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30:13130–7. doi: 10.1523/JNEUROSCI.1039-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]