

Abstract
GGGGCC (G4C2) repeat expansion in a noncoding region of C9ORF72 is the most common cause of sporadic and familial forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)1,2. The basis for pathogenesis is unknown. To capture the consequences of G4C2 repeat expansion in a tractable genetic system, we generated transgenic fly lines expressing 8, 28 or 58 G4C2 repeat-containing transcripts that do not have a translation start site (AUG) but contain an open-reading frame for green fluorescent protein (GFP) to detect repeat-associated non-AUG (RAN) translation. These transgenic animals show dosage-dependent, repeat length-dependent degeneration in neuronal tissues and RAN translation of dipeptide repeat (DPR) proteins as observed in patients. This model was used in a large-scale, unbiased genetic screen ultimately leading to the identification of 18 genetic modifiers that encode components of the nuclear pore complex (NPC) as well as the machinery that coordinates the export of nuclear RNA and the import of nuclear proteins. Consistent with these results we found morphological abnormalities in the architecture of the nuclear envelope in cells expressing expanded G4C2 repeats in vitro and in vivo. Moreover, we identified a substantial defect in RNA export resulting in retention of RNA in the nuclei of Drosophila cells expressing expanded G4C2 repeats and also in mammalian cells, including aged iPSC-derived neurons from C9ORF72 patients. These studies show that a primary consequence of G4C2 repeat expansion is the compromise of nucleocytoplasmic transport through the nuclear pore, revealing a novel mechanism of neurodegeneration.
Keywords: ALS, C9ORF72, DPR, Drosophila, FTD, genetic screen, nuclear pore, RAN translation, repeat expansion
To explore pathogenic mechanisms of disease initiated by C9ORF72 repeat expansion in a genetically tractable model organism, we used PhiC31 integrase mediated insertion of 8, 28 or 58 copies of G4C2 repeats into specific genomic loci of Drosophila. These repeats are not expressed at baseline but are transcribed when GAL4 is introduced in trans by genetic cross (Fig. 1a). The GFP coding sequence without a start codon was placed in frame with a potential poly(GP) dipeptide repeat (Fig. 1a), although RAN translation of the sense strand may also occur in two other reading frames producing poly(GR) and poly(GA).
Figure 1. G4C2 repeats induces length- and dosage-dependent degeneration in Drosophila.

a, Constructs expressing 8, 28, or 58 copies of G4C2 repeats. b, G4C2-58 causes a rough eye phenotype. c, Two copies of G4C2-58 expressed in motor neurons results in a decrease in larval size (left panel) and locomotor activity (right panel). d, Quantification of the distance traveled by larvae expressing repeats in motor neurons. Values are mean ± s.e.m., n = 3 trials, ** p < 0.01, by One Way ANOVA, Tukey’s Post Hoc test. e, Expression of two copies of G4C2-58 in motor neurons reduces bouton number. Scale bar: 25 μm. f, Quantification of bouton number and muscle size in larvae expressing G4C2 repeats. Values are mean ± s.e.m., n = 6, ** p < 0.01, by One Way ANOVA, Tukey’s Post Hoc test.
We expressed these constructs in the Drosophila eye using GMR-GAL4 and observed a length- and dosage-dependent rough eye phenotype (Fig. 1b) similar to that reported recently3,4. Toxicity was also observed when these repeats were expressed in other tissues. Expression of G4C2-58, but not G4C2-8, in motor neurons using OK371-GAL4 led to small larvae with significantly impaired locomotor activity (Fig. 1c, d). The neuromuscular junctions (NMJs) were examined with presynaptic and postsynaptic markers (Fig. 1e). Expression of G4C2-58 resulted in a significant decrease in bouton number and total muscle area when compared with GFP or G4C2-8 (Fig. 1f). Additionally, active zones within the NMJ were markedly reduced in larvae expressing G4C2-58 compared with controls (Extended Data Fig. 1a, b). Expression of G4C2-58 by the pan-neuronal driver elav-GAL4 also resulted in dosage-dependent locomotor defects and NMJ abnormalities (Extended Data Fig. 1c–f). Moreover, expression of G4C2 repeats by the muscle-specific driver MHC-GAL4 led to age-dependent defects in indirect flight muscle resulting in permanent abnormal wing position as compared to controls (Extended Data Fig. 1g).
In this fly model, RAN translation occurs in a G4C2 repeat length-dependent manner. Poly(GP)-GFP DPRs were detected in flies expressing G4C2-58 and to a lesser extent G4C2-28, but not in flies expressing equivalent levels of G4C2-8 (Extended Data Fig. 2a, b). Indeed, nuclear and cytoplasmic inclusions of poly(GP)-GFP were present in tissues expressing G4C2-58 but not G4C2-8 such as brain, muscle and salivary gland cells (Extended Data Fig. 2c–g). To determine whether DPRs are sufficient to drive a degenerative phenotype, we used codons alternate to G4C2 to generate new transgenic flies directly expressing poly(GA), poly(GR), or poly(GP), with an AUG start codon and N-terminal GFP (Supplementary Table 1; Extended Data Fig. 3a). Expression of neither GFP-(GP)47 nor GFP-(GA)50 elicited a degenerative eye phenotype (Extended Data Fig. 3a, b). Thus, while poly(GP)-GFP serves as a useful marker of cells expressing G4C2-58 RNA and producing DPR proteins, it is unlikely to contribute significantly to the degenerative phenotype. By contrast, expression of GFP-(GR)50 was highly toxic resulting in greater than 95% lethality with a few escapers having severely degenerated eyes (Extended Data Fig. 3a). These results are consistent with recent reports indicating that poly(GR) is toxic in cultured cells and Drosophila3,5,6. In fact, immunoblotting with antibodies against DPRs showed that flies expressing G4C2-58 RNA produce poly(GR) in addition to poly(GP) DPRs, although neither poly(GA) nor poly(PR) was detected (Extended Data Fig. 3c, d). Thus, G4C2-58 toxicity may be mediated through toxic RNA or poly(GR) produced in our fly model, or a combination of these mechanisms.
To gain unbiased insight into the pathogenic mechanisms underlying G4C2-58 toxicity, we performed a large-scale genetic screen to identify genetic loci whose partial loss of function may significantly modify the G4C2-58-induced rough eye phenotype. To this end, flies expressing G4C2-58 in the eye were crossed with 372 chromosomal deficiency lines spanning the entirety of the 2nd and 3rd chromosomes, representing ~80% of the fly genome. In total, 5.1% of the deficiencies were found to suppress the rough eye phenotype whereas 8.8% of these enhanced the rough eye phenotype (Extended Data Table 2).
Two deficiency lines on the 2nd chromosome, Df(2R)1725 and Df(2R)1735, each in heterozygotes had no eye phenotype by themselves but led to a marked enhancement of the G4C2-58 rough eye phenotype (Fig. 2a). The genomic regions covered by these deficiencies partially overlap (Fig. 2b), suggesting one or more genes in this overlapping region are responsible for the observed enhancement. After systematic evaluation of this candidate interval with classical loss of function (LOF) alleles and RNAi lines, we identified the gene Nup50, whose partial LOF enhanced the G4C2-58 phenotype (Fig. 2c). The effect of the Nup50 was specific to G4C2-58 since there was no rough eye phenotype present when Nup50 was knocked down in eyes expressing GFP (Fig. 2c). Nup50 is a component of the nuclear pore and also plays a critical role in promoting protein nuclear import through interaction with Importin β and Ran GTPase7, Consistent with the impact of Nup50 LOF on G4C2-58-mediated degeneration, a dominant-negative form of Ran (RanT24N)8 strongly enhanced the G4C2-58 rough eye phenotype (Extended Data Fig. 4a). Moreover, the nuclear import factors Nup153 and Transportin, which work in concert with Nup50 and Ran7, were also identified as enhancers of the G4C2-58 rough eye phenotype (Extended Data Fig. 4a). These genetic analyses suggest that protein import is compromised by G4C2-58 expression.
Figure 2. Genetic screen identifies multiple modifiers of G4C2-58 toxicity in the nucleocytoplasmic transport pathway.
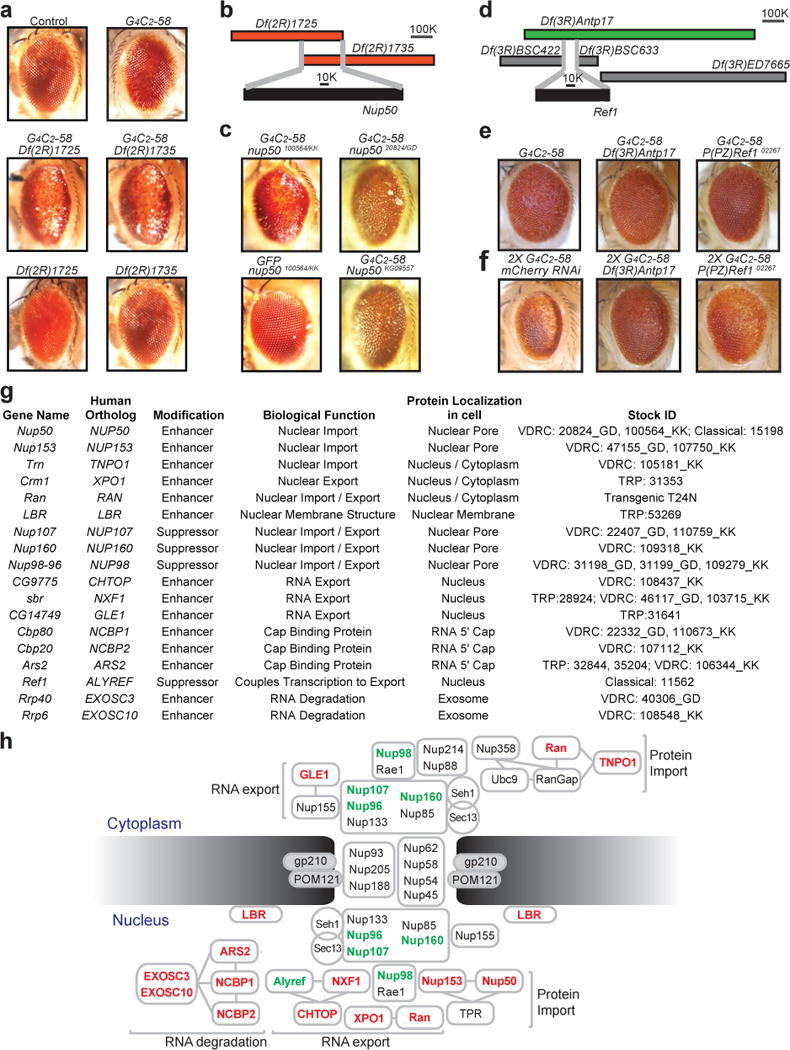
a, G4C2-58 expression driven by GMR-GAL4 causes a rough eye phenotype (top right) that was enhanced by either Df(2R)1725/+ or Df(2R)1735/+(middle). b, Overlapping genomic region of deficiency lines Df(2R)1725 or Df(2R)1735. c, RNAi knockdown (Nup5020824/GD and Nup50100564/KK) and a genetic allele (Nup50KG09557) identify Nup50 as an enhancer. d, The deficiency Df(3R)Antp17 (green) but not others (grey) suppressed the G4C2-58 eye phenotype. e and f, Identification of Ref as a suppressor. g, Table summarizing modifier genes and their known functions. h, Suppressors (green) and enhancers (red) of G4C2-58 toxicity in the nucleocytoplasmic trafficking pathway.
The strongest suppressor identified in the genetic screen was Ref1, revealed by interrogation of the suppressor deficiency Df(3R)Antp17 (Fig. 2d, e). This deficiency also suppressed the strong rough eye phenotype produced by two copies of G4C2-58, as did heterozygosity for the null allele Ref102267 (Fig. 2f). Ref1, and its human ortholog ALYREF, are RNA-binding proteins that associate with the 5′ end of mRNAs to prevent degradation by the nuclear exosome and, together with the TREX component CHTOP, facilitate delivery of fully processed mRNAs to the nuclear pore receptor NXF1 that mediates their export through the nuclear pore9–12. Consistent with the impact of Ref1 LOF on G4C2-58-mediated degeneration, partial LOF of NXF1 or CHTOP enhanced the rough eye phenotype (Fig. 2g, h, Extended Data Fig. 5). The fact that the G4C2-58 phenotype is exacerbated by LOF in NXF1 or CHTOP, but partially rescued by LOF of Ref1 perhaps highlights the dual role played by Ref1 in partitioning target RNAs between the nuclear pore and the nuclear exosome9. Indeed, in support of this interpretation we identified LOF in EXOSC3 and EXOSC10, which encode components of the nuclear exosome, as strong enhancers of G4C2-58-related toxicity (Fig. 2g, h, Extended Data Fig. 5). Notably, LOF mutations in EXOSC3 in humans cause a congenital form of motor neuron disease13.
The genes encoding cap-binding proteins NCBP1, NCBP2, and ARS2, that mediate recruitment of the TREX complex to the 5′ end of RNA to initiate RNA export14, were also identified as enhancers of G4C2-58-mediated degeneration (Fig. 2g, h, Extended Data Fig. 5). Moreover, we found that LOF of either Nup107 or its binding partner Nup160, both of which are nuclear pore components responsible for exporting some RNAs15–17, suppressed the G4C2-58 rough eye phenotype (Extended Data Fig. 5). One particularly notable enhancer was the Drosophila ortholog of human GLE1, a critical mediator of RNA export at the nuclear pore18 (Fig. 2g, h, Extended Data Fig. 5). Interestingly, complete LOF of human GLE1 causes congenital motor neuron disease19, whereas partial LOF of human GLE1 is associated with adult-onset ALS20.
The identification of TREX and nuclear pore proteins as genetic modifiers strongly implies that not only is nuclear import compromised by G4C2-58 expression, but also nuclear export of RNAs and proteins. Consistent with this latter notion we found that LOF of Crm1, a gene encoding a major receptor for the export of RNAs and proteins, resulted in significant enhancement of the G4C2-58 phenotype (Extended Data Fig. 4b). Moreover, expression of G4C2-58 in motor neurons caused exquisite sensitivity to leptomycin B, an inhibitor of nuclear export (Extended Data Fig. 4c). In total, the genetic screen and follow up analyses identified 18 modifier genes within the pathway of nucleocytoplasmic transport, and RNA export in particular, highlighting this system as an important target of G4C2-58-related toxicity (Fig. 2g, h, Supplementary Table 3). In particular, the identification of 4 strong suppressors of G4C2-58 indicates that compromised nucleocytoplasmic transport is an important causal pathway responsible for degeneration.
In follow up to our genetic studies, RFP-Nup107 was expressed in salivary glands using Fhk-GAL4. In cells expressing G4C2-8, Nup107 labels a distinct nuclear boundary that is morphologically indistinguishable from cells expressing GFP (Fig. 3a). In contrast, in cells expressing G4C2-58, the nuclear envelope exhibited a wrinkled appearance and in many nuclei, Nup107 was found to form inclusions near the nuclear envelope (Fig. 3a). To further explore nuclear architecture, the nuclear envelope was visualized by immunostaining of endogenous Lamin C. 43.7% of cells expressing G4C2-58 showed an abnormal, “frayed” nuclear envelope phenotype, whereas this phenotype was observed in only 7.1% of cells expressing G4C2-8 (Fig. 3b, c, Extended Data Fig. 6a). Thus, G4C2-58 expression causes defects in the architecture of the nuclear envelope and mislocalization of nucleoporins. Moreover, cells expressing one copy of G4C2-58 showed a significant increase in the ratio of nuclear to cytoplasmic RNA in comparison to cells expressing GFP or G4C2-8 (Fig. 3d, e). Expression of 2 copies of G4C2-58 resulted in a more pronounced retention of nuclear RNA (Fig. 3d, e). Importantly, depletion of Ref1 by RNAi partially suppressed the nuclear RNA retention phenotype in cells expressing G4C2-58, reducing nuclear RNA density to control levels (Fig. 3d, e). Thus, G4C2 length-dependent degeneration in Drosophila is accompanied by nuclear retention of RNA, consistent with the results of the unbiased genetic screen.
Figure 3. Drosophila salivary gland cells expressing G4C2-58 exhibit nuclear envelope abnormalities and accumulation of nuclear RNA.
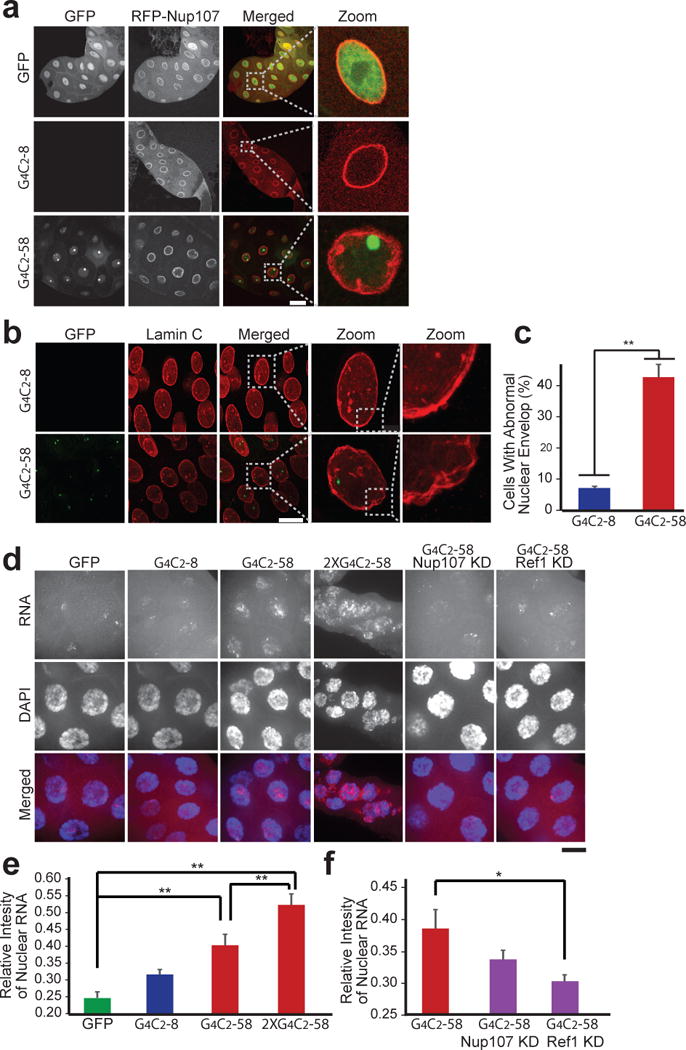
a, The effect of G4C2-58 expression on Nup107 localization. Scale bar: 50 μm. b, G4C2-58 expression causes abnormal nuclear envelope morphology. Scale bar: 50 μm. c, Quantification of the frayed nuclear envelope phenotype in cells expressing either G4C2-8 (n = 251) or G4C2-58 (n = 127). Values are mean ± s.e.m., ** p < 0.001 by Student’s t test. d, Accumulation of total nuclear RNA relative to cytoplasmic RNA (red). Knockdown of Ref1 led to a partial rescue of nuclear RNA accumulation. Scale bar: 25 μm. e and f, Quantification of relative intensity of total nuclear RNA vs. cytoplasmic RNA. Values are mean ± s.e.m., n = 30 cells, * p < 0.01, ** p < 0.001 by One Way ANOVA, Tukey’s Post Hoc test.
Similar retention of nuclear RNA was observed in HeLa and HEK293T cells upon transient expression of G4C2-58, but not G4C2-8 (Fig. 4a–e, Extended Data Fig. 6b). Specifically, this export defect was illustrated using an alternate approach to monitor the fate of newly synthesized RNA by metabolic labeling. In HeLa cells expressing G4C2-58, we observed a significant decrease in the export of nascent RNA in comparison to control cells expressing GFP or G4C2-8, although the total levels of metabolically labeled transcripts were comparable (Fig. 4a–c, Extended Data Fig. 6c). Moreover, polyA+ mRNA as detected by an oligo-dT probe accounts for at least some portion of the retained RNA in mammalian cells expressing G4C2-58 (Fig. 4d, e).
Figure 4. Accumulation of nuclear RNA in human cells expressing G4C2 repeat expansion.

a, Reduced accumulation of newly synthesized RNA in the cytoplasm of cells transfected with G4C2-58 (white arrows) (imaged at 60 min). Yellow arrows: untransfected cells. b, Quantification of cytoplasmic RNA intensity following metabolic labeling of newly synthesized RNA (0 min: n > 24, 30 min: n > 7, 60 min: n > 25). ** p < 0.01, *** p < 0.001 by One Way ANOVA, Tukey’s Post Hoc test. c, Total RNA intensity following labeling of newly synthesized RNA at time point 0 min. d, PolyA+ RNA was measured by FISH in transiently transfected HeLa cells. Scale bar: 50 μm. e, Quantification of cells containing polyA+ RNA puncta, n = 30 cells per condition. f, Total cellular RNA in 2-month old iPSCs-derived cortical neurons. Scale bar: 25 μm. g, Quantification of the cytoplasmic RNA ratio in patient cortical neurons vs. controls. Values are mean ± s.e.m., n > 15 cells per line, ** p < 0.01 by Student’s t test.
Advances in induced pluripotent stem (iPS) cell technology permit the generation of neurons from patients including those with FTD and ALS21. To further examine the impact of expanded G4C2 toxicity on nucleocytoplasmic transport in a more relevant human cell type from C9ORF72 patients, we examined the subcellular distribution of total RNA in 2-month-old cortical neurons, which are affected in FTD, a frequent clinical feature of C9ORF72-related disease22. These neurons were differentiated from previously characterized23–25 as well as newly generated and characterized iPS cell lines derived from 5 C9ORF72 patients and 3 controls (Extended Data Figs. 7 and 8, Supplementary Tables 4 and 5). Consistent with our observations in fly, HEK293T and HeLa cells, this analysis confirmed a significant increase in the nuclear to cytoplasmic ratio of RNA in C9ORF72 patient cells in comparison to controls (Fig. 4f, g). On average, C9ORF72 neurons showed a 35% increase in the nuclear to cytoplasmic ratio of RNA density (Fig. 4g). Interestingly, this difference in the ratio of nuclear to cytoplasmic RNA was not observed in fibroblasts obtained from C9ORF72 patients when compared to controls, consistent with low levels of C9ORF72 expression in fibroblasts25 (Extended Data Fig. 9a–c). Thus, neurons derived from patients with C9ORF72-related disease mirror the nucleocytoplasmic transport defects identified by unbiased approaches in a genetic model of disease.
The NPC is the largest macromolecular complex in eukaryotic cells, consisting of multiple copies of more than 30 different proteins26. Some structural components of the NPC have an exceedingly long half-life measured in years27. Indeed, nuclear pores are a particularly intriguing target for age-related diseases affecting post-mitotic cells such as neurons, putting them at risk of accumulating damage over extended periods of time27. Nucleocytoplasmic transport through the nuclear pore is vital to cell viability, and primary defects in this function cause of a host of human diseases of varying phenotypes ranging from cancer to neurological disease26,28,29.
Important questions regarding the pathogenesis of C9ORF72-related disease remain; in particular the relative contribution of expanded G4C2 RNA vs. DPRs or other mechanisms, which could cause the nuclear pore defect. Also to be determined is whether defects in nucleocytoplasmic transport and other reported defects such as sequestration of specific RNA binding proteins, nucleolar stress and ER stress are parallel processes or directly connected with each other. The rapid pace of discovery in this area of disease biology suggests that answers to these questions will come soon.
Methods
Generation of Drosophila lines
To generate transgenic Drosophila expressing G4C2-8, G4C2-28 and G4C2-58, G4C2 repeats of the respective length were cloned downstream of the UAS promoter and upstream of the EGFP sequence with the start codon removed in the plasmid pUAST-ATTB. Transgenic Drosophila lines were generated by BestGene Inc. such that the transgene was inserted using the PhiC31 integrase into either the attP2 site on chromosome 3 (loci 68A4) or the attP40 site on chromosome 2 (loci 25C6). GFP-(GP)47, GFP-(GA)50, and GFP-(GR)50 plasmids30 were subcloned into the pUAST-ATTB plasmid and inserted into the attP2 site by BestGene Inc. Flies were raised at 25°C on a standard diet. A complete listing of Drosophila stocks that modify the G4C2-58 phenotype are listed in Supplementary Table 3. For genetic interaction studies, the combined stock (GMR-GAL4/Cyo; UAS-G4C2-58/Tm6,Tb) was crossed with deficiency stocks or individual mutants and phenotypic analysis was performed on flies aged 24–48 hours in both males and females. Knock down efficiency of RNAi lines was measured by quantitative RT-PCR (Extended Data Fig. 10, Supplementary Table 6). For nuclear envelope morphology studies, the recombined stocks (Fkh-GAL4, UAS-GFP/TM6,Tb; Fkh-GAL4, UAS-G4C2-8/TM6,Tb; and Fkh-GAL4,UAS-G4C2-58/TM6,Tb) were crossed with UAS-mRFP-Nup107 transgenic flies or immunostained for endogenous Lamin C.
Drosophila stocks
Deficiency stocks were obtained from the Bloomington Drosophila Stock Center deficiency kit. RNAi lines were obtained from either the Bloomington Drosophila Stock Center or the Vienna Drosophila RNAi Center. UAS-RANT24N and Fkh-Gal4 flies were kindly provided by Drs. K. S. McKim and E. Baehrecke respectively.
Drosophila eye, muscle and neuronal phenotype analysis
Phenotypic analysis of G4C2 repeat expression in the Drosophila eye, muscle, and salivary gland was assessed by crossing G4C2-8, G4C2-28 and G4C2-58 lines to GMR-GAL4, MHC-GAL4, and FKH-GAL4, respectively. Neuronal expression was achieved by crossing G4C2-8, G4C2-28 and G4C2-58 lines to elav-GAL4 (pan neuronal driver) or OK371-GAL4 (motor neuron driver). Eye phenotypes were imaged by light microscopy and muscle phenotypes were visually assessed by wing posture (n=30 controls, 21 G4C2-8, 50 G4C2-28 and 24 G4C2-58). Neuromuscular synaptic bouton number, active zone density and crawling ability were measured as previously described31,32 with slight modification. To count type 1b synaptic bouton number, each genotype was double stained presynaptically with anti-HRP-Cy3 (1:200, Jackson Immunoresearch) and postsynaptically with anti-Disc large 1 (1:50, DSHB). Synaptic boutons of muscle 4 in abdominal segment 2, 3 and 4 (A2–A4) were imaged with a Marianas spinning disc microscope and maximum projection images were used to count synaptic boutons. Active zone area and presynaptic area stained by anti-Bruchpilot (NC82, 1:100, DSHB) and anti-HRP-Cy3 were measured with ImageJ and the ratio of active zone area/presynaptic area was calculated. Muscle 4 in abdominal segment 2, 3 and 4 (A2–A4) was analyzed. To examine crawling ability, 4–7 wandering 3rd instar larvae for each group were collected, washed and placed onto a 3% agarose gel in a 10 cm dish. After 5 min acclimation, larval crawling behavior was recorded by a digital camera for 30 sec (15 fps). Each group was tested three times. Moving distances of each larva were manually measured with ImageJ.
Immunofluorescence analysis of salivary gland cells
Dissected salivary glands from wandering third instar larvae were fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, washed three times with phosphate-buffered saline (PBT, 0.1% Triton X-100 in PBS), and blocked for 1 hr at room temperature with 10% normal goat serum (Sigma) diluted in PBT. Tissues were then incubated overnight at 4°C with mouse anti-Lamin C (1:30, LC28.26; Developmental Studies Hybridoma Bank (DSHB)). After three washes in PBT (10 min each), Tissues were incubated for 1 hr at room temperature with secondary antibodies (goat anti-mouse Alexa Fluor 568, 1:200; Invitrogen) diluted in the blocking solution. Tissues were washed three times in PBS and mounted with VECTASHIELD (Vector Laboratories). Fluorescence signals were examined with an Olympus IX70 Microscope or Leica TCS SP5 II laser scanning confocal microscope.
Visualization of RAN products in muscle cells
To visualize poly(GP)-GFP in the thorax expressing G4C2-58, flies were cleared overnight using a modified ScaleA2 (2 M urea, 10% glycerol, 0.1% Triton-X). RAN peptides were visualized using light sheet microscopy. To visualize RAN products at high magnification, thoraxes were dissected and stained with the same immunofluorescence protocol described above for salivary glands but using anti-Lamin (1:100, ADL 67.10; Developmental Studies Hybridoma Bank (DSHB)) to visualize the nucleus and phalloidin (1:40, Life Technologies) to visualize the muscle structure.
Immunoblots
Adult flies were frozen with dry ice and vortexed to remove the heads or thorax. Samples from each genotype were homogenized in RIPA buffer with proteinase inhibitor cocktail added. Urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris pH8.5) was used when indicated to visualize the non-RIPA soluble protein fraction. Sample was mixed with 4X sample buffer (1 M Tris-HCl [pH 6.8], 8% SDS, 40% glycerol, 0.1% bromophenol blue) and boiled for 5 min, separated on a SDS gel and transferred to a membrane. The membrane was blocked, probed with primary antibody, and incubated with secondary antibody. The signal was visualized with either chemiluminescent substrate (SuperSignal West Pico; Pierce) or by using an Odyssey Fc (Li-Cor). Primary western blot antibodies were anti-GFP (AB3080, Millipore or SC-9996 Santa Cruz Biotechnology), anti-poly(GP), poly(GA) or poly(GR) antibodies33,34, anti-β-actin antibody (4967; Cell Signaling Technology) or anti-actin antibody (sc-1616, Santa Cruz Biotechnology).
RNA in situ hybridization
Fixation of dissected salivary glands, cortical neurons, HeLa, or 293T cells (obtained from ATCC) was performed using 1% paraformaldehyde on ice for 5 min, followed by a second fixation in 1% paraformaldehyde plus 0.05% NP40 for an additional 5 min. Samples were then transferred to 70% ethanol and stored at −20°C until needed. The probes for global RNA in either Drosophila or human cells were prepared from genomic DNA derived from w1118 flies or 293T cells. Both probes were labeled by nick translation using either alexafluor 594 dUTP. To hybridize the probes, 70 ng of the labeled probes (the human probe was combined with 2 μg of human cot1 DNA) were suspended in 10 μl of hybridization buffer consisting of 50% formamide, 2X SSC, and 10% dextran sulfate. Fixed cells were dehydrated in 70%, 80%, and 100% ethanol for 2 min each prior to hybridization. Probes suspended in hybridization buffer were denatured at 70°C for 5 min and then applied to the dehydrated cells and hybridized at 37°C overnight. Samples were washed following hybridization in 50% formamide, 2X SSC at 37°C for 5 min, then briefly rinsed in room temperature PBS and mounted on slides with DAPI. To detect poly(A) mRNA, HeLa cells were fixed with 4% paraformaldehyde followed by ice cold methanol and 70% ethanol for 10min each. The cells were treated with 1 ng/ l μL of 5′-labeled Cy3-oligo-dT(20) at 37°C for 1 h. Samples were washed in room temperature PBS and mounted on slides with DAPI. Images were captured using either widefield fluorescence microscopy or a Marianas spinning disc microscope. An extended depth of focus function was used to combine all components of individual microscope fields.
Pulse-chase of newly synthesized RNA
HeLa cells were transfected with plasmids expressing GFP, G4C2-8 or G4C2-58 that had been subcloned into pcDNA 3.1 using Fugene HD (Promega). After 48 h incubation, cells were treated with to 1 mM 5-ethynyl uridine (EU) was for 1 h. RNA was then visualized at indicated time points using the Click-iT detection kit (Invitrogen) as recommended by the manufacturer. To visualize GFP signal, cells were co-stained using monoclonal anti-GFP (SC-9996, Santa Cruz Biotechnology) as a primary antibody.
iPSC lines
iPSC lines from 2 control subjects and 4 G4C2 repeat expansion carriers have been previously characterized23–25,35 (please see Extended Data Table 4). Generation of integration-free induced pluripotent stem cells (iPSCs) from fibroblasts of one G4C2 expansion carrier and one control subject, using episomal plasmids with the reprogramming factors Oct4, Sox2, Klf4, Lin28 and L-Myc were performed as described before36, which was approved by the University of Massachusetts Medical School Institutional Biosafety Committee. The use of human fibroblasts was approved by the UCSF Institutional Review Board and informed consent was obtained from all subjects. After reprogramming, characterization of iPSCs shows that cells have normal karyotype, express pluripotent markers and have the capacity to differentiate into cells of the three germ layers as described in Almeida et al., (2012, 2013)23,35. All the iPSC lines used here were tested regularly with no mycoplasma contamination.
Cortical neuronal cultures
4 iPSC lines from 3 control subjects and 6 iPSC lines from 5 C9ORF72 carriers were differentiated to cortical neurons as described earlier23,35 (please also see Extended Data Table 5). Neuronal cultures were aged for 8-weeks before fixation for RNA FISH.
Immunostaining for iPSCs and iPSCs-derived neurons
For immunostaining cells were fixed in 4% paraformaldehyde for 15 minutes and then permeabilized with 0.3 % Triton X-100 for 5 minutes. Cells were blocked with 5% bovine serum albumin for 30 minutes; cells were incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: mouse anti-OCT4 1:100 (Santa Cruz Biotechnology), goat anti-NANOG 1:100 (R&D Systems), mouse anti-SSEA4 1:100 (Abcam), rabbit anti-desmin 1:100 (Thermo Scientific), mouse anti-βIII-tubulin 1:500 (Promega), mouse anti-α-fetoprotein 1:200 (R&D Systems), mouse anti-MAP2 1:500 (Sigma), goat anti ChAT 1:200 (EMD Millipore), rabbit anti-VGLUT1 1:500 (Synaptic Systems). After incubation with primary antibodies cells were washed with PBS three times and incubated with Alexa Fluor secondary antibodies 1:500 (Invitrogen) for 1 h at room temperature followed by counter staining with DAPI.
Fluorescence in situ hybridization
FISH in iPSCs derived from control and C9ORF72 expansion carrier using a Cy3-conjugated (GGCCCC)4 probe was performed as described in Almeida et al., (2013)23.
Quantification
For salivary glands, 30 cells from at least 5 individual salivary glands for each genotype were used for quantitative analysis. ImageJ software was used to measure the ratio of nuclear to cytoplasmic RNA by comparing the total RNA measured in the nucleus to that of the cytoplasm. DAPI was used to mark the nucleus. For cortical neurons, the ratio of nuclear to cytoplasmic RNA density was calculated by measuring the density of the RNA signal in both the nucleus and cell body using ImageJ software. DAPI was used to mark the nuclear boundary of neurons. For cortical neurons, at least 15 neurons were measured from each individual patient (Controls 1–3 (n=16, 16, 31) and Patients 1–5 (n=16,16,16,30,15)). Error bars for all quantification are standard error.
Extended Data
Extended Data Figure 1. Expression of G4C2 repeats induces length-dependent phenotypes in Drosophila.
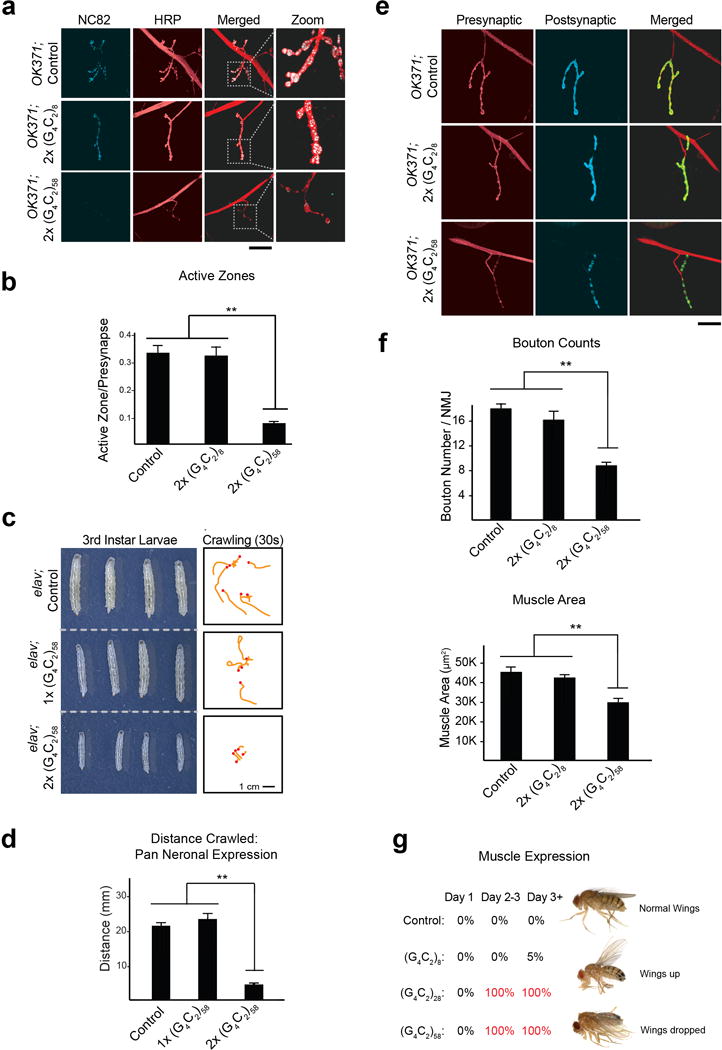
a, G4C2 Drosophila motor neurons using the OK371-GAL4 driver leads to a significant reduction in active zones as immunostained by the anti-Bruchpilot antibody NC82 and anti-HRP. Scale bar: 50 μm. b, Quantification of active zones. Values are mean ± s.e.m. ** p < 0.01, One way ANOVA, Tukey’s Post Hoc test. c, Pan Neuronal expression of G4C2-58 repeats induces dosage-dependent decrease in larval size (left) and locomotor activity measured in 30 seconds (right) when G4C2-58 is expressed in all neurons using the elav-GAL4 driver. d, Quantification of the distance traveled by third instar larvae reveals expressing two copies of G4C2-58 results in a significant deficit in locomotor activity. Values are mean ± s.e.m. n=3 trials, more than 4 larvae per group in each trial. ** p < 0.01, One way ANOVA, Tukey’s Post Hoc test. e, Pan Neuronal expression of G4C2-58 repeats in Drosophila neurons using the elav-GAL4 driver leads to a significant reduction in the bouton number. Bouton number was quantified by examining the presynaptic (anti-HRP) and postsynaptic (anti-DLG1) markers (left). Scale bar: 50 μm. f, Quantification of buoton number (left) and muscle size (right) reveal both are significantly reduced in Drosophila larvae expressing G4C2-58 repeats. Values are mean ± s.e.m. n ≥ 5, ** p < 0.01, One way ANOVA, Tukey’s Post Hoc test. g, Expression of G4C2-28 and G4C2-58 but not G4C2-8 in the muscle using the MHC-GAL4 driver leads to loss of wing control in adult flies (n > 20 for all genotypes). This phenotype was assessed by examining the permanent wing posture of live adult flies.
Extended Data Figure 2. RAN translation is observed in the Drosophila expressing G4C2 repeats.
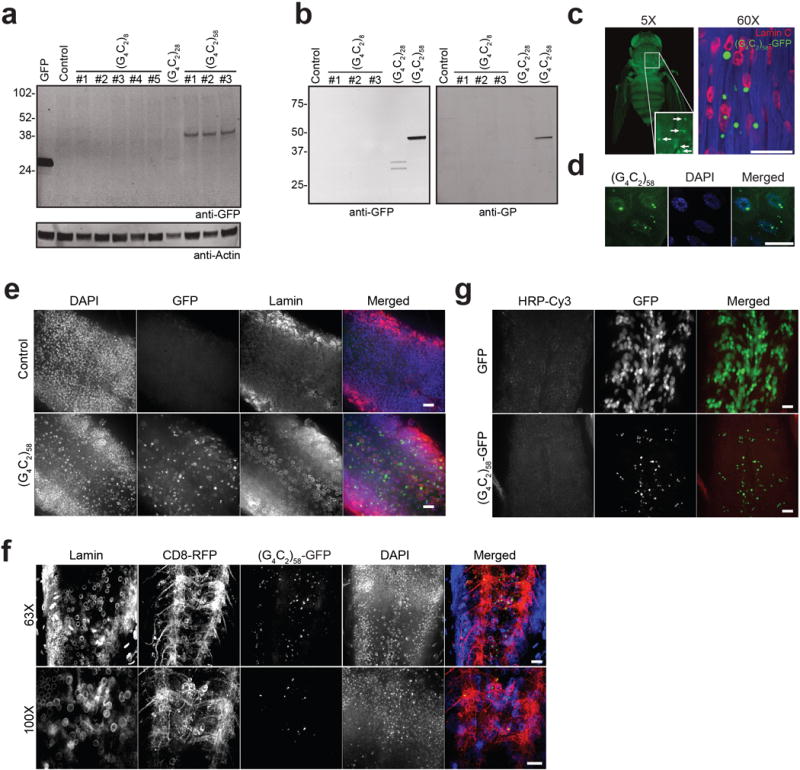
a, Western blot revealing translation of RAN poly-dipeptides in flies expressing G4C2-58 in the eye. RAN poly-dipeptides were not found in flies expressing G4C2-8 or control flies. There was minimal expression of GFP positive product observed in flies expressing G4C2-28. GFP expressing flies (lane 1) were used as a positive control for the anti-GFP antibody. b, Western blot showing production of RAN product when G4C2-28 and G4C2-58 but not G4C2-8 repeats are expressed in the muscle. RAN products were visualized with anti-GFP antibody (left) and anti-poly(GP) antibody (right). c, The RAN product poly-GP-GFP from flies expressing G4C2-58 in the muscle form large visible inclusions as visualized under light sheet fluorescent microscopy (left) and by confocal microscopy (right). Scale bar: 50 μm. d, Expression of G4C2-58 in the salivary gland cells results in the formation of large nuclear inclusions and smaller cytoplasmic inclusions. Scale bar 50 μm. e–f, Expression of G4C2-58 in the ventral ganglion by OK371 driver results in the formation of nuclear and cytoplasmic inclusions, whereas GFP shows diffused nuclear and cytoplasmic localization. Lamin staining shows nuclear membrane, and CD8-RFP shows plasma membrane. Scale bars: 25 μm. g, Expression of G4C2-58 in pan neuronal cells by elav driver results in the nuclear and cytoplasmic inclusions. Scale bar: 25 μm.
Extended Data Figure 3. Ectopic expression of poly(GR) but not poly(GA) or poly(GP) peptides are toxic in Drosophila.
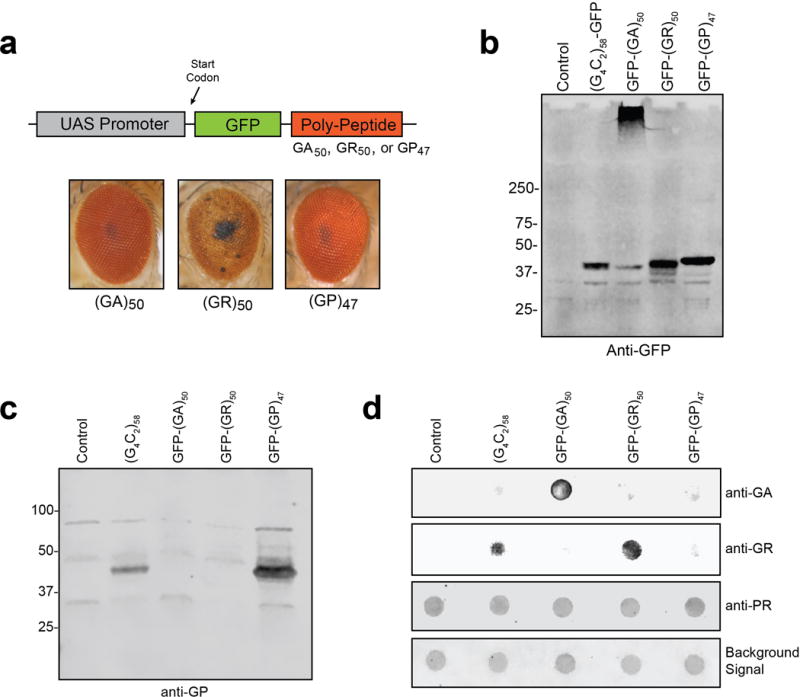
a, Transgenic Drosophila were generated that express ATG driven poly(GA), poly(GR) and poly(GP) peptides with an N-terminal GFP tag (top). Expression of GFP-(GA)50 and GFP-(GP)47 were nontoxic when expressed in the eye with GMR-GAL4 whereas GFP-(GR)50 expression resulted in >95% lethality with surviving adults having severely degenerated eyes (bottom). b, Western blot showing the expression of G4C2-58, GFP-(GA)50, GFP-(GR)50 and GFP-(GP)47 as visualized in muscle by anti-GFP antibody. c, Western blot showing the expression of poly(GP) in muscle of flies expressing G4C2-58 and GFP-(GP)47 but not GFP-(GA)50, GFP-(GR)50 and control flies as visualized by anti-GP antibody. d, Dot blot analysis of RAN peptides in muscle revealing expression of poly(GA) only in GFP-(GA)50 flies, expression of poly(GR) in G4C2-58 and GFP-(GR)50 flies. As expected, anti-sense DPR poly(PR) was not found in any of the lysates. The background protein signal was used as a loading control.
Extended Data Figure 4. Nuclear import and export is altered by G4C2-58 expression.
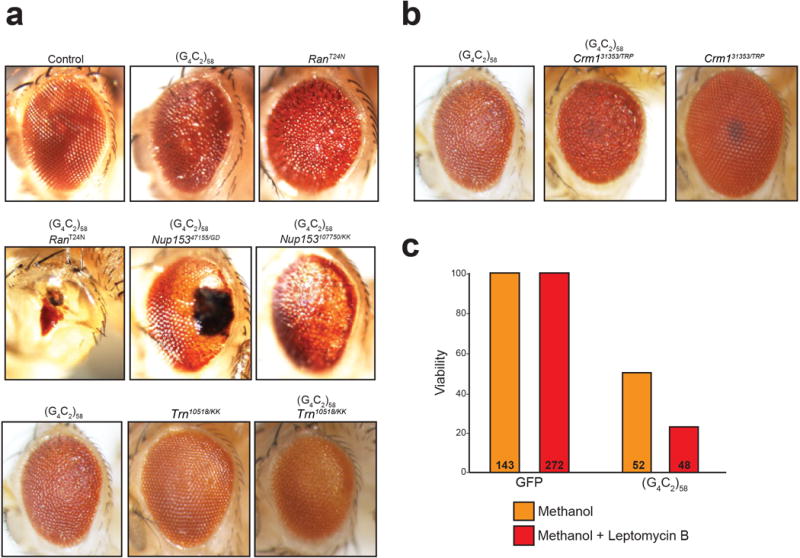
a, A threonine to asparagine substitution at residue 24 in the Ran protein abolishes the affinity for GTP and reduces its affinity for GDP. Hence, the Ran (T24N) is always in either a nucleotide free state or in its inactive, GDP-bound state, and acts as dominant negative. RANT24N expression driven by GMR-GAL4 causes a mild eye phenotype when expressed in the absence of G4C2-58 (upper row, right panel). The G4C2-58 rough eye phenotype is strongly enhanced by dominant negative RANT24N expression (middle row, left panel). The G4C2-58 eye phenotype is significantly enhanced by knockdown of Nup153 by two independent RNAi lines (middle row, two right panels). The G4C2-58 eye phenotype is also mildly enhanced by knockdown of transportin (Trn) (bottom row). b, Knockdown of Crm1 in flies expressing G4C2-58 induces a mild enhancement of the G4C2-58 eye phenotype (left vs. middle). Crm1 knockdown in the absence of G4C2-58 repeats does not produce a rough eye phenotype (left). c, Expression of two copies of G4C2-58 in the Drosophila motor neurons leads to reduced viability (50%). Chemical inhibition of Crm1 with Leptomycin B (500 nM) enhances G4C2-58 toxicity resulting in reduced viability (23%). Leptomycin B does not impede viability (100%) in Drosophila expressing GFP.
Extended Data Figure 5. Phenotypes of additional suppressors and enhancers of G4C2-58.
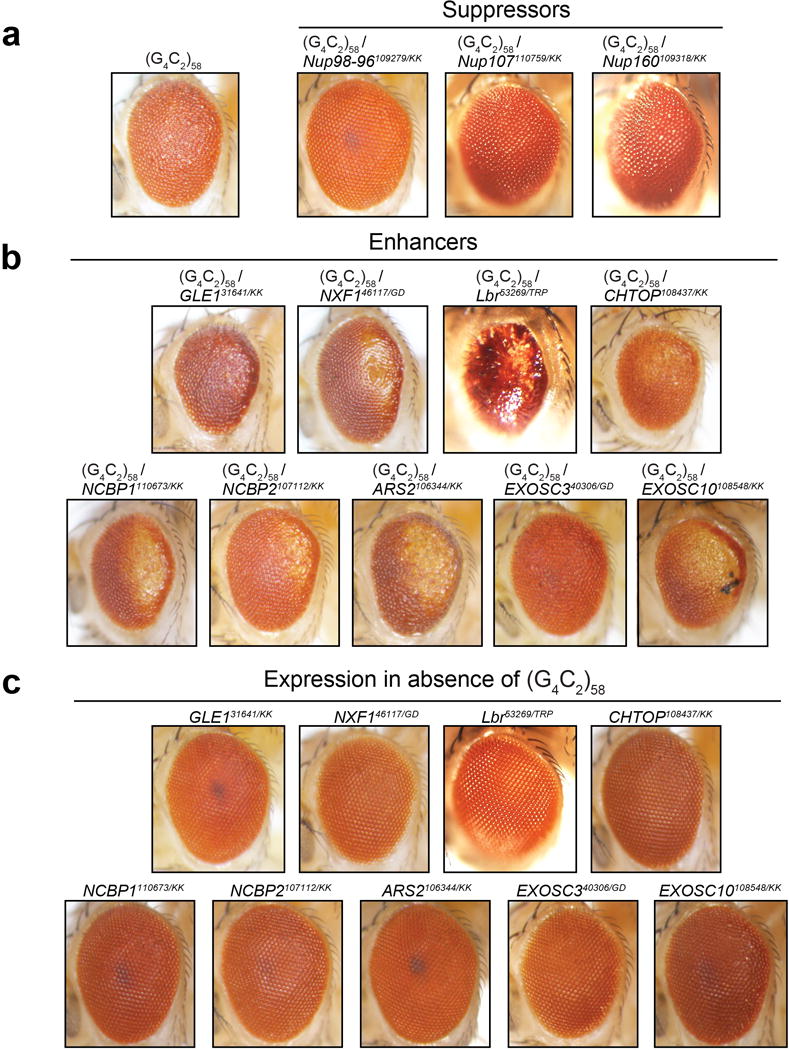
a, Phenotypes demonstrating suppression of the G4C2-58 rough eye phenotype by RNAi knock down of identified genes. b, Phenotypes demonstrating enhancement of the G4C2-58 rough eye phenotype by RNAi knock down of identified genes. c, Knock down of identified modifier genes shows little or no phenotype in the absence of G4C2 repeat expression.
Extended Data Figure 6. Impairment of Nucleocytoplasmic shuttling in Drosophila and cultured human cell lines.
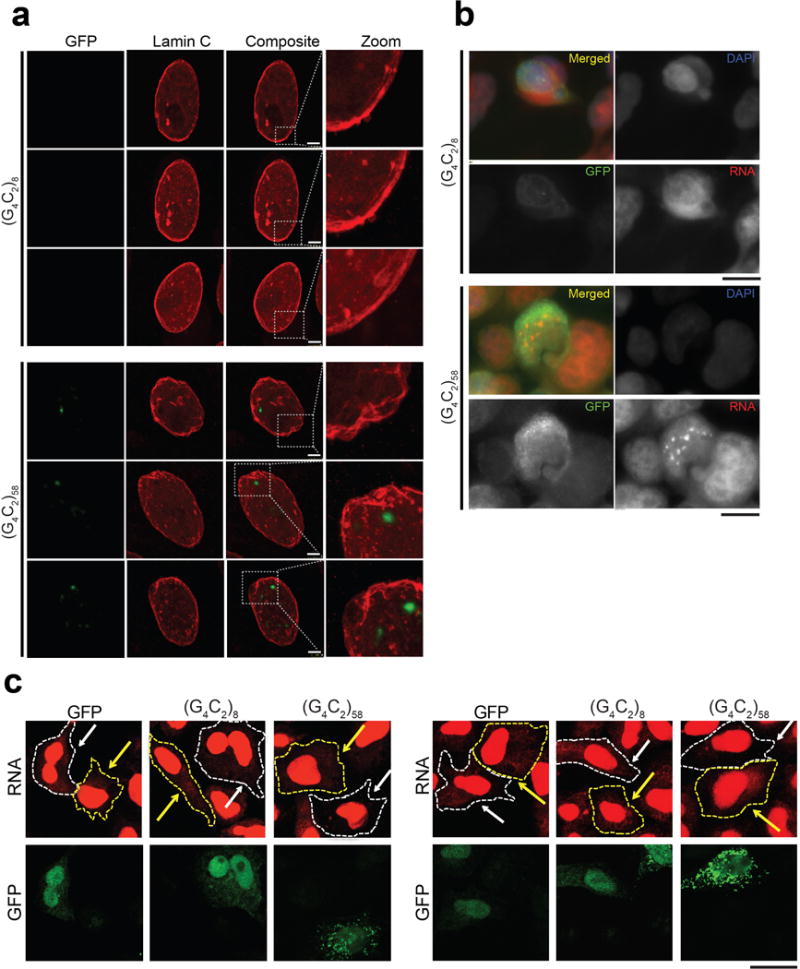
a, G4C2-58 expression driven by FKH-GAL4 causes an abnormal nuclear envelope as shown by Lamin C staining (bottom) in comparison to G4C2-8 (top). Scale bar: 10 μm. b, Transfection of 293T cells with G4C2-58 (bottom) but not G4C2-8 (top) leads to an increase in nuclear RNA puncta as visualized with a total RNA FISH probe. Non-transefected cells (absence of GFP signal) do not show an increase in nuclear RNA in either G4C2-8 or G4C2-58 transfected cell. Scale bars: 25 μm. c, Enlarged images showing slowed accumulation of newly synthesized RNA in the cytoplasm of HeLa cells expressing G4C2-58. Scale bar: 25 μm.
Extended Data Figure 7. Characterization of newly generated integration-free iPSCs lines.
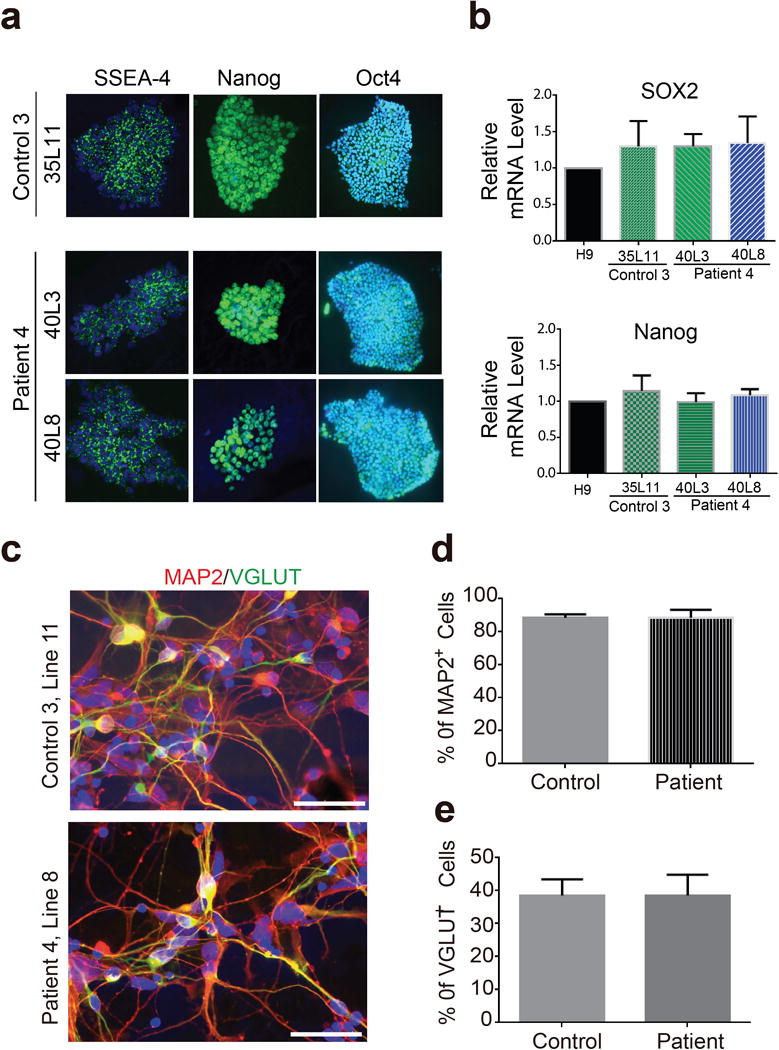
a, iPSC lines from a control subject (Line 11) and a G4C2 repeat expansion carrier (Line 3 and Line 8) express pluripotent markers SSEA-4, Nanog and Oct-4. Bars: 50 μm. b, qRT-PCR analysis ofexpression levels of pluripotent stem cell markers SOX2 and Nanog in these iPSCs lines showing no statistical differences between these lines and human embryonic stem cell line H9. c, After differentiation into cortical neurons about 90% of cells in these cultures are MAP2-positive neurons. d, Quantification of average percentage of MAP2-positive neurons and there is no difference between control and C9ORF72 cultures. e, Quantification of average percentage of VGLUT-positive excitatory neurons among all neurons and there is no difference between control and C9ORF72 cultures. Scale bars in all panels: 50 mm.
Extended Data Figure 8. Karyotyping analysis and pluripotency of newly generated iPSC lines.
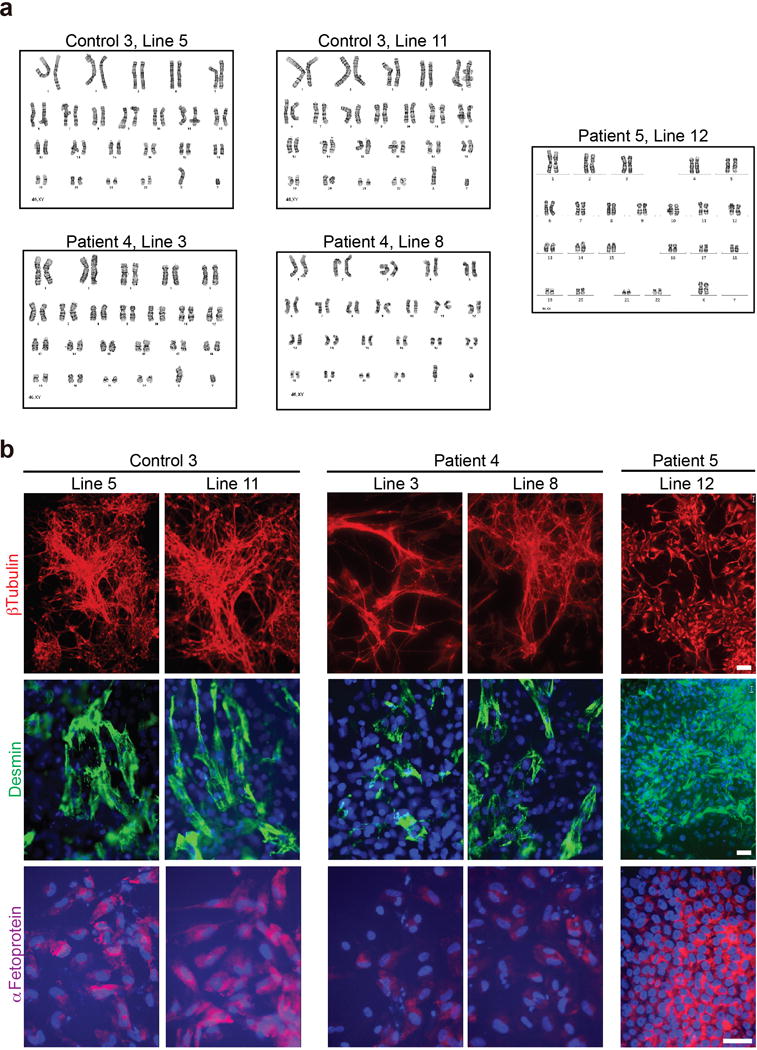
a, G-band staining showing a normal karyotype for all the lines analyzed. b, After in vitro spontaneous differentiation of control and C9 carrier iPSC lines, cells were staining for α-fetoprotein (AFP, endoderm), desmin (mesoderm), βIII-tubulin (ectoderm), and Hoechst (nuclei). All lines showed differentiation towards derivates of three germ layers. Scale bars: 20 mm.
Extended Data Figure 9. Accumulation of nuclear RNA is not seen in fibroblasts derived from patients with G4C2 repeat expansion.
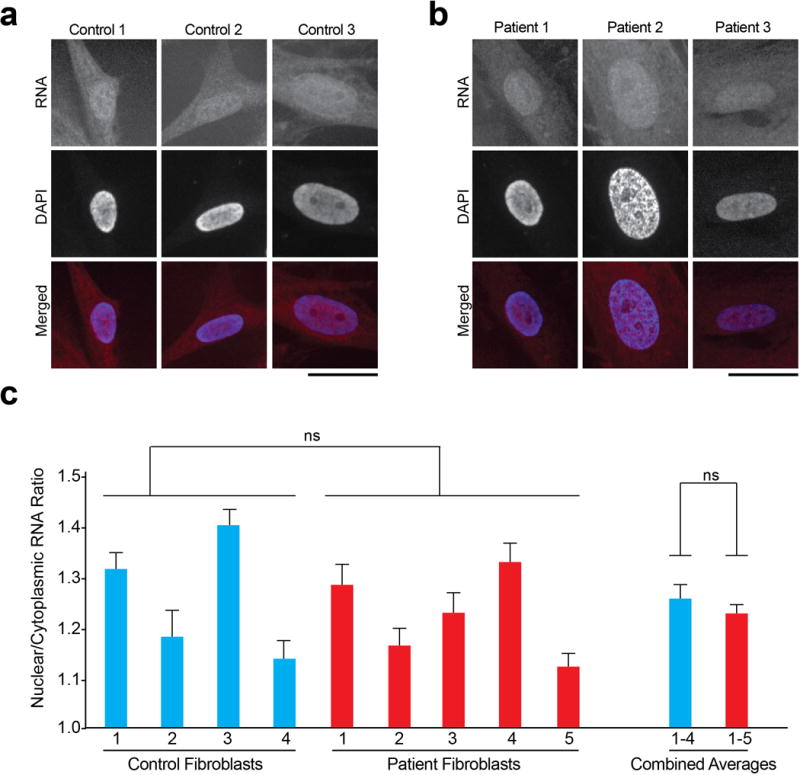
a and b, Total cellular RNA was measured by FISH in fibroblasts derived from either control (a) subjects or patients (b) with G4C2 repeat expansion. Scale bars: 25 μm. c, Quantification shows no statistical difference in the observed nuclear to cytoplasmic RNA ratio in patient vs. control fibroblasts. n=16 for all lines.
Extended Data Figure 10. qRT-PCR Analysis.
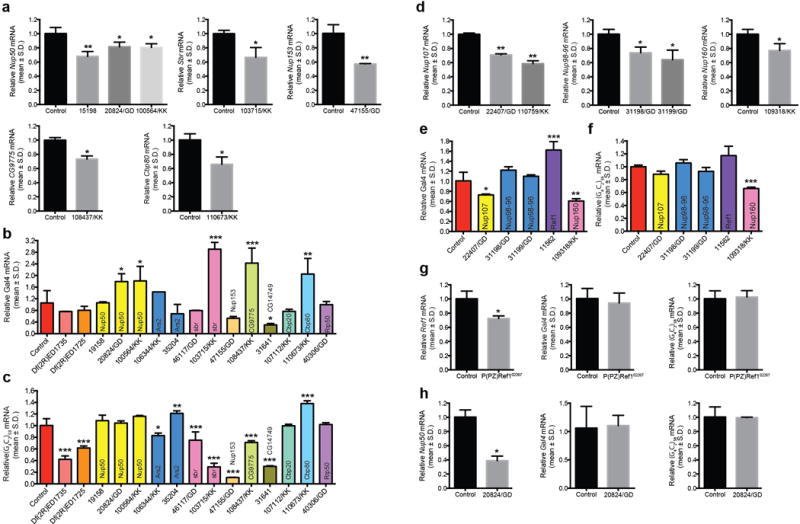
a–f. qRT-PCR analysis demonstrating knockdown of selected modifiers in Drosophila eyes. mRNA levels of selected modifier (a, d), GAL4 (b, e), and G4C2-58 (c, f) assayed by qRT-PCR in progeny resulting from wild type (w1118), classical mutant allele or UAS-RNAi lines of selected modifiers mated with either GMR-GAL4 or GMR-GAL4/Cyo;UAS-G4C2-58-GFP/TM6 to induce knockdown of the selected gene. RNA was obtained from whole Drosophila head lysates. Gene expression levels are mean ± S.D., *p < 0.05, **p<0.01, ***p<0.001 by One Way ANOVA, Tukey’s Post Hoc test. g–h. qRT-PCR analysis demonstrating knockdown of Ref1 and nup50 in Salivary Gland. mRNA levels of selected modifier (left), GAL4 (middle), and G4C2-58 (right) assayed by qRT-PCR in progeny resulting from either P(PZ)Ref102267 (g), or nup5020824/GD (h) mated with FKH-GAL4,UAS-G4C2-58-GFP/TM6. RNA was obtained from salivary gland lysates. Gene expression levels are mean ± S.D., *p < 0.05 by Student’s t-test.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center, the VDRC Stock Center, Drs. K. McKim and E. Baehrecke for fly lines, Dr. V. Budnik for Lamin C antibody, as well as the Cell and Tissue Imaging Core at St. Jude Children’s Research Hospital and the University of Massachusetts Medical School Confocal Core for assistance. This work was supported by grants from Target ALS, The Packard Center for ALS Research at the Johns Hopkins University, and the ALS Association to F.B.G. and J.P.T., and ALS Therapy Alliance, NIH (N079725) to F.B.G and NIH (NS079725 and AG019724) to B.L.M.
Footnotes
Online Content
Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Author Contributions
F.B.G. and J.P.T. conceived and supervised the project. B.D.F., Y.L., H.J.K, F.B.G. and J.P.T. wrote the manuscript. B.D.F. and Y.L. performed the genetic screen and validation. B.D.F., Y.L., N.C.K., N.B. and K.H.L. characterized Drosophila phenotypes and performed the assays characterizing RNA export in human cells. S.A. established human fibroblast cell lines, R.L.G. generated some iPS cell lines, S.A. and R.L.G. performed cortical neuron differentiation. M.V. and B.D.F. conducted FISH experiments. L.P. B.L.M. and P.W. provided key reagents.
References
- 1.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. doi:S0896-6273(11)00828-2[pii]10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Z, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon I, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen X, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makise M, et al. The Nup153-Nup50 protein interface and its role in nuclear import. J Biol Chem. 2012;287:38515–38522. doi: 10.1074/jbc.M112.378893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cesario J, McKim KS. RanGTP is required for meiotic spindle organization and the initiation of embryonic development in Drosophila. J Cell Sci. 2011;124:3797–3810. doi: 10.1242/jcs.084855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reed R. Coupling transcription, splicing and mRNA export. Curr Opin Cell Biol. 2003;15:326–331. doi: 10.1016/s0955-0674(03)00048-6. [DOI] [PubMed] [Google Scholar]
- 10.Viphakone N, et al. TREX exposes the RNA-binding domain of Nxf1 to enable mRNA export. Nat Commun. 2012;3:1006. doi: 10.1038/ncomms2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Z, et al. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]
- 12.Chang CT, et al. Chtop is a component of the dynamic TREX mRNA export complex. The EMBO journal. 2013;32:473–486. doi: 10.1038/emboj.2012.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan J, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng H, et al. Human mRNA export machinery recruited to the 5 end of mRNA. Cell. 2006;127:1389–1400. doi: 10.1016/j.cell.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 15.Boehmer T, Enninga J, Dales S, Blobel G, Zhong H. Depletion of a single nucleoporin, Nup107, prevents the assembly of a subset of nucleoporins into the nuclear pore complex. Proc Natl Acad Sci U S A. 2003;100:981–985. doi: 10.1073/pnas.252749899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasu S, et al. Novel vertebrate nucleoporins Nup133 and Nup160 play a role in mRNA export. J Cell Biol. 2001;155:339–354. doi: 10.1083/jcb.200108007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walther TC, et al. The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell. 2003;113:195–206. doi: 10.1016/s0092-8674(03)00235-6. [DOI] [PubMed] [Google Scholar]
- 18.Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- 19.Nousiainen HO, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaneb HM, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1363–1373. doi: 10.1093/hmg/ddu545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han SS, Williams LA, Eggan KC. Constructing and deconstructing stem cell models of neurological disease. Neuron. 2011;70:626–644. doi: 10.1016/j.neuron.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 22.van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol. 2012;25:689–700. doi: 10.1097/WCO.0b013e32835a3efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One. 2013;8:e76055. doi: 10.1371/journal.pone.0076055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronshaw JM, Matunis MJ. The nuclear pore complex: disease associations and functional correlations. Trends Endocrinol Metab. 2004;15:34–39. doi: 10.1016/j.tem.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Savas JN, Toyama BH, Xu T, Yates JR, 3rd, Hetzer MW. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mor A, White MA, Fontoura BM. Nuclear trafficking in health and disease. Curr Opin Cell Biol. 2014;28:28–35. doi: 10.1016/j.ceb.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnet A, Palancade B. Regulation of mRNA trafficking by nuclear pore complexes. Genes (Basel) 2014;5:767–791. doi: 10.3390/genes5030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YJ, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim NC, et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith R, Taylor JP. Dissection and imaging of active zones in the Drosophila neuromuscular junction. Journal of visualized experiments: JoVE. 2011 doi: 10.3791/2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gendron TF, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta neuropathologica. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ash PE, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almeida S, et al. Induced pluripotent stem cell models of deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012;2:789–798. doi: 10.1016/j.celrep.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.