

Abstract
Palmitoylethanolamide (PEA) is an endogenous lipid modulator in animals and humans, and has been evaluated since the 1970s as an anti-inflammatory and analgesic drug in more than 30 clinical trials, in a total of ~6,000 patients. PEA is currently available worldwide as a nutraceutical in different formulations, with and without excipients. Here we describe the results of all clinical trials evaluating PEA’s efficacy and safety in nerve compression syndromes: sciatic pain and pain due to carpal tunnel syndrome, and review preclinical evidence in nerve impingement models. Both the pharmacological studies as well as the clinical trials supported PEA’s action as an analgesic compound. In total, eight clinical trials have been published in such entrapment syndromes, and 1,366 patients have been included in these trials. PEA proved to be effective and safe in nerve compression syndromes. In one pivotal, double blind, placebo controlled trial in 636 sciatic pain patients, the number needed to treat to reach 50% pain reduction compared to baseline was 1.5 after 3 weeks of treatment. Furthermore, no drug interactions or troublesome side effects have been described so far. Physicians are not always aware of PEA as a relevant and safe alternative to opioids and co-analgesics in the treatment of neuropathic pain. Especially since the often prescribed co-analgesic pregabaline has been proven to be ineffective in sciatic pain in a double blind enrichment trial, PEA should be considered as a new and safe treatment option for nerve compression syndromes.
Keywords: palmitoylethanolamide, sciatic, nerve compression, analgesics, PPAR alpha, anti-inflammatory agents, palmidrol
Video abstract
Introduction
Nerve compression syndromes have different clinical manifestations, dependent on the localization of the compressed nerve, but they share identical pathogenesis and pain symptomatology. We will describe the results of a number of pharmacological experiments and clinical trials conducted to evaluate the efficacy and safety of palmitoylethanolamide (PEA) in nerve compression syndromes, including sciatic pain. We will also review results of trials that were reported as abstracts or as poster presentations at neurological congresses, in order to disclose this information to a broader scientific public.
We will discuss in more detail the data from a pivotal, double blind placebo controlled, dose-finding study in 636 patients, to make the results known to a wider scientific community, as the results of this trial have so far only been published in the Spanish medical journal, Dolor, while the number needed to treat (NNT) analysis was presented by one of the authors in 2011 at the Italian National Congress of Anesthesiology, the SIAARTI.
Pathogenesis in nerve compression syndromes
Nerve pressure induces inflammation of nerves and nerve roots, neuritis and radiculitis. Subsequently, neuritis or radiculitis progresses into a more chronic pathological state due to the induction of a number of cascades of chemical inflammatory reactions.1 Inflammatory cells, such as activated mast cells, play an important role in nerve compression syndromes and are one of the sources of pro-inflammatory prostaglandins (PG)s and cytokines.2 These compounds trigger the synthesis of nitrogen monoxide, which acts as a strong vessel dilator.3 Step-by-step a chronic pathological situation emerges. In the next step, pro-inflammatory compounds such as metalloproteinases are produced, enzymes inducing connective tissue around nerves to expand and to become hyperactivated.4 These initial steps are followed by activation of a number of pro-inflammatory cells, such as mast cells, macrophages, fibroblasts, neutrophils, and Schwann cells.5 This leads to a release of various pro-inflammatory molecules: ATP, cytokines, CCL2, PGs, and various NGFs. This complex cascade is followed by abnormal and enhanced pain sensitivity in peripheral areas.6 This resulting inflammatory state leads to further massive cell migration, edema, erythema, pain, hyperalgesia, and allodynia. Glial cells, mast cells, and related non-neuronal cells contribute to the nerve pain experienced, such as in sciatic compression and carpal tunnel syndrome, due to excitation of neurons transmitting pain signals and by upregulating pain-circuits in the spinal medulla.7,8 In addition to this central windup mechanism, biological windup in the periphery occurs, even in the skin, including mast cell hyperactivation and NGF activation.9,10 Such central and peripheral activation related to mast cell, glia and astrocyte activation, as well as NGF-related inflammation cascades, can be inhibited by PEA.11 This provides the rationale for treating nerve compression syndromes such as sciatic pain and carpal tunnel syndrome with PEA.
PEA: anti-inflammatory and analgesic mechanism of action
PEA is an endogenous fatty acid amide, first described in 1957 and evaluated for the treatment of neuropathic and chronic pain since 1975.12,13 PEA regulates many physiological processes and is effective in a number of animal models such as for nerve compression pain, respiratory inflammation, neuroinflammation, neurotoxicity, and central nervous ischemia.14–16 Activation of inflammatory cascades and of non-neuronal cells, such as glial cells, astrocytes, and mast cells, contributes to the pathogenesis of chronic pain and to peripheral and central sensitization.17,18 PEA reduces mast cell migration and degranulation and reduces over-activation of astrocytes and glial cells.19–22 Both mast cells and glial cells shift under influence of PEA from activated immune cells to resting phenotypes.23 The mast cell however, is not the main pathogenetic factor, and PEA has a number of mechanisms of action, probably more important in nerve compression and impingement syndromes. On the molecular level PEA reduces the activity of the pro-inflammatory enzyme COX, as well as eNOS and iNOS.24 PEA has a number of other properties, related to its affinity for various receptors: the orphan cannabinoid receptors GPR55 and GPR119, the vanilloid receptor TRPV1 and the nuclear PPAR-α.25–28 The latter is clearly expressed in glial cells and neurons, and most probably PEA’s most important mechanism of action. These mechanisms of action of PEA are related to its analgesic and anti-inflammatory actions in nerve compression syndromes.
PEA: effective in models of nerve compression
Fatty acid ethanolamides such as PEA are endogenous agonists of PPAR-α and function as lipid messengers in the regulation of inflammation and chronic pain. Fatty acid ethanolamides are degraded by a number of enzymes, such as lysosomal amidases, fatty acid amide hydrolase (FAAH), and N-acylethanolamine acid amidase (NAAA).29 Sciatic nerve ligation or chemical irritation decreases PEA levels in sciatic nerve and the NAAA inhibitor ARN077 reverses these biochemical effects.30 PEA administered in such a sciatic pain model decreases inflammation and pain. The analgesic properties of PEA are dependent on the modulation of non-neuronal cells in a chronic constriction injury (CCI) model of neuropathic pain in mice.31 Three to 8 days after nerve injury, there was a substantial recruitment and activation of mast cells in the damaged nerve, as well as an upregulation of activated microglia found in the spinal cord. PEA delayed mast cell recruitment and inhibited mast cell degranulation, reduced microglia activation in the spinal cord, and inhibited the increase of NGF in the sciatic nerve and preserved the nerve from degeneration. PEA also significantly reduced the expression of COX-2 and iNOS in sciatic nerves and restored inflammation-induced reductions of the PPAR-α receptor in the dorsal root ganglia.27 PEA further significantly decreased neuropathic mechanical hyperalgesia after 7 days in a rat unilateral sciatic nerve ligation model.32
In the CCI model of the sciatic nerve in the rat, PEA levels were significantly decreased in the spinal cord and other parts of the central nervous system.33 In this CCI model, PEA could prevent pain threshold alterations after 14 days.34 PEA also reduced the presence of intraneural edema and macrophage infiltrates, and led to signs of neuro-regeneration: significantly thicker myelin sheath, increased axonal diameter, and increased number of nerve fibers. In PPAR-α-deficient mice PEA treatment did not lead to analgesia nor did PEA show any sign of neuro-repair. Based on these findings the authors concluded that PEA had disease modifying and neuro-protective properties.
URB597, an inhibitor of PEA hydrolysis by FAAH, was injected intrathecally in the CCI rat model of the sciatic nerve of the CCI rat model.35 URB597 elevated the levels of PEA, and fully inhibited thermal and tactile nociception. PEA derivatives also reduced hypersensitivity to noxious stimuli in the sciatic nerve injury model.36
Data from these pharmacological models support the use of PEA in nerve compression syndromes, and the active dose-range in most of the models was 10–30 mg/kg body weight.
PEA in nerve compression pain: efficacy and safety in patients
The vicious circle of inflammation and increasing pain can be halted by PEA, as has been shown in different animal models. PEA belongs to an entire new class of analgesic products, devoid of addiction potential, without central nervous system side effects, and without clear dose limiting side effects up to a dose of at least 100 mg/kg body weight.37 Drug interactions have so far not been documented, and its use has been described together with a number of different analgesics.
PEA has been explored in a number of clinical trials in sciatic pain and/or chronic back pain and in compression of the median nerve due to carpal tunnel syndrome.11 In total, 1,366 patients were entered in eight clinical trials. These trials have been published in peer reviewed journals, and some additional data were presented at the third International Congress on Neuropathic Pain in Athens (2010), and at special PEA-dedicated symposia at Perugia, Milan, and Naples (Italy). In order to check for completeness, we conducted a MEDLINE, EMBASE, and TRIP search with the word “palmitoylethanolamide”, and one of the following terms: “sciatic”, “hernia”, “nerve”, “compression”, “carpal”, “tarsal”, and “compression” from which we extracted all clinical trials, retrospective studies, and case reports; double reports, as far as being identified as such, were left out. Furthermore, we did a search in the clinical trial database of the US National Institutes of Health. The result of our search did not identify any new study.
One pivotal study was a double blind, placebo controlled study evaluating two different dosages in a 3-week treatment regime.38 The data were presented in the Spanish language in the Spanish anesthesiological journal Dolor in 2010, and the results of this trial as well as an additional NNT analysis were discussed further at the Italian National Congress of Anesthesiology, the SIAARTI, in 2011 by Keppel Hesselink.39 As these clinical data are a key part of PEA’s clinical evidence in nerve compression syndromes we will start presenting and discussing these data in more detail.
Results from a pivotal, randomized double blind, placebo-controlled trial with PEA
In this pivotal trial, two different doses of PEA were compared to placebo, 300 and 600 mg daily. Six hundred thirty-six patients suffering from pain due to radicular compression of the sciatic nerve, between 18 and 75 years with a pain score of ≥5 on the visual analog scale (VAS) were included.38 All patients were allowed to continue their usual treatments. Primary endpoint was the VAS score for pain intensity and a secondary endpoint was the Roland-Morris disability questionnaire (RDQ) to evaluate the quality of life. Treatment period was 3 weeks.
At the final follow-up at day 21, all the patients underwent a clinical investigation, laboratory tests, evaluation of the pain intensity, and quality of life evaluation with the RDQ. Patients and physician gave a subjective judgment about the treatment efficacy. This evaluation was carried out using a 4-point scale, with the following descriptions: excellent, good, some, and no efficacy, respectively. Safety and tolerability were evaluated by means of a full physical investigation, and standard hematological, biochemical, and urine tests were carried out at baseline and the end of treatment period.
Pre/post evaluation was done with the VAS and RDQ between baseline and day 21, using the analysis of variance according to the intention-to-treat last observation carried forward method. The Scheffé test was used for the multiple comparisons between groups, and the chi-squared test was used to analyze the subjective evaluation, considering P<0.05 as statistically significant.
Results of the study: 636 patients entered the study: 336 males (52.8%) and 300 females (47.2%), between 19 and 72 years of age (mean 42.8±11.2 years). All treatment groups were homogeneous with respect to age, sex, height, weight, diagnoses, and severity of pain. None of the dropouts were due to adverse events, though mainly due to absence of efficacy.
At the end of the treatment period the reduction in pain was significantly different between the three groups (P<0.001). The high dose PEA was significantly more effective compared to the low dose (P<0.05); while both doses of PEA were significantly more effective than placebo (P<0.05). The mean decrease on the VAS was largest in the high dose group: a reduction from 7.1 to 2.1, which is more than 50% pain reduction; this is in general considered as a robust clinical response. In the placebo arm the pain VAS score decreased from 6.6 to 4.6 (Table 1).
Table 1.
Mean score values ± SD obtained for the VAS and RDQ at T0 and T21
| Placebo | 300 mg | 600 mg | P-value | |
|---|---|---|---|---|
| Number of patients | 208 | 210 | 214 | |
| Dropouts | 12 | 4 | 1 | |
| VAS T0 (SD) | 6.6 (1.7) | 6.5 (1.9) | 7.1 (1.8) | |
| VAS T21 (SD) | 4.6 (1.7) | 3.6 (1.8) | 2.1 (1.7) | |
| VAS difference (SD) | 2.0 (1.9) | 2.9 (2.3) | 5.0 (2.5) | <0.05* |
| RDQ T0 (SD) | 11.9 (3.8) | 11.7 (4.0) | 12.7 (4.1) | |
| RDQ T21 (SD) | 8.9 (3.2) | 6.7 (3.5) | 3.5 (2.7) | |
| VAS difference (SD) | 3.0 (3.4) | 5.0 (3.3) | 9.2 (4.2) | <0.05* |
Note:
Comparison among groups, calculated with Scheffe test.
Abbreviations: SD, standard deviation; VAS, visual analog scale; RDQ, Roland-Morris disability questionnaire.
The change in quality of life measured with the RDQ differed between groups (P<0.001). Again, the improvement was significantly greater in the high dose group compared to the low dose group and placebo. The improvement in both active treatment arms was superior to placebo (P<0.05).
In a separate post hoc (non-protocol defined) analysis, the NNT to reach a 50% reduction in pain compared to baseline was established. In Figure 1 the NNT for week 2 and 3 for both dose arms are depicted.39
Figure 1.
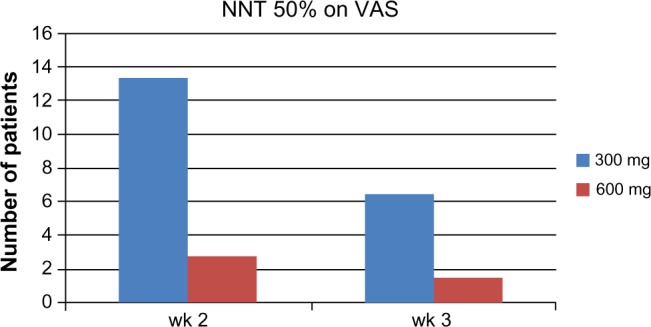
NNT of PEA to reach 50% reduction of pain.
Abbreviations: PEA, palmitoylethanolamide; VAS, visual analog scale; NNT, number needed to treat; wk, week.
The NNT for the PEA 300 mg group was 6.5 and for the PEA 600 mg group was 1.5 after 3 weeks treatment. The NNT was calculated by subtracting the placebo response rate from the PEA 600 mg response. PEA therefore can be considered as a potent analgesic in the league table of neuropathic analgesics.
Clinical trials of PEA in different nerve compression syndromes
Canteri et al40 reported the results of a placebo-controlled, double blind, randomized study in 111 patients suffering from lumbosciatic pain, following the same protocol as Guida et al.38 All patients were allowed to continue their established medication; 53 patients were free from analgesics. After 3 weeks there was a significant decrease of pain in which the high dose (600 mg PEA/day) was most effective (P,0.03) compared to the low dose and placebo group. The use of co-analgesics did not change the outcome. No relevant side effects or drug interactions were reported.
Gatti et al performed an observational study with 610 patients suffering from chronic pain states, among which 331 patients suffered from sciatic pain.41 Other patients were suffering from arthritic pains (n=54), post-herpetic neuralgia (n=44), diabetic neuropathic pain (n=32), pain after failed back surgery (n=76), oncological pain (n=22), and mixed pain states (n=51). Mean VAS was 6.5 at baseline. Patients received 600 mg, twice daily for 3 weeks followed by single daily dosing for 4 weeks. PEA was added to established analgesic therapies, or as single therapy. In total 46 patients dropped out (7.5%). PEA decreased the mean pain on the VAS from 6.4±1.4 to 2.5±1.3 in the patients who completed the study. In patients without concomitant analgesics, PEA was equally efficacious in reducing chronic pain. Neither relevant side effects nor drug-drug interactions were observed.
Desio reported the effects of PEA in an open study in 20 non-responders to previous analgesic pharmacotherapy with sciatic pain, low back pain, hernia, and vertebral stenosis.42 Treatment regime of 30 days was oxycodone 5 mg once daily during the first 5 days; where after 5 mg twice daily; together with PEA 600 mg twice daily. This resulted in a decrease of pain from mean VAS 7.2 to VAS 2.5 at day 30 (P<0.001). No relevant side effects and no drug-drug interactions were reported.
Palomba et al described the effects of PEA in 81 neuropathic low back pain patients in an open group comparison study. One group (n=41) received PEA on top of standard analgesics (pregabalin, gabapentin, amitriptyline, duloxetine) and the other group (n=40) received just standard analgesics.43 For the first 21 days, the PEA group received 600 mg PEA twice daily, thereafter 600 mg PEA once daily. At endpoint, after 51 days treatment, PEA-treated patients had less pain compared to standard care patients (P<0.05); no side effects or drug-drug interactions were observed.
Domenguez et al randomly divided 85 patients suffering from lumbosciatic pain in to two groups, usual care and PEA 300 mg twice daily or usual care.44 The usual care group without PEA had a significant pain reduction of 2.69 compared to baseline (P>0.05), and the PEA group had a pain reduction of 3.85 compared to baseline (P>0.05). No side effects were reported.
Assini et al investigated the effect of 1,200 mg PEA/day in diabetic patients with carpal tunnel syndrome (n=25) and compared the effect with a control group (n=25).45 Results: significant difference in reduction of pain at endpoint between treatment with PEA and control group (P<0.0001). All neurophysiological parameters improved. No side effects were reported.
Congliaro et al treated 26 patients with carpal tunnel syndrome in a group-controlled study of patients suffering from carpal tunnel syndrome, with clear neurophysiological abnormalities and pain.46 The study had three arms: control (standard care), PEA 600 mg/day, PEA 1,200 mg/day during 30 days. This resulted in significant improvement in neurophysiological parameters, such as distal motor latency after PEA treatment, and the higher dose was more effective. No side effects were reported. In Table 2 we have summarized the number of patients entered in these clinical trials.
Table 2.
Eight clinical trials of PEA in nerve compression syndromes, in a total of 1,366 patients
| Publication | Number of patients | Indication |
|---|---|---|
| Guida et al, 201038 | 636 | Sciatic pain |
| Gatti et al, 201241 | 331 | Sciatic pain |
| Gatti et al, 201241 | 76 | Lumbago |
| Desio, 201142 | 20 | Sciatic pain, lumbar stenosis |
| Canteri et al, 201040 | 111 | Sciatic pain |
| Palomba et al, 201043 | 41 | Sciatic pain |
| Dominguez et al, 201044 | 85 | Sciatic pain |
| Assini et al, 201045 | 40 | Carpal tunnel syndrome |
| Conigliaro et al, 201146 | 26 | Carpal tunnel syndrome |
Abbreviation: PEA, palmitoylethanolamide.
Conclusion
PEA has been tested in a variety of animal models for nerve compression and has been evaluated in eight different clinical trials in total, in 1,366 patients with nerve compression syndromes. Both the preclinical as well as the clinical results point in the same direction: PEA acts as a safe analgesic compound in nerve compression. Its safety and efficacy profile supports the clinical use of PEA in compression syndromes such as sciatic pain and carpal tunnel syndrome. PEA is easy to administer. The NNT of PEA for sciatic pain to reach 50% pain reduction is 1.5 and the number needed to harm is at least in the hundreds, but for the time being not calculable due to the absence of serious and troublesome side effects leading to dropouts in clinical trials. The risk–benefit balance of PEA therefore favors its inclusion in the therapeutic armamentarium of chronic pain. PEA can be administered both as a stand-alone analgesic as well as part of a multimodal therapy regime.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mulleman D, Mammou S, Griffoul I, Watier H, Goupille P. Pathophysiology of disk-related sciatica. I. – Evidence supporting a chemical component. Joint Bone Spine. 2006;73(2):151–158. doi: 10.1016/j.jbspin.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Zuo Y, Perkins NM, Tracey DJ, Geczy CL. Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain. 2003;105(3):467–479. doi: 10.1016/S0304-3959(03)00261-6. [DOI] [PubMed] [Google Scholar]
- 3.Wei M, Mo SL, Nabar NR, et al. Modification of Rat Model of Sciatica Induced by Lumber Disc Herniation and the Anti-Inflammatory Effect of Osthole Given by Epidural Catheterization. Pharmacology. 2012;90(5–6):251–263. doi: 10.1159/000340023. [DOI] [PubMed] [Google Scholar]
- 4.Liou JT, Sum DC, Liu FC, Mao CC, Lai YS, Day YJ. Spatial and temporal analysis of nociception-related spinal cord matrix metalloproteinase expression in a murine neuropathic pain model. J Chin Med Assoc. 2013;76(4):201–210. doi: 10.1016/j.jcma.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Nitz AJ, Dobner JJ, Matulionis DH. Structural assessment of rat sciatic nerve following tourniquet compression and vascular manipulation. Anat Rec. 1989;225(1):67–76. doi: 10.1002/ar.1092250110. [DOI] [PubMed] [Google Scholar]
- 6.Saika F, Kiguchi N, Kobayashi Y, Fukazawa Y, Kishioka S. CC-chemokine ligand 4/macrophage inflammatory protein-1β participates in the induction of neuropathic pain after peripheral nerve injury. Eur J Pain. 2012;16(9):1271–1280. doi: 10.1002/j.1532-2149.2012.00146.x. [DOI] [PubMed] [Google Scholar]
- 7.Xu L, Huang Y, Yu X, Yue J, Yang N, Zuo P. The influence of p38 mitogen-activated protein kinase inhibitor on synthesis of inflammatory cytokine tumor necrosis factor alpha in spinal cord of rats with chronic constriction injury. Anesth Analg. 2007;105(6):1838–1844. doi: 10.1213/01.ane.0000287660.29297.7b. [DOI] [PubMed] [Google Scholar]
- 8.Tufek A, Kaya S, Tokgoz O, et al. The protective effect of dexmedetomidine on bupivacaine-induced sciatic nerve inflammation is mediated by mast cells. Clin Invest Med. 2013;36(2):E95–E102. doi: 10.25011/cim.v36i2.19572. [DOI] [PubMed] [Google Scholar]
- 9.Peleshok JC, Ribeiro-da-Silva A. Neurotrophic factor changes in the rat thick skin following chronic constriction injury of the sciatic nerve. Mol Pain. 2012;8:1. doi: 10.1186/1744-8069-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs D, Birklein F, Reeh PW, Sauer SK. Sensitized peripheral nociception in experimental diabetes of the rat. Pain. 2010;151(2):496–505. doi: 10.1016/j.pain.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Keppel Hesselink JM. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. OPJ. 2012;5:12–23. [Google Scholar]
- 12.Kuehl FA, Jacob TA, Ganley OH, Ormond RE, Meisinger MAP. The identification of N-(2-hydroxyethyl)-palmitamide as a naturally occurring anti-inflammatory agent. J Am Chem Soc. 1957;79(20):5577–5578. [Google Scholar]
- 13.Masek K, Perlik F. Letter: Slow encephalopathies, inflammatory responses, and arachis oil. Lancet. 1975;2(7934):558. doi: 10.1016/s0140-6736(75)90939-3. [DOI] [PubMed] [Google Scholar]
- 14.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394(6690):277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 15.Darmani NA, Izzo AA, Degenhardt B, et al. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: Review of the available pre-clinical data, and first human studies. Neuropharmacology. 2005;48(8):1154–1163. doi: 10.1016/j.neuropharm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci. 2003;23(21):7767–7775. doi: 10.1523/JNEUROSCI.23-21-07767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulanova E, Bulfone-Paus S. P2 receptor-mediated signaling in mast cell biology. Purinergic Signal. 2009;6(1):3–17. doi: 10.1007/s11302-009-9173-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohara PT, Vit JP, Bhargava A, et al. Gliopathic pain: when satellite glial cells go bad. Neuroscientist. 2009;15(5):450–463. doi: 10.1177/1073858409336094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esposito E, Paterniti I, Mazzon E, et al. Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav Immun. 2011;25(6):1099–1112. doi: 10.1016/j.bbi.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Scuderi C, Steardo L. Neuroglial Roots of Neurodegenerative Diseases: Therapeutic Potential of Palmitoylethanolamide in Models of Alzheimer’s Disease. CNS Neurol Disord Drug Targets. 2013;12(1):62–69. doi: 10.2174/1871527311312010011. [DOI] [PubMed] [Google Scholar]
- 21.Luongo L, Guida F, Boccella S, et al. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets. 2013;12(1):45–54. doi: 10.2174/1871527311312010009. [DOI] [PubMed] [Google Scholar]
- 22.Benito C, Tolón RM, Castillo AI, et al. β−Amyloid exacerbates inflammation in astrocytes lacking fatty acid amide hydrolase through a mechanism involving PPAR-α, PPAR-γ and TRPV1, but not CB1 or CB2 receptors. Br J Pharmacol. 2012;166(4):1474–1489. doi: 10.1111/j.1476-5381.2012.01889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skaper SD, Facci L. Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos Trans R Soc Lond B Biol Sci. 2012;367(1607):3312–3325. doi: 10.1098/rstb.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costa B, Conti S, Giagnoni G, Colleoni M. Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo-oxygenase systems. Br J Pharmacol. 2002;137(4):413–420. doi: 10.1038/sj.bjp.0704900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godlewski G, Offertáler L, Wagner JA, Kunos G. Receptors for acylethanolamides-GPR55 and GPR119. Prostaglandins Other Lipid Mediat. 2009;89(3–4):105–111. doi: 10.1016/j.prostaglandins.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol. 2002;136(3):452–458. doi: 10.1038/sj.bjp.0704732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Agostino G, La Rana G, Russo R, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-κB nuclear signalling in dorsal root ganglia. Eur J Pharmacol. 2009;613(1–3):54–59. doi: 10.1016/j.ejphar.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 28.LoVerme J, Russo R, La Rana G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol and Exp Ther. 2006;319(3):1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 29.Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4(8):1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- 30.Sasso O, Moreno-Sanz G, Martucci C, et al. Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain. 2013;154(3):350–360. doi: 10.1016/j.pain.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bettoni I, Comelli F, Colombo A, Bonfanti P, Costa B. Non-neuronal cell modulation relieves neuropathic pain: efficacy of the endogenous lipid palmitoylethanolamide. CNS Neurol Disord Drug Targets. 2013;12(1):34–44. doi: 10.2174/1871527311312010008. [DOI] [PubMed] [Google Scholar]
- 32.Helyes Z, Nemeth J, Than M, Bolcskei K, Pinter E, Szolcsanyi J. Inhibitory effect of anandamide on resiniferatoxin-induced sensory neuropeptide release in vivo and neuropathic hyperalgesia in the rat. Life Sci. 2003;73(18):2345–2353. doi: 10.1016/s0024-3205(03)00651-9. [DOI] [PubMed] [Google Scholar]
- 33.Petrosino S, Palazzo E, de Novellis V, et al. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology. 2007;52(2):415–422. doi: 10.1016/j.neuropharm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 34.Di Cesare Mannelli L, D’Agostino G, Pacini A, et al. Palmitoylethanolamide Is a disease-modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR-alpha-mediated mechanism. mediators of inflammation. Mediators Inflamm. 2013;2013:328797. doi: 10.1155/2013/328797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Starowicz K, Makuch W, Korostynski M, et al. Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS One. 2013;8(4):e60040. doi: 10.1371/journal.pone.0060040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallace VC, Segerdahl AR, Lambert DM, et al. The effect of the palmitoylethanolamide analogue, palmitoylallylamide (L-29) on pain behaviour in rodent models of neuropathy. Br J Pharmacol. 2007;151(7):1117–1128. doi: 10.1038/sj.bjp.0707326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esposito E, Cuzzocrea S. Palmitoylethanolamide is a new possible pharmacological treatment for the inflammation associated with trauma. Mini Rev Med Chem. 2013;13(2):237–255. [PubMed] [Google Scholar]
- 38.Guida G, de Martino M, de Fabiani A, et al. La palmitoiletanolamida (Normast) en el dolor neuropatico cronico por lumbociatalgia de tipo compresivo: estudio clinico multicentrico [A multicenter clinical study of palmitoylethanolamide in chronic neuropathic pain: compression lumboischialgia] Dolor. 2010;25(1):35–42. Spanish. [Google Scholar]
- 39.Keppel Hesselink JM. Non-neuronal cells new therapeutic targets for neuropathic pain; X Congresso Nazionale ACD SIAARTI; November 23–25; 2011; Perugia. [Google Scholar]
- 40.Canteri L, Petrosino S, Guida G. Reducción del consumo de antiinflamatorios y analgésicos en el tratamiento del dolor neuropático crónico en pacientes afectados por lumbociatialgia de tipo compresivo y en tratamiento con Normast 300 mg [Reduction in consumption of anti-inflammatory and analgesic medication in the treatment of chronic neuropathic pain in patients affected by compression lumbocischialgia due to the treatment with Normast 300 mg] Dolor. 2010;25(4):227–234. Spanish. [Google Scholar]
- 41.Gatti A, Lazzari M, Gianfelice V, Di Paolo A, Sabato E, Sabato AF. Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med. 2012;13(9):1121–1130. doi: 10.1111/j.1526-4637.2012.01432.x. [DOI] [PubMed] [Google Scholar]
- 42.Desio P. Combination of oxycodone and palmitoylethanolamide for low back pain treatment. AMC. 2011;1(2):62–71. [Google Scholar]
- 43.Palomba RA, Adiletta S, Candiello A, Penimpede M, Bonaccia P, De Martino CJ. Analgesia multimodale per il dolore cronico: directive future e razionale. [Multimodal analgesia for chronic pain: rationale and future directions]; 33rd National Congress of the Italian Association for the Study of Pain; Florence, Italy. 2010. Spanish. [Google Scholar]
- 44.Domínguez CM, Ibáñez Puertas MA, et al. Palmitoiletanolamida (PEA) en lumbociatica en asociacion al tratamiento habitual. [Palmitoylethanolamide in lumbosciatic pain in association with standard therapy] Rev Soc Esp Dolor. 2010;17:35. Spanish. [Google Scholar]
- 45.Assini A, Laricchia D, Pizzo R, et al. P1577: The carpal tunnel syndrome in diabetes: clinical and electrophysiological improvement after treatment with palmitoylethanolamide. Eur J Neurol. 2010;17(S3):295. [Google Scholar]
- 46.Conigliaro R, Drago V, Foster PS, Schievano C, Di Marzo V. Use of palmitoylethanolamide in the entrapment neuropathy of the median in the wrist. Minerva Med. 2011;102(2):141–147. [PubMed] [Google Scholar]