

Abstract
Ovarian cancer is currently the most lethal gynecologic cancer in the United States. There is an urgent need for the development of innovative therapies against ovarian cancer, such as immunotherapy. The toll-like receptor 3 ligand, polyriboinosinic:polyribocytidylic acid (poly(I:C), has emerged as a promising adjuvant for activating the host immune responses for the control of tumors. We reasoned that a strategy to enhance the intracellular uptake of poly(I:C) will likely improve the poly(I:C) adjuvant effect. Since polyethylenimine (PEI) has been shown to increase the transfection efficiency of nucleic acids, we characterized the antitumor effects in mouse ovarian surface epithelial cells (MOSEC) tumor-bearing mice treated intraperitoneally with poly(I:C) and PEI. We observed that tumor-bearing mice treated with poly(I:C) and PEI generated significantly better therapeutic antitumor effects against MOSEC tumors compared with treatment with poly(I:C) alone. Furthermore, we found that NK cells play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI. Intraperitoneal administration of poly(I:C) with PEI led to the uptake of poly(I:C) mainly by CD11b+ macrophages, resulting in the high expression of MHC class II and IL-12 (M1 phenotype). In addition, adoptive transfer of CD11b+ macrophages from mice treated with poly(I:C) and PEI was found to lead to increased number of activated NK cells in the recipient mice. Taken together, our data indicate that PEI can potentially be used to improve the uptake of poly(I:C) by CD11b+ macrophages, leading to the activation of NK cells and the control of murine ovarian tumors.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-011-1013-7) contains supplementary material, which is available to authorized users.
Keywords: Poly(I:C), Polyethylenimine, MOSEC tumors, Toll-like receptor
Introduction
Ovarian cancer is the sixth most common malignancy in women and the most lethal gynecologic cancer in the United States, with approximately 22,000 new cases and 16,000 deaths occurring annually [16]. Current therapies such as surgery, chemotherapy and radiotherapy usually fail to control advanced stages of the disease. This highlights the crucial need to develop novel therapeutic strategies for the control of ovarian cancer. Alternative approaches such as immunotherapeutic strategies to enhance immune responses may serve as an important method to control ovarian tumors.
One approach for the treatment of ovarian cancer by the immune system is the employment of adjuvants such as toll-like receptor (TLR) ligands that can activate both the innate and adaptive immunity (for reviews, see [12]). It is clear that TLR ligands play a crucial role in enhancing innate and adaptive immune responses (for reviews, see [1–3, 15]). TLR 3 ligands, such as viral double-stranded RNA and its synthetic analog, polyriboinosinic:polyribocytidylic acid (poly(I:C)), have been shown to induce inflammatory cytokines and dendritic cell activation (for review see [22]). Specifically, poly(I:C) acts on both TLR3 in the endosome and MDA5 in the cytosol, and both pathways in dendritic cells/macrophages can induce natural killer (NK) cell activation. Recently, intraperitoneal injection of poly(I:C) was found to inhibit lung and liver metastasis of B16 melanoma cells, leading to prolonged survival of the mice [17]. However, since TLR3 is located in the endosomal compartment, it is challenging for poly(I:C) to efficiently penetrate the cell membrane and bind to its receptor. Thus, it is important to develop strategies to enhance the transfection efficiency in order to improve the uptake of (poly(I:C)) into the effector cells in order to enhance the antitumor effects against tumors.
Polyethylenimine (PEI) has emerged as a promising approach to increase the transfection efficiency of nucleic acids such as DNA administered in vivo. PEI has been used in conjugation with several other DNA vectors and has led to a significant enhancement of transfection efficiency [18, 28]. In addition, PEI has been used to enhance the delivery of siRNA [7]. Thus, the employment of polyethylenimine (PEI) may potentially serve as an effective method to improve the transfection efficiency, thus improving the uptake of nucleic acids, such as poly(I:C) into dendritic cells in vivo.
In the current study, we plan to explore the combination of poly(I:C) with PEI to generate significant therapeutic antitumor effects against mouse ovarian surface epithelial cells (MOSEC) ovarian tumors. We observed that mice treated with poly(I:C) in combination with PEI generate significant therapeutic antitumor effects against MOSEC tumors. Furthermore, we found that NK cells play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI. Intraperitoneal administration of poly(I:C) with PEI led to the uptake of poly(I:C) mainly by CD11b+ macrophages, resulting in the high expression of MHC class II and IL-12 (M1 phenotype). Finally, adoptive transfer of CD11b+ macrophages from mice treated with poly(I:C) and PEI was found to lead to increased proliferation and activation of NK cells from recipient mice. The clinical implications of the current study are discussed.
Materials and methods
Animals and cell lines
Female C57BL/6 mice, 5–8 weeks of age, were purchased from National Cancer Institute (Frederick, MD). All animals were maintained under specific pathogen-free conditions, and procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals. The MOSEC cell lines were originally derived from mouse ovarian surface epithelial cells and generated as shown previously [25]. Luciferase-expressing MOSEC cell line (MOSEC/luc) was also previously generated in 2007 as described by Hung et al. [14]. TC-1 cells were obtained by cotransformation of primary C57BL/6 mouse lung epithelial cells with HPV-16 E6 and E7 and an activated ras oncogene as previously described [20]. All cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2 nM glutamine, 1 mM sodium pyruvate, 20 mM HEPES, 50 μM β-mercaptoethanol, 100 IUml−1 penicillin, 100 μg ml−1 streptomycin, and 10% fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA, USA) at 37°C with 5% CO2.
Reagents
Polyriboinosinic:polyribocytidylic acid (poly(I:C)) was purchased from Sigma (Sigma–Aldrich®, USA) and diluted with PBS into 1 mg/ml stack before operating. In vivo-jetPEI™, a linear polyethylenimine(PEI), was purchased from Polyplus transfection (Polyplus transfection, CA, USA) and mixed with poly(I:C) at N/P = 8 as the manufacture suggested. For the PE-labeled-poly(I:C) used in the poly(I:C) transfection experiment, poly(I:C) was labeled using the Label IT® Nucleic Acid Labeling Kit from Mirus (Mirus Bio., WI, USA) based on the manufacture’s instructions.
In vitro tumor killing assay
Groups of C57BL/6 mice (3 per group) were i.p. injected with high-dose poly(I:C) (20 μg/mouse), low-dose poly(I:C) (1 μg/mouse), low-dose poly(I:C) in combination with PEI (0.16 μl/mouse, N/P = 8), PEI(0.16 μl/mouse), and equal amount of PBS (200 μl), killed 3 days before. The mice were euthanized by CO2 asphyxiation, their abdomens were wiped with 70% alcohol, 8–9 ml of cold sterile PBS was injected into the peritoneal cavity, and peritoneal exudate cells (PECs) were harvested through syringe needle dripping. MOSEC/luc cells were seeded into 96-well microplate at 1 × 104/well in complete medium 1 day before, and 5 × 105 PECs were added in triplicates at titrated effecter to target ratios. The plate was imaged for bioluminescence activity (Xenogen IVIS 200) after culturing for 16-h in incubator. For parallel experiments, some mice also received in vivo depletion of peritoneal NK cell as indicated. Those mice were i.p. injected with 200 μl NK1.1mAb (PK136, Harlan bioproducts, WI, USA) every other day, 4 days before poly(I:C) and PEI treatment.
In vivo MOSEC/luc tumor treatment
C57BL/6 mice (5 per group) were injected with 2 × 105/mouse of MOSEC/luc i.p. to induce advanced ovarian cancer (Chang et al., 2007). Four days after tumor inoculation, mice were i.p. injected with high-dose poly(I:C) (20 μg/mouse), low-dose poly(I:C) (1 μg/mouse), low-dose poly(I:C) in combination with PEI (0.16 μl/mouse, N/P = 8), PEI(0.16 μl/mouse), or equal amount of PBS (200 μl). The injections were repeated every 5 days until the mice died. Mice were imaged with IVIS-200 system (Xenogen, Alameda, USA) at baseline and every week to monitor the therapeutic effects. The bioluminescence signals were analyzed using Living Image software (Xenogen).
Antibodies and flow cytometry analysis
Groups of C57BL/6 mice (3 per group) were treated with poly(I:C) and PEI as described earlier. PECs were harvested either 16 h or 3 days after treatment and washed once in FACScan buffer and treated with anti-mouse CD16/CD32 (2.4G2) (BD Pharmingen, CA, USA) before staining with surface antibodies. NK1.1, CD3, CD11c, I-Ab (MHC class II molecule) and FITC-conjugated antibodies were purchased from BD Phamingen (San Jose, CA, USA), and PE-conjugated CD69, FITC-conjugated CD11b were purchased from eBioscience (San Diego, CA, USA). To check the level of MHC class I molecules on MOSEC/luc cell line, MOSCEC/luc were trypsinized, washed, and stained with florescent-conjugated H-2Kb, H-2Db antibodies (BD Pharmingen, CA, USA). For intracellular staining, PECs were washed once in FACScan buffer and treated with anti-mouse CD16/CD32 (2.4G2) before stained with FITC-conjugated CD11b. Cells were subjected to intracellular cytokine staining using the Cytofix/Cytoperm kit according to the manufacturer’s instructions (BD Pharmingen). Intracellular IL-12 was stained with PE-conjugated anti-mouse IL-12 (p40/p70) (BD Pharmingen, CA, USA) to identify the immune response and cytokine levels. Flow cytometry analysis was performed using FACSCalibur with CELLQuest software (BD Biosciences, MountainView, CA, USA).
In vivo antibody depletion experiments
C57BL/6 mice (5 per group) were inoculated with MOSEC/luc cells 2 × 105/mouse at baseline and were treated with PBS or combined treatment (poly(I:C) + PEI) 3 days later and repeated in a 5-day-interval. For in vivo depletion of peritoneal NK cells, mice were i.p. injected with 200 μl NK1.1mAb (PK136, Harlan bioproducts, WI, USA) every other day since one day before injecting MOSEC/luc cells to maintain a low NK cell count through the period. Depletion efficiency was checked by flow cytometry with around 90% depletion of target cells.
Poly(I:C) transfection experiment
Groups of C57BL/6 mice (2 per group) were i.p. injected with PBS, PE-labeled-poly(I:C) (1 μg/mouse) alone, or in combination with PEI (0.16 μl/mouse, N/P = 8). PECs were harvested 16 h after treatment and prepared for surface marker staining for flow cytometry analysis as previously described.
ELISA assay
To determine the level of IL-12, cytokine levels in the supernatant of 5 × 105 PECs harvested one day after treatment were used according to the manufacturer’s instructions (eBioscience, CA, USA).
Adoptive transfer experiment
Naïve C57BL/6 mice were treated with PBS or combined treatment (poly(I:C) + PEI) on day 0, and 200 μl NK1.1mAb (PK136, Harlan Bioproducts, WI, USA) was i.p. injected 3 times every other day, 4 days in advance. On day 1, PECs were harvested and CD11b cells were isolated using CD11b MicroBeads kit from MACS (Miltenyi Biotec.Inc., Auburn, CA, USA); 5 × 105 CD11b cells were then transferred adoptively i.p. into a new group of C57BL/6 mice. PBS and poly(I:C) + PEI were also treated in parallel as negative and positive controls. To assess the effect of adoptively transferred CD11b cells, PECs were collected and NK cell activity and numbers were determined using flow cytometry 3 days later.
Results
Mice vaccinated with poly(I:C) in combination with polyethylenimine generate the best therapeutic antitumor effects against MOSEC tumors
In order to determine whether tumor-bearing mice treated with poly(I:C) in combination with polyethylenimine (PEI) can generate therapeutic antitumor effects, we performed in vivo tumor treatment experiments. C57BL/6 mice (5 per group) were challenged intraperitoneally with MOSEC/luc tumor cells. Four days later, mice were injected i.p. with a low-dose poly(I:C) (1 μg/mouse), a high-dose poly(I:C) (20 μg/mouse), a low-dose poly(I:C) in combination with PEI, PEI alone, or equal amount of PBS repeatedly at 5-day intervals. Tumor growth was monitored by luminescence imaging every week to monitor the therapeutic effects. As shown in Fig. 1, mice vaccinated with poly(I:C) in combination with PEI demonstrated significantly reduced luminescence intensity over time and prolonged survival compared with mice vaccinated with poly(I:C) alone or PEI alone (P < 0.05). We also observed that peritoneal cells isolated from mice treated with poly(I:C) in combination with PEI were capable of killing MOSEC tumor cells in vitro (see Supplementary Figure 1). Thus, our data indicate that mice vaccinated with poly(I:C) in combination with PEI generate the best therapeutic antitumor effects against MOSEC tumors.
Fig. 1.
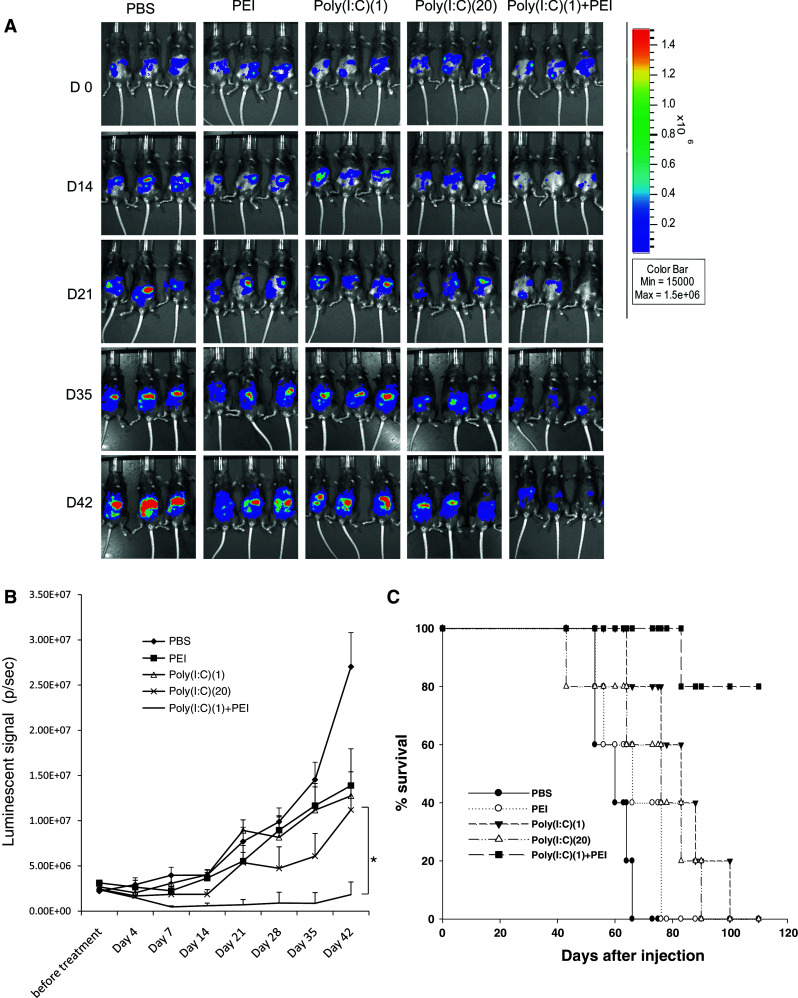
In vivo tumor treatment experiments. C57BL/6 mice (5 per group) were injected with 2 × 105/mouse of MOSEC/luc intraperitoneally on D0. Four days later, mice were injected i.p. with a low-dose poly(I:C) (1 μg/mouse), a high-dose poly(I:C) (20 μg/mouse), a low-dose poly(I:C) in combination with PEI (0.16 μl/mouse, N/P = 8), PEI(0.16 μl/mouse), or equal amount of PBS (200 μl). The injections were repeated every 5 days until the mice died. Mice were imaged with IVIS-200 system at baseline and every week to monitor the therapeutic effects. a Bioluminescence images of the representative MOSEC/luc tumor-bearing mice at different time points after tumor inoculation. b Linear graph depicting the luminescent intensity of tumor-bearing mice treated with the different treatments. Data are represented as mean ± SD. c Kaplan–Meier survival analysis of MOSEC/luc tumor-bearing mice treated with the different treatments (*indicates P < 0.05). Data shown are representative of two experiments conducted
Vaccination with poly(I:C) in combination with polyethylenimine generates significant numbers and activation of NK1.1+ cells in vaccinated mice
We observed that MOSEC tumor cells have a significantly lower level of MHC class I expression compared with other tumor cells such as TC-1 tumors (see Supplementary Figure 2). Since NK1.1+ cells are known to be an important immune effector cells for the control of tumors with low MHC class I expression, we characterized the number of total as well as activated (CD69+) NK1.1+ cells in peritoneal exudate cells (PECs) derived from mice vaccinated with poly(I:C) and PEI by flow cytometry analysis. As shown in Fig. 2a, b, we observed that mice vaccinated with poly(I:C) and PEI generated significantly higher percentage of NK1.1+ cells among the PECs compared with mice vaccinated with poly(I:C) alone or PEI alone (P < 0.05). Furthermore, mice vaccinated with poly(I:C) and PEI generated significantly higher percentage of activated NK cells among the PECs compared with mice vaccinated with poly(I:C) alone or PEI alone (P < 0.05) (Fig. 2c, d). In order to eliminate the concern for contamination of poly(I:C) with LPS, we characterized the number and activation of NK cells generated in mice treated with endotoxin-free poly(I:C) and observed similar results (Supplementary Figure 2). Thus, our data indicate that vaccination with poly(I:C) in combination with PEI generates significant numbers and activation of NK cells in vaccinated mice.
Fig. 2.
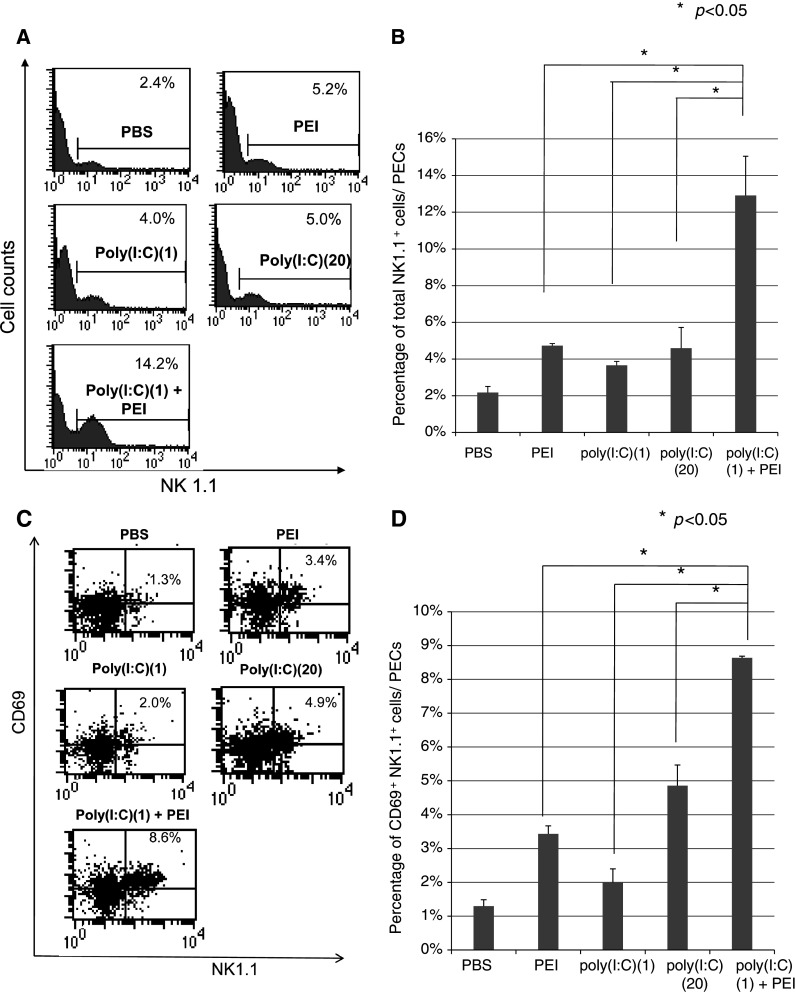
Flow cytometry analysis to determine the number of NK1.1+ cells derived from PECs in vaccinated mice. C57BL/6 mice were injected i.p. with a low-dose poly(I:C) (1 μg/mouse), a high-dose poly(I:C) (20 μg/mouse), a low-dose poly(I:C) in combination with PEI (0.16 μl/mouse, N/P = 8), PEI(0.16 μl/mouse), or equal amount of PBS (200 μl). Peritoneal exudate cells (PECs) were then harvested 3 days later and stained and analyzed by flow cytometry analysis to determine the number of total NK1.1+ cells and CD69 + NK1.1+ cells. a Representative flow cytometry data demonstrating the percentage of NK1.1+ cells in mice immunized with the various regimens. b Bar graph depicting the percentage of total NK1.1+ cells among all PECs (mean ± SD). c Representative flow cytometry data demonstrating the percentage of activated CD69 + NK1.1+ cells in mice immunized with the various regimens (right upper quadrant). d Bar graph depicting the percentage of CD69 + NK1.1+ cells among all PECs (mean ± SD). *indicates P < 0.05. Data shown are representative of two experiments conducted
NK1.1+ cells play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI
In order to confirm the role of NK cells in the antitumor effects caused by treatment with poly(I:C) and PEI, we carried out NK cell depletion experiments. C57BL/6 mice were inoculated with MOSEC/luc cells. Three days later, mice were treated with PBS or combined treatment (poly(I:C) + PEI) repeatedly with 5-day-intervals. One group of mice was depleted of NK cells using NK1.1mAb. Tumor growth was monitored by luminescence imaging. As shown in Fig. 3, mice treated with poly(I:C) and PEI with NK cell depletion demonstrated significantly higher tumor load compared with mice treated with poly(I:C) and PEI without NK cell depletion (P < 0.05). We also carried out in vitro tumor killing experiments using PECs from mice depleted of NK1.1+ cells. We demonstrated approx 90% efficiency of depletion of NK1.1+ cells, using the NK1.1mAb (see Supplementary Figure 3B). We observed that NK1.1 depletion led to a significant reduction in MOSEC tumor cell killing by PECs isolated from mice treated with poly(I:C) and PEI compared with mice treated with PECs without NK depletion (see Supplementary Figure 3C). Thus, our data indicate that NK cells play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI. However, other factors may also potentially contribute to the observed effect.
Fig. 3.
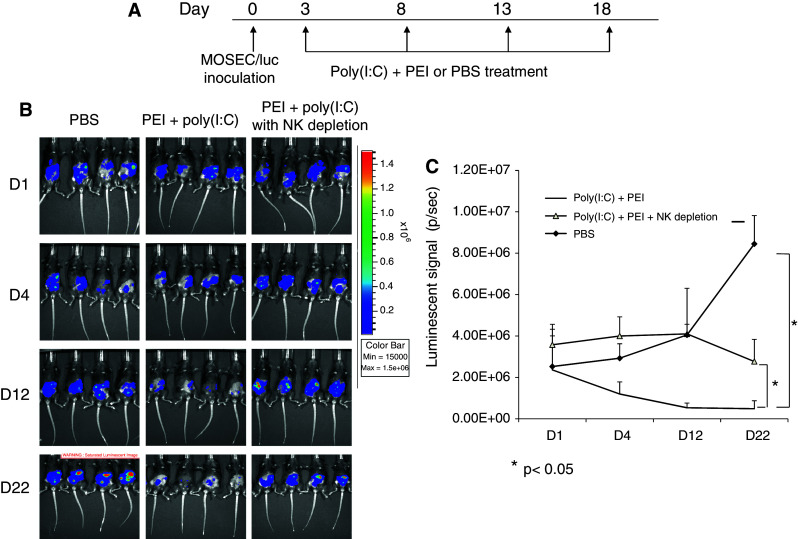
In vivo antibody depletion experiments. a Schematic diagram to depict the treatment regimen in MOSEC/luc tumor-bearing mice. C57BL/6 mice (5 per group) were inoculated with MOSEC/luc cells 2 × 105/mouse on D0. Three days later, mice were treated with PBS or combined treatment (poly(I:C) + PEI) repeatedly with 5-day-intervals. Mice were imaged with IVIS-200 system at regular intervals. b Bioluminescence images of the representative MOSEC/luc challenged mice treated with poly(I:C) + PEI with or without NK1.1 depletion on days 1, 4, 12, and 22 after tumor inoculation. c Linear graph depicting the luminescent intensity of tumor-bearing mice treated with poly(I:C) + PEI with or without NK1.1 depletion. Data are represented as mean ± SD. *indicates P < 0.05. Data shown are representative of two experiments conducted
Co-administration of PEI with poly(I:C) leads to increased uptake of poly(I:C) by PECs, especially by CD11b+ cells
In order to investigate the other cells that may play a role in the effect of poly(I:C) and PEI on tumor cells, we fluorescently labeled poly(I:C) and characterized the transfection of poly(I:C) into various different cell types by flow cytometry analysis. Groups of C57BL/6 mice (2 per group) were i.p. injected with PBS, PE-labeled-poly(I:C) alone or in combination with PEI. PECs were harvested 16 h after treatment and stained for NK1.1, CD11b, CD11c and CD3 surface markers and analyzed by flow cytometry analysis. As shown in Fig. 4, PECs isolated from mice treated with poly(I:C) and PEI showed increased uptake of poly(I:C) into CD11b+ cells (2.28% of all PECs and 5.07% of CD11b+ cells) compared with PECs from mice treated with poly(I:C) alone (P < 0.05). Thus, our data indicate that CD11b+ cells (macrophages) may play an important role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI.
Fig. 4.
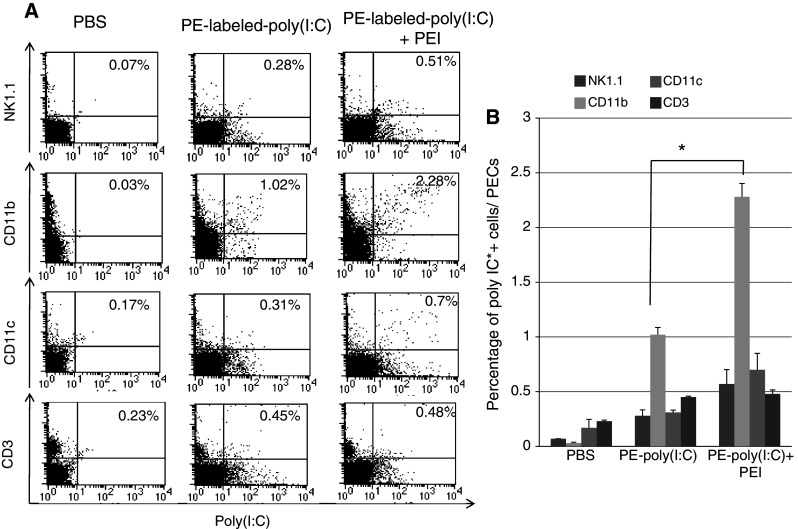
Flow cytometry analysis to determine the uptake of PE-labeled poly(I:C) into various types of peritoneal cells with or without PEI. Groups of C57BL/6 mice were i.p. injected with PBS, PE-labeled-poly(I:C) (1 μg/mouse) alone or in combination with PEI (0.16 μl/mouse, N/P = 8). PECs were harvested 16 h after treatment and stained for NK1.1, CD11b, CD11c and CD3 surface markers and analyzed by flow cytometry analysis. a Representative flow cytometry data demonstrating the percentage of NK1.1, CD11b, CD11c and CD3+ cells transfected with poly(I:C) among all PECs (right upper quadrant). Significant amounts of CD11b+ cells (5.07% of CD11b+ cells and 2.28% of all PECs) were positive for poly(I:C). b Bar graph depicting the percentage of poly(I:C)+ cells among all PECs. Data are represented as mean ± SD. *indicates P < 0.05. Data shown are representative of two experiments conducted
Treatment with poly(I:C) and PEI induces high MHC class II and IL-12 expression in CD11b+ cells derived from PECs from vaccinated mice
In order to further characterize the CD11b+ cells that were induced in mice treated with poly(I:C) and PEI, we analyzed the MHC class II and IL-12 expression on the CD11b+ cells from treated mice. Groups of C57BL/6 mice (2 per group) were treated with poly(I:C) and/or PEI or PBS as described in Fig. 1. Sixteen hours later, PECs were harvested and stained for MHC class II or intracellular IL-12 and analyzed by flow cytometry analysis. As shown in Fig. 5, CD11b+ cells isolated from mice treated with poly(I:C) and PEI demonstrated significantly higher levels of MHC class II and IL-12 expression as well as the IL-12p40 subunit expression compared with cells from mice treated with poly(I:C) alone or PEI alone. Thus, our data indicate that treatment with poly(I:C) and PEI induces high IL-12 and MHC class II expression in the CD11b+ macrophages indicating the “classically” activated phenotype of macrophage activation (M1 phenotype) [9, 13].
Fig. 5.
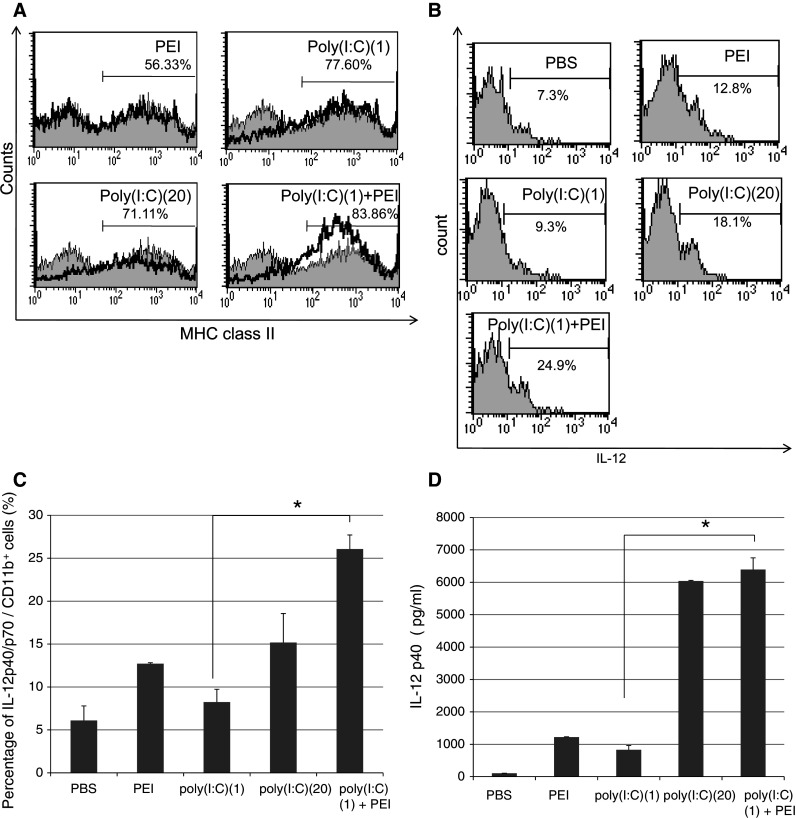
Flow cytometry analysis to determine the level of MHC class II and IL-12 expression among the CD11b+ peritoneal cells from treated mice. Groups of C57BL/6 mice were treated with poly(I:C) and/or PEI or PBS as described in Fig. 1. Sixteen hours later, PECS were harvested and stained with CD11b and I-Ab (MHC class II molecule in C57BL/6 mice) or intracellular IL-12 and analyzed by flow cytometry analysis. Shaded area indicates PBS-treated controls. a Representative flow cytometry data demonstrating the percentage of MHC class II+ cells in CD11b+ PECs. b Representative flow cytometry data demonstrating the percentage of IL-12+ cells in CD11b+ PECs. c Bar graph depicting the percentage of IL-12+ cells among CD11b+ PECs. d ELISA assay depicting the IL-12 p40 levels in the supernatant of 5 × 105 PECs harvested 1 day after treatment with the various treatments. All data above were presented as mean ± SD. *indicates P < 0.05. Data shown are representative of two experiments conducted
Adoptive transfer of NK1.1-CD11b+ cells from mice treated with poly(I:C) and PEI leads to increased number of total NK1.1+ cells and CD69 + NK1.1+ cells in the PECs of recipient mice
In order to further investigate the effects of treatment with poly(I:C) and PEI on CD11b+ cells, we carried out an adoptive transfer experiment. Naïve C57BL/6 mice were treated with PBS or poly(I:C) and PEI on day 0 and depleted of NK cells using the regimen as shown in Fig. 6a. On day 1, PECs were harvested and CD11b+ cells were isolated; 5 × 105 isolated cells were then transferred adoptively i.p. into a new group of C57BL/6 mice. Three days later, PECs from the recipient mice were collected and NK cell activity and numbers were determined using flow cytometry analysis. Figure 6b demonstrates the efficiency of isolation of the NK1.1-CD11b+ cells. As shown in Fig. 6c, d, PECs isolated from recipient mice that received adoptive transfer of CD11b+ cells from donor mice treated with poly(I:C) and PEI demonstrated a significantly percentage of total NK cells and CD69 + NK cells compared with PECs isolated from recipient mice that received adoptive transfer of CD11b+ cells from donor mice treated with PBS. Thus, our data indicate that adoptive transfer of CD11b+ cells from mice treated with poly(I:C) and PEI leads to increased proliferation and activation of NK cells from recipient mice.
Fig. 6.

Characterization of the number of CD69 + NK1.1+ cells in the PECs of mice receiving adoptive transfer of CD11b+ cells from mice treated with or without poly(I:C) and PEI. a Schematic diagram to depict the depletion of NK1.1, treatment regimen and adoptive transfer of CD11b+ cells in mice. Naïve C57BL/6 mice were treated with PBS or combined treatment (poly(I:C)+ PEI) on day 0. Mice were depleted of NK1.1 cells using PK136 Ab injected i.p. 3 times every other day, 4 days before treatment. On day 1 after treatment, PECs were harvested and CD11b+ cells were isolated; 5 × 105 of isolated CD11b+ cells were then adoptively transferred i.p. into a new group of naïve C57BL/6 mice. Three days later, PECs from the recipient mice were collected and analyzed for the number of total NK1.1+ cells and CD69 + NK1.1+ cells using flow cytometry. b Representative flow cytometry data to characterize the isolated NK1.1-CD11b+ in the PECs. c Representative flow cytometry data demonstrating the percentage of CD69 + NK1.1+ cells following adoptive transfer of CD11b+ cells from mice treated with PBS (upper panel) or poly(I:C) + PEI (lower panel). d Bar graph depicting the percentage of total NK1.1+ cells and CD69 + NK1.1+ cells among all PECs following adoptive transfer of CD11b+ cells from mice treated with poly(I:C) + PEI or PBS. *indicates P < 0.05. Data shown are representative of two experiments conducted
Discussion
In the current study, we observed that mice vaccinated with poly(I:C) in combination with PEI generate significant therapeutic antitumor effects against MOSEC tumor challenge. Furthermore, NK cells play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI. Treatment with poly(I:C) and PEI was found to induce the M1 activation phenotype (high MHC class II and IL-12 expression) in CD11b+ macrophages. Finally, adoptive transfer of CD11b+ macrophages from mice treated with poly(I:C) and PEI was found to lead to increased proliferation and activation of NK cells from recipient mice.
In our study, we found that co-administration of PEI can lead to increased uptake of poly(I:C) and result in improved antitumor effects against murine ovarian tumors. This is consistent with our previous study that used an antimicrobial polypeptide LL-37 to enhance the immunostimulatory effects of the TLR9 ligand, CpG-ODN by increasing the uptake of CpG-ODN into the immune cells, thus enhancing the antitumor effects against ovarian cancer [6]. We found that treatment with the combination of CpG-ODN and LL-37 enhanced proliferation and activation of NK cells in the peritoneal cavity. Both these studies have employed a strategy to improve the uptake of the intracellular TLR ligands 3 and 9, thus leading to the activation of NK cells. The success of these two studies indicates that the strategy to enhance the uptake of ligands for intracellular TLRs could be potentially used as a general approach for enhancing the immune responses to control tumors and/or infectious diseases.
IL-12 is known to be a potent NK cell activator [5, 19, 27] and is composed of p35 and p40 subunits. It has been shown that the heterodimer IL-12 p70 can induce IFN-gamma and TNF production while the p35 or the p40 subunit alone cannot [5, 19, 27]. However, the level of IL-12 p40 protein appears to correlate with the level of biologically active IL-12 [5, 19]. Thus, in Fig. 5b–d, we show IL-12 p70 intracellular staining together with IL-12 p40 ELISA to strengthen our claim. Furthermore, the effect of IL-12 on NK cells can also be observed in mice similar to that in human [5]. In addition, macrophages are also known to be a major IL-12 source [8]. Thus, our data indicate that treatment with poly(I:C) and PEI induces high IL-12 and MHC class II expression in the CD11b+ macrophages indicating the “classically” activated phenotype of macrophage activation (M1 phenotype), resulting in NK cell activation.
We observed that treatment with poly(I:C) and PEI led to the high expression of MHC class II and IL-12 (M1 phenotype) in CD11b+ macrophages in the peritoneal cavity (Fig. 5). Macrophages are an extremely heterogeneous lineage displaying a range of both pro- and anti-inflammatory phenotypes, the “classically” activated (or M1) macrophages, which are considered pro-inflammatory, and the other (M2) macrophages, which are considered anti-inflammatory (for reviews see [10, 11, 21]). The M1 phenotype is characterized by high IL-12 and MHC class II and inducible nitric oxide synthase (iNOS) expression [9, 13], whereas the M2 phenotype is characterized by elevated expression of anti-inflammatory cytokines such as IL-10. In general, M1 participate as inducer and effecter cells in polarized Th1 responses and mediate resistance against intracellular parasite and tumor. M2, on the other hand, participate in polarized Th2 reactions, promote killing and encapsulation of parasites [23]. Thus, the co-administration of poly(I:C) with PEI leads to the induction of macrophages with pro-inflammatory phenotype, resulting in potent antitumor effects against ovarian tumors.
In our study, we found that co-administration of PEI with poly(I:C) mainly led to increased uptake of poly(I:C) by CD11b+ macrophages. However, we also observed a slight increase in the uptake of poly(I:C) by CD11c+ dendritic cells (see Fig. 4). Although the current study focuses on the effect of poly(I:C) on macrophages, we cannot exclude the contribution of CD11c+ cells to the antitumor effect. It has been shown that poly(I:C) can lead to an increased number of IFN-producing killer dendritic cells (IKDCs) in tumor-bearing mice [17]. Furthermore, PEI has also been shown to lead to the reprogramming of tumor-associated dendritic cells from immunosuppressive cells to efficient antigen-presenting cells via the TLR5 pathway [7]. Thus, CD11c+ dendritic cells may also play a role in the observed antitumor effects generated by co-administration of poly(I:C) with PEI.
It has been shown that NK cells are particularly effective in killing tumors, which have a low MHC class I expression. In our study, we have shown that the MOSEC tumor cell line has a low MHC class I expression and is thus susceptible to NK cell-mediated killing (see Supplementary Figure 3). Thus, our approach will be particularly effective in ovarian cancers that demonstrate low MHC class I expression levels. For clinical translation, it is important to address concerns regarding the potential toxicity associated with administration of poly(I:C) and PEI. In this regard, both poly(I:C) [24, 26] and PEI [4] have been previously used in several clinical trials and have been proven to be safe. Thus, the current strategy employing poly(I:C) and PEI has significant potential for future clinical translation in the treatment of tumors with low MHC class I expression.
Our study indicates that NK1.1+ cells and macrophages play a significant role in the antitumor effects generated by treatment with poly(I:C) in combination with PEI. However, we cannot exclude the possibility of the direct effect of poly(I:C) and PEI on tumor cells. Nevertheless, our NK cell depletion experiments support the conclusion that the observed antitumor effects generated by treatment with poly(I:C) and PEI are mainly mediated by NK cells (Fig. 3; Supplementary Figure 4). Furthermore, our data in Supplementary Figure 1 show that peritoneal cells, including NK cells and macrophages isolated from mice treated with poly(I:C) in combination with PEI, were capable of killing tumor cells without direct treatment on tumor cells. Thus, our data suggest that intraperitoneal administration of poly(I:C) with PEI leads to significant antitumor immunity against tumors mainly mediated by NK cells and/or macrophages.
In summary, our data indicate that PEI can potentially be used to improve the uptake of poly(I:C) by CD11b+ macrophages, leading to the activation of NK cells and the control of murine ovarian tumors. Our study serves as an important foundation for future clinical translation in patients with ovarian cancer. Furthermore, this approach may potentially be used for the control of other cancers and/or infectious diseases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the American Cancer Society (C.F. Hung).
References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/S0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 4.Anwer K, Barnes MN, Fewell J, Lewis DH, Alvarez RD. Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther. 2010;17:360–369. doi: 10.1038/gt.2009.159. [DOI] [PubMed] [Google Scholar]
- 5.Chan SH, Perussia B, Gupta JW, Kobayashi M, Pospisil M, Young HA, et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173:869–879. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuang CM, Monie A, Wu A, Mao CP, Hung CF. Treatment with LL-37 peptide enhances antitumor effects induced by CpG oligodeoxynucleotides against ovarian cancer. Hum Gene Ther. 2009;20:303–313. doi: 10.1089/hum.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest. 2009;119:2231–2244. doi: 10.1172/JCI37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, et al. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J Exp Med. 1992;176:1387–1398. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J Exp Med. 2008;205:1269–1276. doi: 10.1084/jem.20080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 11.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 12.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 13.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hung CF, Tsai YC, He L, Wu TC. Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8+ T cells. Gene Ther. 2007;14:921–929. doi: 10.1038/sj.gt.3302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 16.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 17.Jiang Q, Wei H, Tian Z. IFN-producing killer dendritic cells contribute to the inhibitory effect of poly I:C on the progression of murine melanoma. J Immunother. 2008;31:555–562. doi: 10.1097/CJI.0b013e31817d8e75. [DOI] [PubMed] [Google Scholar]
- 18.Kakimoto S, Hamada T, Komatsu Y, Takagi M, Tanabe T, Azuma H, et al. The conjugation of diphtheria toxin T domain to poly(ethylenimine) based vectors for enhanced endosomal escape during gene transfection. Biomaterials. 2009;30:402–408. doi: 10.1016/j.biomaterials.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–845. doi: 10.1084/jem.170.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin K-Y, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August T, Pardoll DM, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 21.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Noel W, Raes G, Hassanzadeh Ghassabeh G, De Baetselier P, Beschin A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004;20:126–133. doi: 10.1016/j.pt.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Robinson RA, DeVita VT, Levy HB, Baron S, Hubbard SP, Levine AS. A phase I-II trial of multiple-dose polyriboinosic-polyribocytidylic acid in patients with leukemia or solid tumors. J Natl Cancer Inst. 1976;57:599–602. doi: 10.1093/jnci/57.3.599. [DOI] [PubMed] [Google Scholar]
- 25.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 26.Salazar AM, Levy HB, Ondra S, Kende M, Scherokman B, Brown D, et al. Long-term treatment of malignant gliomas with intramuscularly administered polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose: an open pilot study. Neurosurgery. 1996;38:1096–1104. [PubMed] [Google Scholar]
- 27.Tripp CS, Wolf SF, Unanue ER. Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci USA. 1993;90:3725–3729. doi: 10.1073/pnas.90.8.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshihara C, Ito T, Shew CY, Koyama Y. Improvement of transcription- and transfection-efficiency by synthetic polyampholytes. Nucleic Acids Symp Ser (Oxf) 2008;52:713–714. doi: 10.1093/nass/nrn360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.