

Abstract
The presence of excess fluid in the interstitium and air spaces of the lung presents severe restrictions to gas exchange. The pulmonary endothelial barrier regulates the flux of fluid and plasma proteins from the vascular space into the underlying tissue. The integrity of this endothelial barrier is dynamically regulated by transitions in cAMP (3′,5′-cyclic adenosine monophosphate), which are synthesized in discrete subcellular compartments. Cyclic AMP generated in the subplasma membrane compartment acts through PKA and Epac (exchange protein directly activated by cAMP) to tighten cell adhesions, strengthen cortical actin, reduce actomyosin contraction, and decrease permeability. Confining cAMP within the subplasma membrane space is critical to its barrier-protective properties. When cAMP escapes the near membrane compartment and gains access to the cytosolic compartment, or when soluble adenylyl cyclases generate cAMP within the cytosolic compartment, this second messenger activates established cytosolic cAMP signaling cascades to perturb the endothelial barrier through PKA-mediated disruption of microtubules. Thus the concept of cAMP compartmentalization in endothelial barrier regulation is gaining momentum and new possibilities are being unveiled for cytosolic cAMP signaling with the emergence of the bicarbonate-regulated mammalian soluble adenylyl cyclase (sAC or AC10).
Keywords: adenylyl cyclase; compartmentalization; Epac, PKA; sAC
Emerging Concepts of cAMP Signaling
In 1957, while studying adrenaline regulation of glycogen metabolism, Sutherland and coworkers isolated a heat-stable factor generated in membrane fractions of liver homogenates. Subsequently, the factor was identified as cAMP (150). These seminal findings led to the emergence of the second messenger concept: extracellular hormone stimulation, adrenaline, generates an intracellular mediator, cAMP (149). Currently, cAMP has been implicated in the regulation of many physiological responses and cellular processes, including modification of the immune response, cardiac contractility, cell cycle control, and endothelial barrier regulation. Specificity of the physiological response was linked to hormone-specific receptor expression on the particular tissue or cell. However, it soon became apparent that individual cells express multiple hormone-specific adenylyl cyclase-linked receptors, combinations of G protein subunits, and several adenylyl cyclase isoforms to catalyze cAMP production. The finding that two different agonists generated a cAMP signal yet imparted a different physiological response gave rise to the idea that cAMP signals are spatially segregated into isolated subcellular compartments within a single cell (72, 79, 118, 170). Thus, although our interpretation of cAMP signaling specificity began at the tissue level, we now consider cAMP signaling specificity at the subcellular compartment level.
In 1968, Krebs identified a kinase whose activity was dependent on cAMP, thus providing a mechanism whereby cAMP exerted its cellular actions; cAMP-dependent protein kinase (PKA) provided a link between stimulation of adenylyl cyclase and activation of downstream effectors (162). Yet PKA does not account for all physiological effects of cAMP. In the search to explain PKA-independent cAMP signals, exchange protein directly activated by cAMP (Epac) was identified, which acts as a cAMP-activated guanine nucleotide exchange factor (cAMP-GEF) for Rap1 and Rap2 (36, 80). Whereas downstream functions of cAMP are mediated through PKA and Epac, temporal changes in cAMP are modulated by phosphodiesterase activity, which hydrolyzes cAMP to 5′-AMP halting activation of cAMP effectors (29, 95, 148).
Although cAMP is a highly hydrophobic second messenger with the potential to rapidly diffuse throughout the cytosolic environment creating a homogenous signal, probes that track the spatial migration of cAMP suggest that it is unevenly distributed and concentrates in microdomains (41, 107, 112, 120, 121, 178). It is possible that the perimeters of the microdomains are lined by obstacles, such as the endoplasmic reticulum, to physically block cAMP diffusion, and phosphodiesterases, which hydrolyze cAMP before it escapes the microdomain (120). Thus barricades encircling the microdomain restrict cAMP diffusion and create a subcellular compartment. This nonuniform rise in cAMP creates gradients, with elevated near membrane cAMP in close proximity to the adenylyl cyclase and lower cAMP concentrations in the cytosol distal to the source (120). Further discrimination of cAMP signals is achieved through A kinase anchoring proteins (AKAPs), which are targeted to subcellular compartments and coordinate cAMP signaling elements such as PKA, Epac, phosphodiesterases, and phosphatases (42, 100, 172). This review focuses on cAMP signaling pathways that regulate endothelial barrier integrity and highlights spatially segregated cAMP signals, which have opposing effects on endothelial permeability (57).
Pulmonary Edema and the cAMP Connection
The pathogenesis of edema associated with inflammation was of keen interest during the nineteenth century. In 1873, J. Cohnheim reported that blood vessels could become leaky during inflammation (as summarized in Ref. 97). Following his isolation of histamine from the fungus, ergot, Sir Henry Dale (35) revealed that histamine induced a “loss of blood volume owing to morbid permeability of the endothelium.” Over time, the concept that increased vascular permeability could initiate edema became more widely accepted. A few years after Sutherland and Rall's description of cAMP (150), Majno and Palade (97) demonstrated histamine and serotonin induced vascular hyperpermeability involved “numerous endothelial openings…in blood vessels” and cellular contraction. They explained, “…endothelial leak was a result of endothelial cells [that] become partially disconnected.” It is now widely accepted that edema can be mediated by increased vascular permeability resulting from interendothelial cell gaps, which are generated by loss of cell adhesions, cytoskeletal contraction, and endothelial cell retraction (101, 114).
Within the lung, R. T. H. Laennec described the complications of edema as “an infiltration of serum into the pulmonary tissue, carried to a degree such that it significantly diminishes its permeability to air” (as summarized in Ref. 144). Indeed, the pathogenesis of pulmonary edema, provoked by various insults such as bacterial induced sepsis, air embolism, acid injury, and inflammatory mediators, is a multifactorial disease including increased pulmonary vascular permeability (144, 145, 164). Agents that increase cAMP, such as isoproterenol, prostaglandin, forskolin, and cholera toxin, attenuate edema in whole animal, isolated lung, and clinical studies of lung injury (2, 52, 82, 99, 103). However, it was not until the isolation of pulmonary endothelial cells, the identification of the cAMP signaling axis in the endothelium, and treatment of cells in culture that conclusive studies were performed linking elevations in cAMP with increased endothelial barrier integrity (23, 110, 125, 146).
The Pulmonary Endothelial Barrier
The pulmonary vasculature is lined by a nonfenestrated endothelium characterized by its continuous basement membrane and closely apposed, caveolae-rich endothelial cells. The endothelial monolayer has multiple homeostatic functions including acting as semipermeable barrier. In quiescent cells, this barrier functions as a molecular sieve permitting diffusion of low molecular weight molecules across the vessel wall and into the underlying tissue via paracellular transport, whereas larger molecules, such as proteins, traverse the endothelial barrier via the transcellular pathway in the process of transcytosis (101, 109). Multiple agonists perturb the normal homeostasis of the barrier, expanding the repertoire of molecules and volume of fluid that can gain access to the underlying tissue. The route of increased permeability is primarily through the formation of intercellular gaps or paracellular transport (46, 101, 102). Endothelial hyperpermeability leads to flooding of the underlying tissue and alveoli leading to life-threatening pulmonary edema due to hindered gas exchange. This review addresses cAMP signals that regulate the paracellular permeability pathway.
Endothelial barrier integrity is determined by endothelial cell shape, which was described by the tensegrity model as a balance between competing forces: barrier-protective adhesive forces impart centrifugal tension whereas barrier-disruptive antiadhesive forces provide centripetal tension (77). Adhesive forces are dependent on transmembrane cell adhesion molecules that anchor adjacent cells to each other as well as anchoring cells into the underlying matrix. To perform their adhesive function and maintain endothelial barrier integrity, adhesion molecules are tethered into the cortical actin cytoskeleton by adaptor molecules, such as α-, β-, and γ-catenin, plakoglobin, and α-actinin, which, together with p120, form junctional complexes (37, 167). This cortical rim (or dense peripheral band) appears as the cell monolayer reaches confluence and is composed of F-actin bundles stabilized by actin-binding proteins (171). Spatially, the dense peripheral band lies just beneath the spectrin cytoskeleton and circumnavigates the entire cell (114). In contrast to adhesive forces, barrier-disruptive, nonadhesive forces arise when cortical actin filaments disperse and stress fibers, extending throughout the cytoplasm, become more prominent. Actomyosin contraction of these stress fibers increases centripetal tension and is proposed to physically pull adjacent cells apart, changing cell shape and reducing endothelial barrier integrity (46, 105). Other cytoskeletal elements, such as microtubules, communicate with actin; thus microtubule dynamics also play an integral role in endothelial cell shape and vascular integrity (19, 91, 114, 160).
Barrier-protective or disruptive agents adjust the balance of adhesive vs. nonadhesive forces influencing endothelial cell shape and paracellular transport. In a quiescent endothelium and in endothelial monolayers with enhanced barrier properties, actin filaments distribute primarily in cortical actin with few stress fibers (10, 19, 114, 166). In contrast, barrier-disruptive agonists shift the balance of forces to reduced tethering of adhesion molecules, promote actin reorganization into stress fibers, and induce actin-myosin contraction leading to cell shape changes. Indeed, disruption of junctional complexes, stress fiber formation, and actomyosin contraction are hallmarks of endothelial barrier disruption. These events lead to retraction of cell-cell borders that permits the paracellular movement of fluid and macromolecules from the blood into the underlying tissue. Agents that reduce actomyosin contraction, stabilize cortical actin, and strengthen cell adhesions complexes improve the endothelial barrier integrity and/or facilitate repair. Multiple reports document that subplasma membrane cAMP, acting via PKA or Epac, plays a key role in modulating cytoskeletal dynamics and cell adhesions to improve endothelial barrier integrity (46, 103, 104, 114). Indeed, the use of PKA inhibitors (e.g., RpcAMPs) demonstrated the role of PKA in maintenance of basal endothelial monolayers as well as attenuating agonist-induced hyperpermeability (110, 147).
Targets of Barrier-Protective cAMP Mediated by PKA
In certain models of endothelial barrier disruption actomyosin contraction is triggered by phosphorylation of the regulatory subunit of myosin light chain (MLC) (46). Indeed, both thrombin and histamine elevate MLC phosphorylation, which is reversed by elevated cAMP (60, 104). When phosphorylated, MLC interacts with actin to promote cross-bridge cycling and tension development. In turn, the level of MLC phosphorylation is a balance between phosphorylation by calcium-calmodulin-activated MLC kinase (MLCK) and dephosphorylation by MLC phosphatase (Fig. 1) (161, 173). Thus decreasing MLCK activity, increasing MLC phosphatase activity, or easing MLC phosphatase inhibition can attenuate endothelial cell contraction and alleviate endothelial barrier disruption (51, 60). As the signaling molecules implicated in regulation of MLC phosphorylation were elucidated, they became potential targets to explain the barrier-protective roles of cAMP signaling.
Fig. 1.
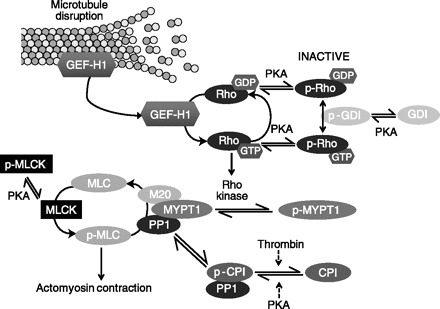
Transmembrane adenylyl cyclase activates barrier-protective PKA targets. Myosin light chain (MLC) phosphorylation promotes actomyosin contraction. MLC kinase (MLCK) phosphorylates MLC whereas MLC phosphatase (M20, MYPT1, and PP1) dephosphorylates MLC. MLC phosphorylation can be relieved by PKA phosphorylation of MLCK. PKA phosphorylation attenuates MLCK activity, thereby acting as a potential barrier-protective strategy. In contrast, dephosphorylation of MLC through activation of MLC phosphatase attenuates actomyosin contraction. PKA-mediated Rho phosphorylation at serine 188 in either the GTP or GDP form enhances Rho affinity for Rho-GDI (guanosine nucleotide dissociation inhibitor), inactivating Rho by sequestering Rho in the cytosol. The affinity of Rho for Rho-GDI is further enhanced by PKA phosphorylation of GDI. Inactivation of Rho decreases Rho kinase activity, thus relieving inhibition of the regulatory subunit, MYPT1, of MLC phosphatase. Improved MLC phosphatase activity decreases actomyosin contraction. The catalytic subunit of MLC phosphatase, PP1, can be sequestered and inhibited by the phosphorylated form of CPI-17 (p-CPI). Although thrombin promotes phosphorylation of CPI-17, PKA promotes its dephosphorylation, releasing PP1 to its active form to promote dephosphorylation of MLC and relieve actomyosin contraction and endothelial relaxation. Upon microtubule disassembly, GEF-H1 is released from microtubules to activate Rho.
The nonmuscle MLCK isoform identified in endothelial cells by Garcia et al. (61) exhibits a unique amino terminus and multiple conserved phosphorylation sequences including a conserved PKA phosphorylation sequence adjacent to the calcium-calmodulin binding region. Treatment of bovine pulmonary artery endothelial cells with cholera toxin, to elevate cAMP levels, increased MLCK phosphorylation with associated decrease in kinase activity (61). Subsequent studies using phosphatase inhibitors supported that MLCK phosphorylation reduced its kinase activity although the specific kinase was not identified (160). In bovine pulmonary artery endothelial cells, loss of PKA activity by RpcAMPS or overexpression of the PKA peptide inhibitor (PKI) promoted stress fiber formation and decreased electrical resistance across the monolayer, implicating PKA in cytoskeletal reorganization necessary to maintain baseline barrier integrity (110). However, in these studies PKA inhibition failed to increase MLC phosphorylation under basal or thrombin treated conditions, suggesting that PKA does not play a role in regulating MLCK activity, yet the possibility still remains for other endothelial cell types or models of elevated permeability (94, 110).
As stated above, another mechanism to reduce MLC phosphorylation and decrease contraction is to promote MLC dephosphorylation. MLC phosphatase is a trimeric molecule composed of a regulatory/targeting subunit, MYPT1, a catalytic subunit, PP1, and a subunit of unknown function, M20. PKA signaling modulates MLC phosphatase activity through the regulatory and catalytic subunits. First, Rho kinase phosphorylation of the regulatory subunit, MYPT1, inhibits MLC phosphatase activity. The small GTPase, Rho, which relays signals for cytoskeletal reorganization, activates Rho kinase. In its active GTP-bound state, Rho translocates to the membrane where it activates Rho kinase, whereas in the inactive GDP-bound state it is sequestered in the cytoplasm. The level of active Rho is a balance between Rho-guanine nucleotide exchange factors (Rho-GEFs) and Rho-GTPase activating proteins (Rho-GAPs). Rho-GEFs activate Rho by promoting GDP for GTP exchange, whereas Rho-GAPS catalyze the slow intrinsic GTPase activity of Rho hydrolyzing Rho-GTP to inactive Rho-GDP. In addition, Rho-GDI (guanosine nucleotide dissociation inhibitor) can sequester Rho-GDP within the cytoplasm preventing its activation. PKA phosphorylation of RhoA at serine 188 increases its affinity for Rho-GDI even in the GTP-bound state, thereby extracting active RhoA from the membrane and promoting its translocation to the cytosol or retaining Rho-GDP in the cytosol (43, 48, 90). Forskolin and IBMX activation of bovine pulmonary artery endothelial cells increases RhoA phosphorylation at serine 188 and corresponds to decreased RhoA activity, decreased MLC phosphorylation, and protection against thrombin-induced hyperpermeability (65). Another level of PKA barrier protection comes through Rho-GDI phosphorylation, which negatively regulates RhoA activation (115); however, the significance of PKA-mediated Rho-GDI phosphorylation in regulation of the endothelial barrier has not been resolved. Thus PKA phosphorylation negatively regulates Rho through direct phosphorylation of Rho or its inhibitor, Rho-GDI. PKA phosphorylation renders Rho unable to activate Rho kinase thereby improving MLC phosphatase activity and attenuating MLC phosphorylation (65, 116). In addition to MLC phosphatase, Rho kinase targets Lim kinase, which subsequently phosphorylates and inactivates the actin binding protein, cofilin. In its active unphosphorylated state, cofilin promotes actin depolymerization and F-actin severing. Currently, how active, unphosphorylated cofilin affects the actin cytoskeleton related to endothelial barrier is not clear (65). Thus there are multiple implications of PKA-induced inhibition of Rho and downstream Rho kinase activity, including release of MLC phosphatase inhibition to promote MLC dephosphorylation and attenuate actomyosin stress fiber contraction. Collectively, cAMP-PKA-mediated inhibition of Rho promotes protection against endothelial barrier disruption via inactivation of the contractile machinery.
A second mechanism whereby cAMP-PKA signaling modulates MLC phosphatase activity is through regulation of the catalytic subunit, PP1, by CPI-17 (PKC-potentiated inhibitory protein for PP1 of 17 kDa). Following phosphorylation, CPI-17 forms an inhibitory complex with PP1. Thrombin- and histamine-induced hyperpermeability is mediated in part by phosphorylation of CPI-17 (84), whereas forskolin activation of adenylyl cyclase activity promotes CPI-17 dephosphorylation, loss of CPI-17-PP1 complex formation, and improved PP1 activity (5). Thus elevated cAMP-PKA activity releases MLC phosphatase from its inhibitor to decrease MLC phosphorylation and promote endothelial cell relaxation leading to endothelial barrier enhancement.
Stabilization of cortical actin by actin binding proteins attenuates agonist induced endothelial barrier disruption. The actin binding protein filamin facilitates high-angle F-actin branching and localizes to cell-cell borders in endothelial cells (106, 163). Loss of filamin is embryonically lethal with dramatic increases in vascular permeability (55). Through its protein-binding domain, filamin binds transmembrane proteins, such as integrins, thus anchoring membrane-tethered proteins into the cortical actin rim (56). The PKA phosphorylation site, serine 2152, lies within the protein-binding domain (78). Filamin expression at the plasma membrane fluctuates with the level of endothelial monolayer confluence, with greatest membrane distribution at confluence and increased cytosolic expression in pre- and postconfluent monolayers (163). Barrier-disruptive agonists such as bradykinin and hydrogen peroxide induce filamin translocation into the cytosol. However, when pretreated with cAMP-PKA stimulating agents, which protect against agonist-induced barrier disruption, filamin translocation is prevented (69, 163). Furthermore, increasing subplasma membrane cAMP with isoproterenol increases filamin phosphorylation at a consensus PKA phosphorylation site, serine 2152, while decreasing subplasma membrane cAMP with thrombin decreases phosphorylation at this site (27, 129, 168). Thus cAMP-PKA retains filamin at the periphery and protects against endothelial barrier disruption.
Vasodilator-stimulated phosphoprotein (VASP) is also an actin filament-associating protein involved in regulation of actin dynamics, cell adhesions, cell migration, and regulation of the endothelial permeability (73, 132). VASP is a target for PKA, PKG, and AMP kinase phosphorylation at serine 157, serine 239, and threonine 278, respectively (18, 66, 141). VASP localizes to stress fibers, cell-cell contacts, and focal adhesions by its NH2- and COOH-terminal Ena/VASP homology domains, which includes threonine 278, whereas the central proline-rich region has been linked to F-actin polymerization and includes serine 157 and serine 239 residues (28). PKA phosphorylation of VASP at serine 157 has been implicated in adenosine-mediated endothelial barrier protection (28) and attenuation of LPS hyperpermeability by transelectrical resistance measurements (20). PKA-induced VASP phosphorylation attenuates LPS-induced VASP translocation from the cell periphery to focal adhesions and suppresses stress fiber formation (20). The mechanisms of PKA-induced VASP endothelial protection have not been fully resolved, yet emerging evidence suggests that VASP is involved in cAMP-mediated activation of the small GTPase, Rac (131–133).
Not all permeability-inducing agonists promote stress fiber formation though MLCK-mediated phosphorylation of MLC. Indeed, phorbol 12-myristate 13-acetate (PMA) activates a PKC-Raf-1 pathway leading to ERK activation and phosphorylation of the regulatory cytoskeletal protein, caldesmon, which is involved in restructuring the actin cytoskeleton (46, 94). Elevations in cAMP attenuate the PMA-induced permeability and gap formation. Furthermore, PKA inhibitors (PKI and Rp-cAMP) transiently activate Raf-1 and ERK to promote caldesmon phosphorylation that was inhibited by the upstream ERK kinase, MEK. Thus these data suggest a role for cAMP-PKA in restricting caldesmon phosphorylation to enhance endothelial barrier integrity, although the target of PKA has not been resolved (94).
The microtubule cytoskeleton is tightly linked to endothelial permeability, such that disassembly of peripheral microtubules increases endothelial permeability whereas microtubule stabilization attenuates permeability (11, 142, 159). Indeed, treatment with microtubule-disrupting, anticancer vinca alkaloids leads to sudden pulmonary edema (24). During TNF-α-induced endothelial cell barrier injury, disruption of microtubules precedes actin cytoskeletal reorganization suggesting interplay between the cytoskeletal elements (111). Microtubules originate at the microtubule-organizing center where they have the greatest density of fibers, with lower fiber density as they emanate toward the cell periphery (142). Functionally, microtubule filaments stabilize cell shape, transport intracellular membrane-bound organelles and act as storage sites for signaling proteins, such as the Rho family GTPases. For example, GEF-H1 becomes active upon release from microtubules whereby it promotes Rho activation and increases MLC phosphorylation and actin-myosin contraction (Fig. 1) (8). Thus GEF-H1 provides cross talk between microtubule dynamics, actomyosin contraction, and endothelial barrier disruption (8, 14, 87, 157). However, under these experimental conditions, forskolin- or cholera toxin-mediated stimulation of PKA activity attenuates nocodozole-induced stress fiber formation and increase in transelectrical resistance (14).
Thus the second messenger cAMP signals through PKA-mediated phosphorylation of RhoA, Rho-GDI, and CPI-17 to attenuate actomyosin contraction and endothelial tension whereas phosphorylation of filamin and VASP stabilizes cortical actin. Furthermore, PKA-mediated regulation of RhoA activity also modulates cofilin. Currently, it is unclear whether all these phosphorylation events occur simultaneously or independently to attenuate agonist-induced endothelial barrier disruption. For example, subcellular compartments could separate some events from others such that only a subset of PKA targets is activated at any one time.
Targets of Barrier-Protective cAMP Mediated by Epac
In 1998, Epac was identified as a novel cAMP target that directly activates Rap1 and Rap2, small GTPases of the Ras superfamily, and the only downstream effectors of Epac that have currently been identified (36, 80). Rap1 had been shown to suppress Ras oncogenic transformation by maintenance of cell adhesions (81). By utilizing the Epac-specific agonist 8-pCPT-2′-O-Me-cAMP (also known as 007), the barrier-protective properties of Epac-mediated Rap1 activation have been linked to improved cell-cell contacts through enhanced VE-cadherin expression at cell-cell borders, enrichment of cortical actin, and decreased RhoA activation (1, 32, 59, 85) and explained cAMP-mediated, PKA-independent events that regulate vascular permeability (21, 64, 122). Currently, the mechanisms of Epac-Rap1 enhanced VE-cadherin accumulation at cell-cell borders and improved adhesive properties are unresolved, although recent studies suggest that forskolin-induced stabilization of VE-cadherin to cortical actin bundles is dependent on α- and β-catenin (108). Indeed, Epac has been shown to form a complex at cell-cell junctions with phosphodiesterase 4, VE-cadherin, and β-catenin to regulate endothelial permeability (119). Furthermore, Epac localizes to the plasma membrane, nuclear membrane, mitochondria, and microtubules (96, 117). In human umbilical vein endothelial cells, Epac association with microtubules has been linked to microtubule growth in a Rap1-independent but AKAP9-dependent manner (137, 138). Although a second Epac isoform 2 has been identified, Epac 1 is the isoform abundant in blood vessels (64).
Atrial natriuretic peptide (ANP), PGE2, PGI2, isoproterenol, and forskolin in combination with the phosphodiesterase 4 inhibitor rolipram (or forskolin and the pan phosphodiesterase inhibitor, IBMX) elevate subplasma membrane cAMP to activate Epac-GEF function in the endothelium (7, 16, 17, 32, 59). Although Epac promotes Rap1 activation through GDP for GTP exchange, GAPs, such as Spa-1, enhance intrinsic GTP to GDP hydrolysis and inactivate Rap1 (88) (Fig. 2). A growing body of evidence implicates Epac in reduced baseline endothelial permeability as well as attenuated thrombin (7, 32, 85), PAF (1) and ventilator-induced lung injury (VILI) (13) hyperpermeability in a Rap1-dependent manner. In turn, Rap1 activates the Rac-specific GEFs, Vav2 and Tiam1, promoting Rac and Cdc42 activation (17). Rac-GTP acts on downstream targets, p21-activated kinase 1 (PAK1), p190-Rho-GAP and cortactin, to enhance endothelial barrier integrity (15). The Rac downstream target, PAK1, inhibits p115Rho-GEF, thus repressing RhoA activity (9, 123). In fibroblasts, Rac also stimulates p190Rho-GAP to further promote Rho downregulation and attenuate actomyosin contraction (169). Indeed, PGE2, PGI2, and ANP induced human pulmonary artery or microvascular endothelial barrier enhancement and protection from thrombin-induced hyperpermeability is mediated through Rap-Vav2 and -Tiam1 activation of Rac (13, 16, 17). Loss of any of these signaling molecules leads to diminished Rac and PAK1 activation, whereas small interfering RNA (siRNA) to either Rap1 or Rac leads to loss of PGE2- and iloprost-induced VE-cadherin recruitment to cell-cell borders (13, 17). The PGI2 analog, iloprost, acts through a similar cAMP activated Epac-Rap1 pathway to protect against VILI (13). Using the Rac and Cdc42 agonist, CNF-1, Waschke et al. (165) demonstrated Rac and Cdc42 enhanced cortical actin by recruiting cortactin and VASP and reduced PAF-mediated increase in hydraulic conductivity in perfused mesenteric microvessels. IQGAP, which lacks GTPase activity, binds and stabilizes Rac and Cdc42 in their active GTP-bound form (22, 68). Furthermore, Rac and Cdc42 binding, prohibits IQGAP from binding β-catenin. Thus Rac and Cdc42 strengthen cell-cell junctions by release of β-catenin from IQGAP, promoting β-catenin association with junctional complexes.
Fig. 2.
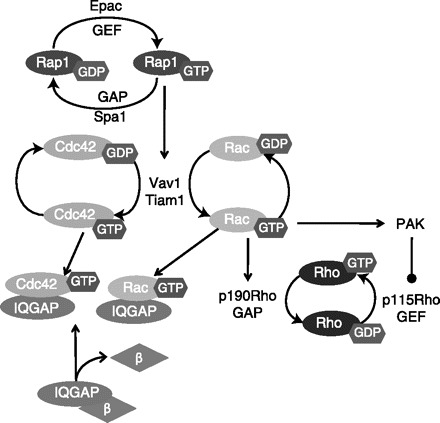
Activation of transmembrane adenylyl cyclase activates barrier-protective exchange protein directly activated by cAMP (Epac) targets. Epac-GEF activates Rap1, which activates the GEFs Vav1 and Tiam. In turn, Vav1 and Tiam activate both Rac and Cdc42. In the active GTP-bound form, both Rac and Cdc42 sequester IQGAP, which prohibits the formation of an IQGAP-β-catenin complex. When released from IQGAP, β-catenin stabilizes cell-cell junctions. In addition, Rac decreases Rho activation through stimulation of p190RhoGAP and PAK. In turn, PAK inhibits p115RhoGEF, further inactivating Rho to attenuate endothelial contraction and promote endothelial relaxation.
A recently identified target of Epac-Rap1 signaling is Krit1 (Krev1 interaction trapped gene), a protein lost in the endothelial junction disease cerebral cavernous malformations (45). Active Rap1 enhances Krit1 association with β-catenin at cell-cell junctions and Krit1 expression at junctions is essential for Rap1 barrier stabilization (63). Krit1 depletion abolishes Epac-mediated protection against thrombin hyperpermeability (63).
Recently, ANP has been implicated in protection against thrombin hyperpermeability. ANP elevates cAMP to activate both Epac-Rap1 and PKA signaling cascades leading to decreased MLC phosphorylation (16). Subsequent studies reveal Rac integrates cAMP-Epac and -PKA-mediated endothelial barrier protection (12). Thus, although Epac and PKA events are described independently here, their actions are intertwined. Both cAMP effectors modulate similar pathways and indeed both effectors can be integrated into the same subcellular complexes. However, the signaling events described above implicate cAMP in improved barrier integrity, yet cAMP has similarly been linked to endothelial barrier disruption.
Bacterial Soluble Adenylyl Cyclases Disrupt the Endothelial Barrier
It is well known that microorganisms are a risk factor for endothelial cell dysfunction (91). Early reports describe a toxin produced by the bacterium Bacillus anthracis, which increases endothelial permeability (31, 140, 143). This edema-inducing agent, later named edema factor, is a calcium- and calmodulin-dependent adenylyl cyclase, which elevates intracellular cAMP when delivered into eukaryotic cells, increases endothelial permeability, and impairs host immunity (4, 25, 92). The cytoplasmic actions of edema factors are mediated through both PKA and Epac to alter the cytoskeleton and cell shape, leading to cell rounding (75, 76). Although transmembrane adenylyl cyclase activity generates cAMP gradients with decreasing concentrations away from the subplasma membrane space toward the cell center, edema factor-cAMP was focused around the cell center and diminished toward the cell periphery (33, 34). Thus reversing the endogenous cAMP gradient by localizing adenylyl cyclase to the cytosolic compartment leads to edema.
Other bacteria have evolved strategies to transfer exotoxins into eukaryotic cells, such as the type three-secretion system (T3SS) of Pseudomonas aeruginosa (176). The T3SS uses an injection type needle to deliver exotoxins across bacterial and host membrane barriers into the cytosol of eukaryotic cells (70). P. aeruginosa is a ubiquitous pathological bacterium associated with acute infections in patients with sepsis, in postoperative and burn patients, and in ventilator-associated pneumonia and cystic fibrosis patients (49). Strains of P. aeruginosa expressing a functional T3SS are correlated with severity of disease, increased mortality, poor disease outcomes in patients with ventilator-associated pneumonia, and bacterial persistence (47, 71, 124, 158). In general, 89% of strains that possess the T3SS have the exoY gene, which encodes for ExoY, a soluble adenylyl cyclase (53, 177). To avoid unregulated cAMP production within the pathogen, ExoY is stimulated by an as yet unknown soluble eukaryotic factor (177). ExoY lacks membrane localization domains expressed on other exotoxin proteins, and in pulmonary microvascular endothelial cells it is excluded from the subplasma membrane space but localizes to the cytosolic compartment (38, 86, 130). Thus T3SS-competent, exoY-encoded P. aeruginosa deliver ExoY directly into the cytosol of infected cells where it manipulates the host cellular machinery to elevate cytosolic cAMP concentrations (130, 156, 177). This cytosolic cAMP pool leads to endothelial cell gaps as cell-cell borders retract and increases permeability in the isolated lung (126, 130). However, a criticism of these studies was that the genetically engineered strains of P. aeruginosa used to deliver ExoY permitted the accumulation of nonphysiological levels of cAMP. Toward this end, we exploited a forskolin-sensitive mammalian adenylyl cyclase chimera (sACI/II), which generated significantly lower cytosolic cAMP levels compared with plasma membrane cAMP levels (128).
The central cytosolic loop and carboxy terminus of eukaryotic transmembrane adenylyl cyclase merge to create the catalytic domain of the enzyme. The catalytic domains of adenylyl cyclase I and II have been excised from the membrane-spanning regions and linked to form a functional forskolin-sensitive, soluble adenylyl cyclase chimera (sACI/II) (40, 151). Forskolin stimulation of pulmonary endothelial cells expressing the sACI/II chimera activates not only sACI/II but also transmembrane adenylyl cyclase, thus elevating cAMP in both the cytosolic and membrane compartments (128). These elevations in cAMP provoke retraction of cell-cell borders and endothelial cell gaps despite simultaneous production of barrier-protective subplasma membrane cAMP. Thus even low levels of cytosolic cAMP are sufficient to disrupt the endothelial barrier and, indeed, this minimal cytosolic cAMP overwhelms barrier-protective plasmalemma cAMP. Cytosolic cAMP barrier disruption appears to be mediated through reorganization of microtubules.
Microtubule-associated proteins (MAPs) bind and stabilize microtubule structures and provide docking sites for enzymes (155). Indeed, MAPs promote microtubule assembly from tubulin subunits and create a more stable structure that resists disassembly (39, 44). In turn, phosphorylation events regulate the association of MAPs with microtubules. Nonneuronal tau is a MAP expressed in pulmonary endothelium with a PKA phosphorylation target at serine 214 (30, 152). In the phosphoserine 214 configuration, tau dissociates from microtubules and promotes microtubule disassembly (98, 136, 155). Although forskolin activation of transmembrane adenylyl cyclase did not phosphorylate tau at serine 214, the cytosolic cAMP generated by either ExoY or sACI/II increased tau phosphoserine 214 (30, 113, 129) (Fig. 3). In contrast, whereas isoproterenol increased filamin phosphorylation at serine 2152, ExoY did not, demonstrating that cytosolic cAMP cannot traverse into the near membrane compartment (129). Thus tau is a target of cytosolic cAMP, which initiates microtubule disruption and endothelial barrier disruption. Surprisingly, microtubule disruption did not instigate remodeling of actin filaments into stress fibers as has been described for other mechanisms of microtubule-related endothelial barrier disruption, suggesting the possibility of two independent mechanisms of microtubule induced endothelial barrier disruption (113).
Fig. 3.
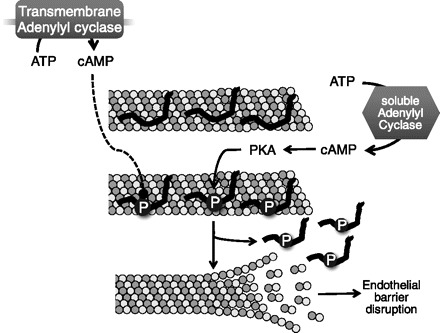
Cytosolic cAMP disrupts the endothelial barrier. The microtubule-associated protein (MAP) nonneuronal tau stabilizes microtubules (top). Although soluble adenylyl cyclase activity of ExoY or the sACI/II chimera generates cytosolic cAMP that leads to the phosphorylation of nonneuronal tau at serine 214, activation of transmembrane adenylyl cyclase activity does not lead to tau serine 214 phosphorylation (middle). In the phosphoserine 214 state, tau dissociates from microtubules, which favors microtubule disassembly and leads to endothelial gap formation (bottom).
The inability of transmembrane adenylyl cyclase activity to induce tau phosphorylation suggests that spatially restrictive cAMP compartments exist within the pulmonary endothelium. cAMP elevations in each distinct compartment are linked to endothelial barrier-protective (membrane cAMP) or disruptive outcomes (cytosolic cAMP). Phosphodiesterases play a key role in defining cAMP signaling microdomains functioning as a barricade to prevent cAMP diffusion between near plasma membrane and cytosolic compartments and shaping intracellular cAMP gradients (58, 120, 121). Interestingly, in pulmonary endothelial cells, phosphodiesterase 4D4 is anchored to the cell periphery through its association with the spectrin membrane skeleton. By use of a dominant-negative peptide to inhibit phosphodiesterase 4D4 activity, forskolin stimulation of plasmalemma adenylyl cyclase induced PKA-mediated tau serine 214 phosphorylation that was accompanied by microtubule reorganization and endothelial barrier disruption (30). Interestingly, these effects were analogous to the barrier-disruptive effects of soluble ExoY or sACI/II adenylyl cyclase activity. When tau serine 214 is mutated to alanine, loss of phosphodiesterase D4D activity failed to reorganize microtubules and disrupt the barrier (179). Thus, in pulmonary endothelium, phosphodiesterase 4D4 localizes to the subplasma membrane space to spatially limit migration of the cAMP signal into the cytosol. Furthermore, cytosolic cAMP leads to phosphorylation of tau and decreased endothelial barrier integrity (30, 179).
Thus accumulating evidence supports the idea that subplasma membrane cAMP is critically retained within near membrane compartments to maintain the endothelial barrier. Once cAMP escapes into the cytosolic compartment, it activates cytosolic cAMP signaling cascades that disrupt the barrier. These observations raise several intriguing questions. First, if cAMP is highly compartmentalized to the subplasma membrane compartment, then why are cAMP targets localized to the cytosol? Indeed, the cAMP signaling machinery for microtubule-mediated endothelial barrier disruption is in place, but the endogenous cAMP source to activate these targets has not been defined. Second, these findings bring into question the impact of the endogenous soluble adenylyl cyclase (sAC or AC10) on endothelial permeability.
Emerging Story of Endogenous Soluble Adenylyl Cyclase Isoform 10
Unlike transmembrane adenylyl cyclases, sAC lacks transmembrane domains and is expressed in the cytosol (Fig. 4). sAC is insensitive to G protein signaling and forskolin, yet activated by physiological transitions in bicarbonate, which is potentiated by calcium (26, 67, 93, 154). In contrast to membrane localization, sAC localizes to discrete compartments in the cell such as microtubules, mitochondria, the microtubule-organizing center, and the nucleus (54, 62, 174, 180, 181). Expression of sAC was initially found in sperm, yet distribution in somatic tissues is now evident (139). Currently, sAC activity has been in implicated biological processes such as sperm motility, cilia beat frequency, apoptosis, regulation of the electron transport chain, cell migration, and gene transcription (50, 74, 134, 135, 175). Thus sAC is involved in regulation of microtubule functions, i.e., sperm motility and cilia beat frequency, providing the possibility that bacterial adenylyl cyclase hijack the endogenous sAC signaling systems to regulate endothelial microtubule dynamics.
Fig. 4.
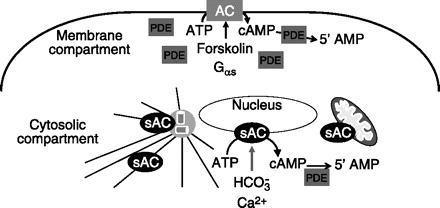
Mammalian cells possess 2 adenylyl cyclase (AC) systems. Transmembrane adenylyl cyclases are anchored into the plasma membrane and when stimulated by forskolin or Gαs generate cAMP in the subplasma membrane compartment. Phosphodiesterases (PDE), critically located at the periphery of the compartment, hydrolyze cAMP to 5′-AMP. In contrast, mammalian soluble adenylyl cyclase 10 (sAC) lacks transmembrane domains and localizes to the centrosome, microtubules, nucleus, and mitochondria. When stimulated by bicarbonate and calcium, sAC generates a phosphodiesterase sensitive cAMP pool in the cytosolic compartment (adapted from Ref. 182).
We have identified sAC in pulmonary microvascular endothelial cells where it generates a bicarbonate-sensitive cAMP pool. Unlike the ExoY- and forskolin-stimulated sACI/II-cAMP pool, the sAC-cAMP pool is regulated by phosphodiesterase 4. Furthermore, this sAC-cAMP pool appears to increase endothelial permeability. These data have been published in abstract form (127). The bicarbonate transitions that induce increased permeability are relevant to bicarbonate therapy in the management of ventilator induced hypercapnic acidosis. The ARDSnet (http://www.ards.org/clinicalnetwork/) suggests sodium bicarbonate infusion to correct acidosis when pH falls below pH 7.3 (3, 83, 158a). Yet within the clinic bicarbonate infusion to correct acidosis is controversial, with some reports suggesting it actually worsens outcomes (89, 153). The role of bicarbonate-stimulated sAC activity in this setting is currently under investigation.
Summary
Understanding of the mechanisms of cAMP regulation of the endothelial barrier has evolved significantly over the last 15–20 years. The emergence of the new cAMP effector, Epac, has recruited many new players into regulation of endothelial barrier. Facilitated by the development of PKA vs. Epac selective analogs, these two cAMP signaling branches have emerged independently only to finally be reunited through common effectors. Yet, still new players are emerging. From the organ level to the tissue level and now to the cellular level, all aspects of cAMP signaling are highly compartmentalized such that the physiological and cellular response to cAMP depends on where it is made. This has been eloquently demonstrated within the endothelium, where plasma membrane cAMP protects the endothelial barrier whereas cytosol cAMP disrupts the barrier. To facilitate signaling specificity, cAMP signaling platforms are localized throughout the cellular environment and are not restricted to the plasma membrane. Thus it appears that the signaling components are already in place for cytosolic cAMP production. One of the future challenges to resolve the physiological function of compartmentalized cAMP signals is to understand the cytosolic bicarbonate-stimulated sAC.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1. Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FE. Epac/Rap1 pathway regulates microvascular hyperpermeability induced by PAF in rat mesentery. Am J Physiol Heart Circ Physiol 294: H1188–H1196, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Adkins WK, Barnard JW, May S, Seibert AF, Haynes J, Taylor AE. Compounds that increase cAMP prevent ischemia-reperfusion pulmonary capillary injury. J Appl Physiol 72: 492–497, 1992. [DOI] [PubMed] [Google Scholar]
- 3. Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338: 347–354, 1998. [DOI] [PubMed] [Google Scholar]
- 4. Ascenzi P, Visca P, Ippolito G, Spallarossa A, Bolognesi M, Montecucco C. Anthrax toxin: a tripartite lethal combination. FEBS Lett 531: 384–388, 2002. [DOI] [PubMed] [Google Scholar]
- 5. Aslam M, Hartel FV, Arshad M, Gunduz D, Abdallah Y, Sauer H, Piper HM, Noll T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: role of CPI-17. Cardiovasc Res 87: 375–384, 2010. [DOI] [PubMed] [Google Scholar]
- 7. Baumer Y, Drenckhahn D, Waschke J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem Cell Biol 129: 765–778, 2008. [DOI] [PubMed] [Google Scholar]
- 8. Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 290: L540–L548, 2006. [DOI] [PubMed] [Google Scholar]
- 9. Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J 21: 2776–2786, 2007. [DOI] [PubMed] [Google Scholar]
- 10. Birukova AA, Arce FT, Moldobaeva N, Dudek SM, Garcia JG, Lal R, Birukov KG. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomedicine 5: 30–41, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J 18: 1879–1890, 2004. [DOI] [PubMed] [Google Scholar]
- 12. Birukova AA, Burdette D, Moldobaeva N, Xing J, Fu P, Birukov KG. Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc Res 79: 128–138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birukova AA, Fu P, Xing J, Birukov KG. Rap1 mediates protective effects of iloprost against ventilator-induced lung injury. J Appl Physiol 107: 1900–1910, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Birukova AA, Liu F, Garcia JG, Verin AD. Protein kinase A attenuates endothelial cell barrier dysfunction induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol 287: L86–L93, 2004. [DOI] [PubMed] [Google Scholar]
- 15. Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol 211: 608–617, 2007. [DOI] [PubMed] [Google Scholar]
- 16. Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol 215: 715–724, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 313: 2504–2520, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blume C, Benz PM, Walter U, Ha J, Kemp BE, Renne T. AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem 282: 4601–4612, 2007. [DOI] [PubMed] [Google Scholar]
- 19. Bogatcheva NV, Verin AD. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc Res 76: 202–207, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bogatcheva NV, Zemskova MA, Kovalenkov Y, Poirier C, Verin AD. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol 221: 750–759, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Borland G, Smith BO, Yarwood SJ. EPAC proteins transduce diverse cellular actions of cAMP. Br J Pharmacol 158: 70–86, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brill S, Li S, Lyman CW, Church DM, Wasmuth JJ, Weissbach L, Bernards A, Snijders AJ. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol Cell Biol 16: 4869–4878, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buonassisi V, Venter JC. Hormone and neurotransmitter receptors in an established vascular endothelial cell line. Proc Natl Acad Sci USA 73: 1612–1616, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cattan CE, Oberg KC. Vinorelbine tartrate-induced pulmonary edema confirmed on rechallenge. Pharmacotherapy 19: 992–994, 1999. [DOI] [PubMed] [Google Scholar]
- 25. Chaudry GJ, Moayeri M, Liu S, Leppla SH. Quickening the pace of anthrax research: three advances point towards possible therapies. Trends Microbiol 10: 58–62, 2002. [DOI] [PubMed] [Google Scholar]
- 26. Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289: 625–628, 2000. [DOI] [PubMed] [Google Scholar]
- 27. Cioffi DL, Moore TM, Schaack J, Creighton JR, Cooper DM, Stevens T. Dominant regulation of interendothelial cell gap formation by calcium-inhibited type 6 adenylyl cyclase. J Cell Biol 157: 1267–1278, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J 16: 583–585, 2002. [DOI] [PubMed] [Google Scholar]
- 29. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481–511, 2007. [DOI] [PubMed] [Google Scholar]
- 30. Creighton J, Zhu B, Alexeyev M, Stevens T. Spectrin-anchored phosphodiesterase 4D4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J Cell Sci 121: 110–119, 2008. [DOI] [PubMed] [Google Scholar]
- 31. Cromartie WJ, Bloom WL, Watson DW. Studies on infection with Bacillus anthracis; a histopathological study of skin lesions produced by B. anthracis in susceptible and resistant animal species. J Infect Dis 80: 1–13, 1947. [DOI] [PubMed] [Google Scholar]
- 32. Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105: 1950–1955, 2005. [DOI] [PubMed] [Google Scholar]
- 33. Dal Molin F, Tonello F, Ladant D, Zornetta I, Zamparo I, Di Benedetto G, Zaccolo M, Montecucco C. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J 25: 5405–5413, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dal Molin F, Zornetta I, Puhar A, Tonello F, Zaccolo M, Montecucco C. cAMP imaging of cells treated with pertussis toxin, cholera toxin, and anthrax edema toxin. Biochem Biophys Res Commun 376: 429–433, 2008. [DOI] [PubMed] [Google Scholar]
- 35. Dale HH, Laidlaw PP. Histamine shock. J Physiol 52: 355–390, 1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474–477, 1998. [DOI] [PubMed] [Google Scholar]
- 37. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121: 2115–2122, 2008. [DOI] [PubMed] [Google Scholar]
- 38. Deng Q, Barbieri JT. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu Rev Microbiol 62: 271–288, 2008. [DOI] [PubMed] [Google Scholar]
- 39. Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83–117, 1997. [DOI] [PubMed] [Google Scholar]
- 40. Dessauer CW, Gilman AG. Purification and characterization of a soluble form of mammalian adenylyl cyclase. J Biol Chem 271: 16967–16974, 1996. [DOI] [PubMed] [Google Scholar]
- 41. DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA 101: 16513–16518, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437: 574–578, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem 273: 22554–22562, 1998. [DOI] [PubMed] [Google Scholar]
- 44. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141–1154, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dubovsky J, Zabramski JM, Kurth J, Spetzler RF, Rich SS, Orr HT, Weber JL. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet 4: 453–458, 1995. [DOI] [PubMed] [Google Scholar]
- 46. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001. [DOI] [PubMed] [Google Scholar]
- 47. El Solh AA, Akinnusi ME, Wiener-Kronish JP, Lynch SV, Pineda LA, Szarpa K. Persistent infection with Pseudomonas aeruginosa in ventilator-associated pneumonia. Am J Respir Crit Care Med 178: 513–519, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 278: 19023–19031, 2003. [DOI] [PubMed] [Google Scholar]
- 49. Engel J, Balachandran P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol 12: 61–66, 2009. [DOI] [PubMed] [Google Scholar]
- 50. Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, van Duin M, Conti M, Gossen JA. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA 101: 2993–2998, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem 273: 21867–21874, 1998. [DOI] [PubMed] [Google Scholar]
- 52. Farrukh IS, Gurtner GH, Michael JR. Pharmacological modification of pulmonary vascular injury: possible role of cAMP. J Appl Physiol 62: 47–54, 1987. [DOI] [PubMed] [Google Scholar]
- 53. Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 147: 2659–2669, 2001. [DOI] [PubMed] [Google Scholar]
- 54. Feng Q, Zhang Y, Li Y, Liu Z, Zuo J, Fang F. Two domains are critical for the nuclear localization of soluble adenylyl cyclase. Biochimie 88: 319–328, 2006. [DOI] [PubMed] [Google Scholar]
- 55. Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci USA 103: 19836–19841, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol 6: 1034–1038, 2004. [DOI] [PubMed] [Google Scholar]
- 57. Fischmeister R. Is cAMP good or bad? Depends on where it's made. Circ Res 98: 582–584, 2006. [DOI] [PubMed] [Google Scholar]
- 58. Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res 99: 816–828, 2006. [DOI] [PubMed] [Google Scholar]
- 59. Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol 25: 136–146, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 163: 510–522, 1995. [DOI] [PubMed] [Google Scholar]
- 61. Garcia JG, Lazar V, Gilbert-McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol 16: 489–494, 1997. [DOI] [PubMed] [Google Scholar]
- 62. Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW. Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol 288: C1305–C1316, 2005. [DOI] [PubMed] [Google Scholar]
- 63. Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 179: 247–254, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 50: 355–375, 2010. [DOI] [PubMed] [Google Scholar]
- 65. Goeckeler ZM, Wysolmerski RB. Myosin phosphatase and cofilin mediate cAMP/cAMP-dependent protein kinase-induced decline in endothelial cell isometric tension and myosin II regulatory light chain phosphorylation. J Biol Chem 280: 33083–33095, 2005. [DOI] [PubMed] [Google Scholar]
- 66. Halbrugge M, Friedrich C, Eigenthaler M, Schanzenbacher P, Walter U. Stoichiometric and reversible phosphorylation of a 46-kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J Biol Chem 265: 3088–3093, 1990. [PubMed] [Google Scholar]
- 67. Han H, Stessin A, Roberts J, Hess K, Gautam N, Kamenetsky M, Lou O, Hyde E, Nathan N, Muller WA, Buck J, Levin LR, Nathan C. Calcium-sensing soluble adenylyl cyclase mediates TNF signal transduction in human neutrophils. J Exp Med 202: 353–361, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO J 15: 2997–3005, 1996. [PMC free article] [PubMed] [Google Scholar]
- 69. Hastie LE, Patton WF, Hechtman HB, Shepro D. H2O2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J Cell Physiol 172: 373–381, 1997. [DOI] [PubMed] [Google Scholar]
- 70. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7: 654–665, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hauser AR, Cobb E, Bodi M, Mariscal D, Valles J, Engel JN, Rello J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit Care Med 30: 521–528, 2002. [DOI] [PubMed] [Google Scholar]
- 72. Hayes JS, Brunton LL, Brown JH, Reese JB, Mayer SE. Hormonally specific expression of cardiac protein kinase activity. Proc Natl Acad Sci USA 76: 1570–1574, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Henes J, Schmit MA, Morote-Garcia JC, Mirakaj V, Kohler D, Glover L, Eldh T, Walter U, Karhausen J, Colgan SP, Rosenberger P. Inflammation-associated repression of vasodilator-stimulated phosphoprotein (VASP) reduces alveolar-capillary barrier function during acute lung injury. FASEB J 23: 4244–4255, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, Levin LR, Williams CJ, Buck J, Moss SB. The “soluble” adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev Cell 9: 249–259, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hong J, Beeler J, Zhukovskaya NL, He W, Tang WJ, Rosner MR. Anthrax edema factor potency depends on mode of cell entry. Biochem Biophys Res Commun 335: 850–857, 2005. [DOI] [PubMed] [Google Scholar]
- 76. Hong J, Doebele RC, Lingen MW, Quilliam LA, Tang WJ, Rosner MR. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J Biol Chem 282: 19781–19787, 2007. [DOI] [PubMed] [Google Scholar]
- 77. Ingber DE. Cellular tensegrity: defining new rules of biological design that govern the cytoskeleton. J Cell Sci 104: 613–627, 1993. [DOI] [PubMed] [Google Scholar]
- 78. Jay D, Garcia EJ, Lara JE, Medina MA, de la Luz Ibarra M. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Arch Biochem Biophys 377: 80–84, 2000. [DOI] [PubMed] [Google Scholar]
- 79. Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci USA 93: 295–299, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science 282: 2275–2279, 1998. [DOI] [PubMed] [Google Scholar]
- 81. Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras-related gene with transformation suppressor activity. Cell 56: 77–84, 1989. [DOI] [PubMed] [Google Scholar]
- 82. Kobayashi H, Kobayashi T, Fukushima M. Effects of dibutyryl cAMP on pulmonary air embolism-induced lung injury in awake sheep. J Appl Physiol 63: 2201–2207, 1987. [DOI] [PubMed] [Google Scholar]
- 83. Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med 332: 27–37, 1995. [DOI] [PubMed] [Google Scholar]
- 84. Kolosova IA, Ma SF, Adyshev DM, Wang P, Ohba M, Natarajan V, Garcia JG, Verin AD. Role of CPI-17 in the regulation of endothelial cytoskeleton. Am J Physiol Lung Cell Mol Physiol 287: L970–L980, 2004. [DOI] [PubMed] [Google Scholar]
- 85. Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett 579: 4966–4972, 2005. [DOI] [PubMed] [Google Scholar]
- 86. Krall R, Zhang Y, Barbieri JT. Intracellular membrane localization of pseudomonas ExoS and Yersinia YopE in mammalian cells. J Biol Chem 279: 2747–2753, 2004. [DOI] [PubMed] [Google Scholar]
- 87. Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 4: 294–301, 2002. [DOI] [PubMed] [Google Scholar]
- 88. Kurachi H, Wada Y, Tsukamoto N, Maeda M, Kubota H, Hattori M, Iwai K, Minato N. Human SPA-1 gene product selectively expressed in lymphoid tissues is a specific GTPase-activating protein for Rap1 and Rap2. Segregate expression profiles from a rap1GAP gene product. J Biol Chem 272: 28081–28088, 1997. [DOI] [PubMed] [Google Scholar]
- 89. Laffey JG, Engelberts D, Kavanagh BP. Buffering hypercapnic acidosis worsens acute lung injury. Am J Respir Crit Care Med 161: 141–146, 2000. [DOI] [PubMed] [Google Scholar]
- 90. Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 15: 510–519, 1996. [PMC free article] [PubMed] [Google Scholar]
- 91. Lee JS, Gotlieb AI. Understanding the role of the cytoskeleton in the complex regulation of the endothelial repair. Histol Histopathol 18: 879–887, 2003. [DOI] [PubMed] [Google Scholar]
- 92. Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA 79: 3162–3166, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem 278: 15922–15926, 2003. [DOI] [PubMed] [Google Scholar]
- 94. Liu F, Verin AD, Borbiev T, Garcia JG. Role of cAMP-dependent protein kinase A activity in endothelial cell cytoskeleton rearrangement. Am J Physiol Lung Cell Mol Physiol 280: L1309–L1317, 2001. [DOI] [PubMed] [Google Scholar]
- 95. Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther 109: 366–398, 2006. [DOI] [PubMed] [Google Scholar]
- 96. Magiera MM, Gupta M, Rundell CJ, Satish N, Ernens I, Yarwood SJ. Exchange protein directly activated by cAMP (EPAC) interacts with the light chain (LC) 2 of MAP1A. Biochem J 382: 803–810, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol 11: 571–605, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging 16: 355–362; discussion 362–353, 1995. [DOI] [PubMed] [Google Scholar]
- 99. Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc 2: 206–213, 2005. [DOI] [PubMed] [Google Scholar]
- 100. McConnachie G, Langeberg LK, Scott JD. AKAP signaling complexes: getting to the heart of the matter. Trends Mol Med 12: 317–323, 2006. [DOI] [PubMed] [Google Scholar]
- 101. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006. [DOI] [PubMed] [Google Scholar]
- 102. Michel CC, Curry FE. Microvascular permeability. Physiol Rev 79: 703–761, 1999. [DOI] [PubMed] [Google Scholar]
- 103. Moore TM, Chetham PM, Kelly JJ, Stevens T. Signal transduction and regulation of lung endothelial cell permeability. Interaction between calcium and cAMP. Am J Physiol Lung Cell Mol Physiol 275: L203–L222, 1998. [DOI] [PubMed] [Google Scholar]
- 104. Moy AB, Shasby SS, Scott BD, Shasby DM. The effect of histamine and cyclic adenosine monophosphate on myosin light chain phosphorylation in human umbilical vein endothelial cells. J Clin Invest 92: 1198–1206, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Moy AB, Van Engelenhoven J, Bodmer J, Kamath J, Keese C, Giaever I, Shasby S, Shasby DM. Histamine and thrombin modulate endothelial focal adhesion through centripetal and centrifugal forces. J Clin Invest 97: 1020–1027, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nakamura F, Osborn TM, Hartemink CA, Hartwig JH, Stossel TP. Structural basis of filamin A functions. J Cell Biol 179: 1011–1025, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem 279: 37215–37218, 2004. [DOI] [PubMed] [Google Scholar]
- 108. Noda K, Zhang J, Fukuhara S, Kunimoto S, Yoshimura M, Mochizuki N. Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through alpha- and beta-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol Biol Cell 21: 584–596, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pappenheimer JR, Renkin EM, Borrero LM. Filtration, diffusion and molecular sieving through peripheral capillary membranes; a contribution to the pore theory of capillary permeability. Am J Physiol 167: 13–46, 1951. [DOI] [PubMed] [Google Scholar]
- 110. Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium 7: 287–308, 2000. [DOI] [PubMed] [Google Scholar]
- 111. Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol 28: 574–581, 2003. [DOI] [PubMed] [Google Scholar]
- 112. Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep 5: 1176–1180, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Prasain N, Alexeyev M, Balczon R, Stevens T. Soluble adenylyl cyclase-dependent microtubule disassembly reveals a novel mechanism of endothelial cell retraction. Am J Physiol Lung Cell Mol Physiol 297: L73–L83, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res 77: 53–63, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Qiao J, Holian O, Lee BS, Huang F, Zhang J, Lum H. Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am J Physiol Cell Physiol 295: C1161–C1168, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 284: L972–L980, 2003. [DOI] [PubMed] [Google Scholar]
- 117. Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem 277: 26581–26586, 2002. [DOI] [PubMed] [Google Scholar]
- 118. Rall TW. Opening remarks. Adv Cyclic Nucleotide Res 5: 1–2, 1975. [Google Scholar]
- 119. Rampersad SN, Ovens JD, Huston E, Umana MB, Wilson LS, Netherton SJ, Lynch MJ, Baillie GS, Houslay MD, Maurice DH. Cyclic AMP phosphodiesterase 4D (PDE4D) tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J Biol Chem 285: 33614–33622, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol 116: 147–161, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA 98: 13049–13054, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Roscioni SS, Elzinga CR, Schmidt M. Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol 377: 345–357, 2008. [DOI] [PubMed] [Google Scholar]
- 123. Rosenfeldt H, Castellone MD, Randazzo PA, Gutkind JS. Rac inhibits thrombin-induced Rho activation: evidence of a Pak-dependent GTPase crosstalk. J Mol Signal 1: 8, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis 183: 1767–1774, 2001. [DOI] [PubMed] [Google Scholar]
- 125. Ryan US, Clements E, Habliston D, Ryan JW. Isolation and culture of pulmonary artery endothelial cells. Tissue Cell 10: 535–554, 1978. [DOI] [PubMed] [Google Scholar]
- 126. Sayner S, Stevens T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem Soc Trans 34: 492–494, 2006. [DOI] [PubMed] [Google Scholar]
- 127. Sayner SL. Bicarbonate stimulation of endogenous soluble adenylyl cyclase, AC10, in pulmonary microvascular endothelial cells disrupts the endothelial cell barrier. Am J Respir Crit Care Med 181: A3415, 2010. [Google Scholar]
- 128. Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res 98: 675–681, 2006. [DOI] [PubMed] [Google Scholar]
- 129. Sayner SL, Balczon R, Frank DW, Cooper DM, Stevens T. Targets of subplasma membrane versus cytosolic adenylyl cyclase activity demonstrate the bidirectional nature of the camp diffusion barricade that maintains endothelial barrier integrity. Am J Respir Crit Care Med 181: A1037, 2010. [Google Scholar]
- 130. Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res 95: 196–203, 2004. [DOI] [PubMed] [Google Scholar]
- 131. Schlegel N, Waschke J. Impaired integrin-mediated adhesion contributes to reduced barrier properties in VASP-deficient microvascular endothelium. J Cell Physiol 220: 357–366, 2009. [DOI] [PubMed] [Google Scholar]
- 132. Schlegel N, Waschke J. Vasodilator-stimulated phosphoprotein: crucial for activation of Rac1 in endothelial barrier maintenance. Cardiovasc Res 87: 1–3, 2010. [DOI] [PubMed] [Google Scholar]
- 133. Schlegel N, Waschke J. VASP is involved in cAMP-mediated Rac 1 activation in microvascular endothelial cells. Am J Physiol Cell Physiol 296: C453–C462, 2009. [DOI] [PubMed] [Google Scholar]
- 134. Schmid A, Bai G, Schmid N, Zaccolo M, Ostrowski LE, Conner GE, Fregien N, Salathe M. Real-time analysis of cAMP-mediated regulation of ciliary motility in single primary human airway epithelial cells. J Cell Sci 119: 4176–4186, 2006. [DOI] [PubMed] [Google Scholar]
- 135. Schmid A, Sutto Z, Nlend MC, Horvath G, Schmid N, Buck J, Levin LR, Conner GE, Fregien N, Salathe M. Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J Gen Physiol 130: 99–109, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: 3549–3558, 1999. [DOI] [PubMed] [Google Scholar]
- 137. Sehrawat S, Cullere X, Patel S, Italiano J, Jr, Mayadas TN. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell 19: 1261–1270, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sehrawat S, Ernandez T, Cullere X, Takahashi M, Ono Y, Komarova Y, Mayadas TN. AKAP9 regulation of microtubule dynamics promotes Epac1 induced endothelial barrier properties. Blood 117: 708–718, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sinclair ML, Wang XY, Mattia M, Conti M, Buck J, Wolgemuth DJ, Levin LR. Specific expression of soluble adenylyl cyclase in male germ cells. Mol Reprod Dev 56: 6–11, 2000. [DOI] [PubMed] [Google Scholar]
- 140. Smith H, Keppie J, Stanley JL, Harris-Smith PW. The chemical basis of the virulence of Bacillus anthracis IV Secondary shock as the major factor in death of guinea-pigs from anthrax. Br J Exp Pathol 36: 323–335, 1955. [PMC free article] [PubMed] [Google Scholar]
- 141. Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem 275: 25723–25732, 2000. [DOI] [PubMed] [Google Scholar]
- 142. Smurova KM, Biriukova AA, Verin AD, Alieva IB. [The microtubule system in endothelial barrier dysfunction: disassembly of peripheral microtubules and microtubules reorganization in internal cytoplasm]. Tsitologiia 50: 49–55, 2008. [PubMed] [Google Scholar]
- 143. Stanley JL, Smith H. Purification of factor I and recognition of a third factor of the anthrax toxin. J Gen Microbiol 26: 49–63, 1961. [DOI] [PubMed] [Google Scholar]
- 144. Staub NC. Pulmonary edema. Physiol Rev 54: 678–811, 1974. [DOI] [PubMed] [Google Scholar]
- 145. Staub NC. “State of the art” review. Pathogenesis of pulmonary edema. Am Rev Respir Dis 109: 358–372, 1974. [DOI] [PubMed] [Google Scholar]
- 146. Stelzner TJ, Weil JV, O'Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 139: 157–166, 1989. [DOI] [PubMed] [Google Scholar]
- 147. Stevens T, Nakahashi Y, Cornfield DN, McMurtry IF, Cooper DM, Rodman DM. Ca2+-inhibitable adenylyl cyclase modulates pulmonary artery endothelial cell cAMP content and barrier function. Proc Natl Acad Sci USA 92: 2696–2700, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Strada SJ, Thompson WJ. Multiple forms of cyclic nucleotide phosphodiesterases: anomalies or biologic regulators? Adv Cyclic Nucleotide Res 9: 265–283, 1978. [PubMed] [Google Scholar]
- 149. Sutherland EW. Studies on the mechanism of hormone action. Science 177: 401–408, 1972. [DOI] [PubMed] [Google Scholar]
- 150. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem 232: 1077–1091, 1958. [PubMed] [Google Scholar]
- 151. Tang WJ, Gilman AG. Construction of a soluble adenylyl cyclase activated by Gs alpha and forskolin. Science 268: 1769–1772, 1995. [DOI] [PubMed] [Google Scholar]
- 152. Tar K, Birukova AA, Csortos C, Bako E, Garcia JG, Verin AD. Phosphatase 2A is involved in endothelial cell microtubule remodeling and barrier regulation. J Cell Biochem 92: 534–546, 2004. [DOI] [PubMed] [Google Scholar]
- 153. Tobin MJ. Mechanical ventilation. N Engl J Med 330: 1056–1061, 1994. [DOI] [PubMed] [Google Scholar]
- 154. Townsend PD, Holliday PM, Fenyk S, Hess KC, Gray MA, Hodgson DR, Cann MJ. Stimulation of mammalian G-protein-responsive adenylyl cyclases by carbon dioxide. J Biol Chem 284: 784–791, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell 6: 1887–1902, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Vallis AJ, Finck-Barbancon V, Yahr TL, Frank DW. Biological effects of Pseudomonas aeruginosa type III-secreted proteins on CHO cells. Infect Immun 67: 2040–2044, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Van Horck FP, Ahmadian MR, Haeusler LC, Moolenaar WH, Kranenburg O. Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J Biol Chem 276: 4948–4956, 2001. [DOI] [PubMed] [Google Scholar]
- 158. Vance RE, Rietsch A, Mekalanos JJ. Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect Immun 73: 1706–1713, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158a. Ventilation with lower tidal volumes compared with traditional tidal volumes for acute lung injury, and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 342: 1301–1308, 2000. [DOI] [PubMed] [Google Scholar]
- 159. Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am J Physiol Lung Cell Mol Physiol 281: L565–L574, 2001. [DOI] [PubMed] [Google Scholar]
- 160. Verin AD, Gilbert-McClain LI, Patterson CE, Garcia JG. Biochemical regulation of the nonmuscle myosin light chain kinase isoform in bovine endothelium. Am J Respir Cell Mol Biol 19: 767–776, 1998. [DOI] [PubMed] [Google Scholar]
- 161. Verin AD, Wang P, Garcia JG. Immunochemical characterization of myosin-specific phosphatase 1 regulatory subunits in bovine endothelium. J Cell Biochem 76: 489–498, 2000. [PubMed] [Google Scholar]
- 162. Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem 243: 3763–3765, 1968. [PubMed] [Google Scholar]
- 163. Wang Q, Patton WF, Chiang ET, Hechtman HB, Shepro D. Filamin translocation is an early endothelial cell inflammatory response to bradykinin: regulation by calcium, protein kinases, and protein phosphatases. J Cell Biochem 62: 383–396, 1996. [DOI] [PubMed] [Google Scholar]
- 164. Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med 353: 2788–2796, 2005. [DOI] [PubMed] [Google Scholar]
- 165. Waschke J, Burger S, Curry FR, Drenckhahn D, Adamson RH. Activation of Rac-1 and Cdc42 stabilizes the microvascular endothelial barrier. Histochem Cell Biol 125: 397–406, 2006. [DOI] [PubMed] [Google Scholar]
- 166. Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol 288: H1296–H1305, 2005. [DOI] [PubMed] [Google Scholar]
- 167. Weis WI, Nelson WJ. Re-solving the cadherin-catenin-actin conundrum. J Biol Chem 281: 35593–35597, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Werthmann RC, von Hayn K, Nikolaev VO, Lohse MJ, Bunemann M. Real-time monitoring of cAMP levels in living endothelial cells: thrombin transiently inhibits adenylyl cyclase 6. J Physiol 587: 4091–4104, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127: 1027–1039, 2006. [DOI] [PubMed] [Google Scholar]
- 170. Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev 87: 965–1010, 2007. [DOI] [PubMed] [Google Scholar]
- 171. Wong MK, Gotlieb AI. Endothelial cell monolayer integrity. I. Characterization of dense peripheral band of microfilaments. Arteriosclerosis 6: 212–219, 1986. [DOI] [PubMed] [Google Scholar]
- 172. Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol 5: 959–970, 2004. [DOI] [PubMed] [Google Scholar]
- 173. Wysolmerski RB, Lagunoff D. Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci USA 87: 16–20, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Xie F, Conti M. Expression of the soluble adenylyl cyclase during rat spermatogenesis: evidence for cytoplasmic sites of cAMP production in germ cells. Dev Biol 265: 196–206, 2004. [DOI] [PubMed] [Google Scholar]
- 175. Xie F, Garcia MA, Carlson AE, Schuh SM, Babcock DF, Jaiswal BS, Gossen JA, Esposito G, van Duin M, Conti M. Soluble adenylyl cyclase (sAC) is indispensable for sperm function and fertilization. Dev Biol 296: 353–362, 2006. [DOI] [PubMed] [Google Scholar]
- 176. Yahr TL, Goranson J, Frank DW. Exoenzyme S of Pseudomonas aeruginosa is secreted by a type III pathway. Mol Microbiol 22: 991–1003, 1996. [DOI] [PubMed] [Google Scholar]
- 177. Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci USA 95: 13899–13904, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295: 1711–1715, 2002. [DOI] [PubMed] [Google Scholar]
- 179. Zhu B, Zhang L, Creighton J, Alexeyev M, Strada SJ, Stevens T. Protein kinase A phosphorylation of tau-serine 214 reorganizes microtubules and disrupts the endothelial cell barrier. Am J Physiol Lung Cell Mol Physiol 299: L493–L501, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17: 82–84, 2003. [DOI] [PubMed] [Google Scholar]
- 181. Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA, Levin LR, Buck J. Bicarbonate-responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J Cell Biol 164: 527–534, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zippin JH, Levin LR, Buck J. CO2/HCO3−-responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol Metab 12: 366–370, 2001. [DOI] [PubMed] [Google Scholar]