

Abstract
Epidemiologic studies associate exposure to ambient particulate matter (APM) with increased cardiovascular mortality. Since both pulmonary inflammation and systemic circulation of ultrafine particles are hypothesized as initiating cardiovascular effects, we examined responses of potential target cells in vitro. Human aortic endothelial cells (HAEC) were exposed to 10 μg/ml fine and ultrafine APM collected in an urban setting in summer 2006 or winter 2007 in the San Joaquin Valley, California. RNA isolated after 3 h was analyzed with high-density oligonucleotide arrays. Summer APM treatment affected genes involved in xenobiotic and oxidoreductase activity, transcription factors, and inflammatory responses in HAEC, while winter APM had a robust xenobiotic but lesser inflammatory response. Real-time polymerase chain reaction analysis confirmed that particulate matter (PM)-treated HAEC increased mRNA levels of xenobiotic response enzymes CYP1A1, ALDH1A3, and TIPARP and cellular stress response transcription factor ATF3. Inflammatory response genes included E-selectin, PTGS2, CXCL-2 (MIP-2α), and CCL-2 (MCP-1). Multiplex protein assays showed secretion of IL-6 and MCP-1 by HAEC. Since induction of CYP1A1 is mediated through the ligand-activated aryl hydrocarbon receptor (AhR), we demonstrated APM induced AhR nuclear translocation by immunofluorescence and Western blotting and activation of the AhR response element using a luciferase reporter construct. Inhibitor studies suggest differential influences of polycyclic aromatic hydrocarbon signaling, ROS-mediated responses and endotoxin alter stress and proinflammatory endothelial cell responses. Our findings demonstrate gene responses correlated with current concepts that systemic inflammation drives cardiovascular effects of particulate air pollution. We also demonstrate a unique pattern of gene responses related to xenobiotic metabolism in PM-exposed HAEC.
Keywords: oligonucleotide arrays, environmental particulate matter, inflammation, aryl hydrocarbon receptor, cytochrome P450
epidemiologic studies indicate that peaks of ambient particulate matter (APM) air pollution are associated with an increase in pulmonary and cardiovascular morbidity and mortality. Recent studies suggest a greater risk for cardiovascular than respiratory consequences during episodes of urban air pollution (55). Several lines of evidence suggest APM causes cardiovascular effects through induction of a systemic proinflammatory and procoagulant state (10, 46, 47, 71). The mechanism connecting inhalation of environmental particulate matter (PM) and systemic thrombotic and inflammatory effects remains an enigma. One hypothesis suggests that translocation of PM, especially the ultrafine portion, into the systemic circulation would interact with vascular endothelium to augment activation of circulating leukocytes and platelets. Alternatively, PM deposited in the lung might interact with pulmonary endothelium to enhance activation of circulating elements of blood.
APM is a complex mixture of liquid and/or solid materials suspended in the air. Its chemical composition varies greatly and depends on many factors including combustion sources, climate, season, distance from source and type of urban or industrial pollution. Regulatory agencies classify APM in relative size ranges as coarse (2.5–10 μm) fine (<2.5 μm) and ultrafine (<0.1 μm). Anthropogenic APM is generally composed of a particle core of carbonaceous material, mainly from combustion processes and vehicular exhaust particles. A variety of compounds can be absorbed on these including volatile or semivolatile organic species [e.g., polycyclic aromatic hydrocarbons (PAHs), nitro-PAHs, quinones], transition metals (iron, nickel, vanadium, copper, etc.), ions (sulfate, nitrate, acidity), and reactive gases (ozone, peroxides, aldehydes). Other components of APM include materials of biologic origin (endotoxins, bacteria, viruses, animal and plant debris) and salts and minerals (ammonium, nitrate, sodium sulfate, silicates, and other crustal elements). Coarse particles consist mainly of insoluble crust-derived minerals, sea salt, and material of biologic origin, while fine and ultrafine particles (UFP) are mainly salts admixed with carbonaceous aggregates with metals and organic species adsorbed on their surface (4, 66).
Regulation of PM exposure is largely based on size and concentration with relatively little consideration given to composition. A recurring hypothesis is that induction of reactive oxygen species catalyzed by transition metals in Fenton-like reactions is a key cellular stress pathway inducing pulmonary and perhaps systemic responses. The role of a variety of more complex, potentially biologically active, organic components has received less attention. Some evidence suggests a significant role for endotoxin in stimulating effects in pulmonary epithelial cells and alveolar macrophages (6).
A variety of PAHs are also found in APM, especially in urban areas. These compounds result from a combination of incomplete combustion of petroleum-based fuels, burning of biomass, and atmospheric chemical reactions with short chain hydrocarbons (57). While such compounds are readily detected through air pollution monitoring, their cellular or biological effects relative to PM inhalation have received little investigation.
Several investigations demonstrate transport of ultrafine PM from the lungs to the systemic circulation and their accumulation in extrapulmonary organs (i.e., liver, heart, spleen, brain) (24, 49, 50, 62, 65). These findings suggest that in addition to pulmonary inflammation (59), UFP in circulation could directly or indirectly influence hemostasis or vascular inflammation and thus induce adverse cardiovascular endpoints more directly (46–48).
Much of the research evaluating pulmonary response to environmental particulates has focused on upregulation of pulmonary inflammatory cytokines such as interleukin (IL)-6 or induction of oxidative stress and its associated response elements (5, 39). Additional studies of cytokines in subjects exposed to a wide variety of PM pollutants demonstrated that several proinflammatory cytokines are elevated in the circulation (67). These subjects showed significant increases in blood levels of IL-6, IL-1β, macrophage inflammatory protein 1α (MIP-1α), and granulocyte-macrophage colony-stimulating factor (GM-CSF), leading the authors to suggest that acute exposure to air pollution induces a systemic inflammatory response contributing to cardiopulmonary disease (67).
Cellular interactions with and responses to APM have largely been investigated in cultured lung cells. Particles of differing sources and sizes elicit a variety of responses in cultured airway epithelial cells that generally relate to inflammation or oxidant stress responses. Coarse particles are associated more with proinflammatory responses, which may be correlated with endotoxin and Toll-like receptor activity. Fine and ultrafine particles more characteristically elicit oxidant stress responses (6). Microarray-based gene response studies are beginning to expand the range and specificity of responses to PM. They have been used to distinguish responses to zinc and vanadium in airway epithelial cell cultures (27). A similar approach identified membrane transport mechanisms, extracellular matrix proteins, adhesion molecules, and signal transduction responses in umbilical endothelial cell cultures treated with residual oil fly ash (42).
In the present study, we asked whether seasonal variation in environmental PM composition affected the global gene response patterns in cultured human cells representing systemic vascular targets. We also asked whether selective gene transcription identified response pathways are consistent with systemic inflammation or characteristic of PM exposure.
MATERIAL AND METHODS
Reagents
PCR primers were from Operon (Huntsville, AL). Antibodies were purchased from the following sources: aryl hydrocarbon receptor (AhR, Sc-5579) from Santa Cruz Biotechnology (Santa Cruz, CA); donkey-anti-rabbit antibody conjugated to Alexa Fluor 488 from Molecular Probes (Eugene, OR). Monoclonal anti-β-actin (A5441); dimethyl sulfoxide (DMSO), α-napthoflavone (α-NF, N5757), resveratrol (R5010), and polymyxin B (pol B) sulfate salt (P4932) were purchased from Sigma Chemical (St. Louis, MO); cell culture reagents and media from Lonza (Walkersville, MD). Apo-ONE homogeneous caspase-3/7 assay reagents (catalog no. G7790) was from Promega (Madison, WI), and nuclear-ID blue/green cell viability assay reagent (catalog no. ENZ-53004-C100) was from Enzo Life Sciences (Plymouth Meeting, PA). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was a gift from Dr. S. Safe (Texas A&M University).
Collection of Fine and Ultrafine Concentrated APM
The University of California (UC) Center Fresno sampling site is located in a mixed retail commercial and residential setting. The site is bound by residences to the north, a commercial property to the east, a principal arterial road to the south, and a collector road to the west. The site is approximately two blocks east of Highway 41 and 6 miles north of its intersection with Highway 99. This places the site in the center of an urban area on the east side of the San Joaquin Valley near the junction of two major transportation routes with heavy use for truck transport. APM were collected using a high-volume sampler combined with a PM2.5 cutoff cascade impactor in summer of 2006 (sum06) and winter of 2007 (win07). The summer sampling was done during a period with high potential for photochemical modification of emissions. The winter sampling was done during a typical winter temperature inversion associated with high PM concentrations in the San Joaquin Valley. Bulk APM samples were collected with three colocated reference ambient air monitor filter samplers loaded with prebaked (48 h at 550°C) Teflon-coated quartz fiber filters. Cumulative samples were collected during a field exposure lasting 2 wk. Samples were collected for 6 h a day for 5 days each week. Collected filters were sealed in an argon atmosphere and stored at −80°C until extracted. Particles for cell treatment were extracted by probe sonication in H2O. Preliminary experiments with laboratory generated 100 nm particles demonstrated efficient recapitulation of particle size with this approach as determined by dynamic light scattering (data not shown). Estimates of extracted particle mass were made by weighing filters before and after extraction using a humidity control chamber to control for water weight. Extracted particles suspended in H2O were refrozen at −80°C until diluted for cell culture experiments.
Particle Characterization
Size-resolved PM samples were collected with six identical micro-orifice uniform deposit impactors with size cuts of 0.056, 0.1, 0.18, 0.32, 0.56, 1.0, and 1.8 μm particle diameter. Particle characterization, detailed in a separate manuscript consisted of elemental carbon and organic carbon concentrations determined by thermo-optical analysis, water-soluble ions quantified with ion chromatography analysis and trace elements quantified with inductively coupled plasma mass spectrometry (Table 1). Analysis of a panel of PAHs from sum06 was done by gas chromatography (47).
Table 1.
Characterization of Summer 2006 and Winter 2007 APM in Fresno, CA
| Summer 2006 |
Winter 2007 |
|||
|---|---|---|---|---|
| Percent Total Mass | PM1.8 | PM0.1 | PM1.8 | PM0.1 |
| Major species (ppm = %) | ||||
| OC | 23.58% | 41.03% | 16.30% | 27.03% |
| EC | 5.43% | 17.95% | 2.54% | 8.11% |
| NO3 | 6.13% | 15.38% | 31.56% | NA |
| SO4 | 13.76% | 10.26% | 5.66% | 2.70% |
| NH4+ | 3.53% | 0.00% | 10.81% | NA |
| Trace species (ppm = % * 10,000) | ||||
| Fe/57 | 6.99 | 3.33 | 12.95 | NA |
| Cu/63 | 1.24 | 11.54 | 0.65 | 9.73 |
| Zn/66 | 1.29 | 8.72 | 1.12 | 8.38 |
| As/75 | 0.06 | 0.26 | 0.13 | NA |
| Benzo(e)pyrene | 0.0049 | 0.0460 | 0.0143 | NA |
| Benzo(a)pyrene | 0.0042 | NA | 0.0118 | 0.0433 |
| Indeno(1,2,3-cd)pyrene | 0.0090 | 0.0640 | 0.0134 | 0.1441 |
| Benzo(GHI)perylene | 0.0120 | 0.1102 | 0.0098 | 0.0816 |
| Dibenz(ah)anthracene | 0.0112 | NA | 0.0097 | NA |
| Coronene | 0.0114 | 0.1142 | 0.0160 | 0.1058 |
APM, ambient particulate matter; PM, particulate matter; OC, organic carbon; EC, elemental carbon; NA, not available. For details see Ref. 57.
Cell Culture and Treatments
Human aortic endothelial cells (HAEC) were purchased from Lonza (Portland, OR). HAEC were cultured in EBM basal media supplemented with EGM-2 bullet kit (EBM plus Single Quotes growth supplements) (CC-3124). Experiments were conducted with cells at passages 4–6 that were maintained at 37°C in 5% CO2 and 95% air incubator. APM collected in sum06 and win07 was used at estimated concentration of 10 μg/ml for cell-based AhR-transcription bioassay, caspase 3/7 assay, and microarray analysis. For inhibitor studies, 10 μg/ml sum06 APM were used. In immunofluorescence studies 10 or 50 μg/ml sum06 APM were used. Particle extracts were probe sonicated immediately before treatment for uniform dispread. Cells were treated with control (H2O) or APM for 3 h unless otherwise stated.
Time-Course Studies in HAEC Exposed to Sum06 APM
To characterize the time course of gene responses, HAEC were grown to confluence on six-well plates and then treated with control (H2O) or sum06 APM (10 μg/ml) for 30 min, 1 h, 2 h, 3 h, and 4 h. After incubation, cells were washed with PBS (without Ca and Mg), and RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA) including the DNA digestion step as described by the manufacturer. mRNA expression of specific genes were analyzed by quantitative RT-PCR (qRT-PCR) as described below.
Cell Viability Assay
To determine the toxicity of different APM sources, HAEC were grown to confluence on fibronectin-coated 12-mm round coverslips placed in 24-well medical-grade polystyrene plates (BD Falcon) and were treated with control (H2O) or 10 μg/ml of sum06 APM or win07 APM for 3 h. The samples were stained with Nuclear -ID Blue Green cell viability reagent as described by the manufacturer. Coverslips were mounted, and digital micrographs of six randomly chosen fields per coverslip obtained with an Olympus BX61 fluorescence microscope. A live-dead ratio was determined by counting green fluorescent nuclei (dead cells) relative to all nuclei in each field.
Apoptosis Assays
HAEC were grown to confluence on 96-well plate and then treated with control (H2O) or 10 μg/ml of sum06 APM or win07 APM for 3 h. After incubation, caspase 3/7 activity was measured by Apo-ONE Homogeneous Caspase-3/7 assay (Promega) as described by the manufacturer. The samples were measured by Fluorescence microplate reader (Molecular Devices, SpectraMax M2) with a 485 nm excitation filter and 530 nm emission filter.
RNA Extraction and Synthesis of Biotin-Labeled RNA
Total RNA was extracted from control and APM-treated HAEC in T-75 flasks using TRIzol Reagent (Invitrogen, Carlsbad, CA) followed by RNeasy Mini Kit (Qiagen) including the DNA digestion step as described by the manufacturer. Microarray experiments were performed with pooled RNA isolates from each treatment group. Confirmatory RT-PCR analysis was done on individual aliquots of RNA from each replicate flask.
GeneChip analyses.
GeneChip analyses of the pooled total RNA samples (n = 4 for sum06 treated HAEC and n = 3 for win07 treated HAEC) were performed as previously described (3). A 9 μg aliquot of total RNA from each pooled sample was reverse-transcribed, followed by second-strand synthesis, cRNA amplification in the presence of biotin-labeled nucleotides, and fragmentation as described in the Affymetrix One Cycle Sample Preparation protocol (Affymetrix, Santa Clara, CA). The fragmented, biotin-labeled cRNA samples were hybridized to Human Genome U133A 2.0 Array high-density oligonucleotide arrays with ∼22,000 probe sets representing 14,500 well-characterized human genes (Affymetrix). The hybridization, washing, labeling, and scanning of the GeneChips were performed as described in the Affymetrix protocols by the Microarray Core Facility in the UC Davis Genome and Biomedical Sciences Facility. Complete microarray data are available at http://www.ncbi.nlm.nih.gov/geo/ (Gene Expression Omnibus accession number GSE18593).
Analysis and interpretation of GeneChip data.
The data were analyzed by GeneChip Operating System (GCOS) 1.4 (Affymetrix). The upper limit of P value for statistically reliable detection of an mRNA transcript was 0.05 (except for “batch analysis,” see below), independent of its signal intensity (usually >10), because the detected mRNA can be confirmed by an independent method such as qRT-PCR. The P value for detection of mRNAs discussed here ranged from 0.0001 to 0.05 (except for “batch analysis,” see below), and the signal intensity ranged from 10 to 10,000 units; the signal intensities approximate the abundance of mRNAs.
Lists of APM-responsive genes were obtained by using the “batch analysis” function in GCOS 1.4. Data for all of the 22,277 probe sets from HAEC treated with control were used as baseline and compared with those from the HAEC treated with APM. The lists (in Excel format) of sensitive genes were then sorted to satisfy three requirements: 1) the mRNA must be detectable with a P value of ≤0.05 in at least one of the two samples being compared, 2) the fold-change be ≥2, and 3), the mRNA have an annotation that suggests either a known or a predicted function. The lower limit of twofold change was selected and selected gene expression was confirmed by qRT-PCR. This may be particularly desirable in microarray experiments performed on pooled RNA samples, an analytical strategy that results in loss of statistical information (discussed in results). Affymetrix GCOS software was used to facilitate visualization and interpretation of GeneChip Data. The functional groups were assigned using PubMed Gene Data base.
Validation of changes in mRNA expression by qRT-PCR.
Since GeneChip analysis was done on pooled samples from each experimental group, confirming qRT-PCR analyses were performed on individual aliquots of total RNA samples from each treatment replicate. The purpose of these analyses was to evaluate the reliability of GeneChip data and develop statistical data to validate the changes suggested by the GeneChip assay of pooled RNA samples as our previous studied (3). Frequently, the two assays do not match quantitatively but they always match qualitatively. Several analytical procedures may contribute to these discrepancies. 1) Total RNA samples used for the PCR assay are different from those used for the microarray assays. 2) The method of hybridization and detection for the two assays are very different; the microarray assay utilizes hybridization of fragmented cRNA generated by Affymetrix protocols, whereas the RT-PCR utilizes amplication of DNA fragments using specific primer sets designed by a different method. These PCR primers are usually different from those on GeneChips.
An aliquot equivalent to 5 μg of total RNA extracted from each sample was reverse-transcribed to obtain cDNA in a final volume of 20 μl solution consisting of buffer, oligo-dT primer, DTT, dNTPs, and Superscript-II reverse transcriptase (Invitrogen). RT-PCR with SYBR as fluorescent reporter was used to quantify the expression of selected genes identified by GeneChip analysis. Specific primers (Table 2) were designed with Primer Express 1.0 software (Applied Biosystems) using the gene sequences obtained from Affymetrix Probeset IDs. Reactions were carried out in 384-well optical plates containing 25 ng RNA in each well. The quantity of applied RNA was normalized by simultaneously amplifying cDNA samples with glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific primers. The transcript levels were measured by real-time RT-PCR using the ABI PRISM 7700 Sequence detection system (PE Applied Biosystems, Foster City, CA). The PCR amplification parameters were: initial denaturation step at 95°C for 10 min followed by 40 cycles, each at 95°C for 15 s (melting) and 60°C for 1 min (annealing and extension). A comparative threshold cycle (Ct) method (30) was used to calculate relative changes in gene expression determined from real-time quantitative PCR experiments [Applied Biosystems user bulletin no. 2 (P/N4303859)]. The Ct, which correlates inversely with the target mRNA levels, was measured as the cycle number at which the SYBR Green emission increases above a preset threshold level. The specific mRNA transcripts were expressed as fold difference in the expression of the specific mRNAs in RNA samples from the APM-treated cells compared with those from the control-treated cells.
Table 2.
Primer sequences for quantitative real-time PCR
| Gene | Primer sequence (5′-3′) |
|---|---|
| GAPDH | Sense-CACCAACTGCTTAG |
| Antisense-TGGTCATGAGTCCT | |
| CYP1A1 | Sense-TCACATTCCTCTGCCCTTCC |
| Antisense-CAGAAGGCAGCCCTGTTTGT | |
| TIPARP | Sense-GGAAATTGTGCCAGTGGTCC |
| Antisense-CAGCATATTCACCAAACGTTGTG | |
| ALDH1A3 | Sense-GCCCGTAACAGAACCAGTGTG |
| Antisense-GCCCATTTTATACATGGTTTTCGT | |
| PTGS2 | Sense-CTGAATGTGCCATAAGACTGACCT |
| Antisense-TCCACAGATCCCTCAAAACATTT | |
| ATF3 | Sense-TTCTCCCAGCGTTAACACAAAA |
| Antisense-AGAGGACCTGCCATCATGCT | |
| CEBPD | Sense-TGCAGCAGAAGTTGGTGGAG |
| Antisense-GCTGGTGCAGCTTCTCGTTC | |
| SELE | Sense-TGGCAATGAAAAATTCTCAGTCA |
| Antisense-TCAAGGCTAGAGCAGCTTTGG | |
| CCL2 | Sense-CAGCAGCAAGTGTCCCAAAG |
| Antisense-TTGGCCACAATGGTCTTGAA | |
| CXCL-2 | Sense-GAGAGACACAGCTGCAGAGGC |
| Antisense-TGCTCAAACACATTAGGCGC | |
| TNFAIP3 | Sense-TCTTTCTGTTGCTGGCTTACAGTT |
| Antisense-TGACAGCAACCACAAAGCACA | |
| SOCS3 | Sense-ATGGAGAGGGACCCAGCATA |
| Antisense-CCACCCATCCAGGCTGAGTAT | |
| PLA2G4A | Sense-CATGTACTGGAAATGGCAGCA |
| Antisense-GGGATTGCAAACTGCCTCAG | |
| APOLD1 | Sense-GAATCCTAACTGCTTTGATGCACTT |
| Antisense-ATTGGAAATGACAGGTGCCC | |
| SLC7A2 | Sense-TGCAGACACTTGGGTCAGATTC |
| Antisense-ATCAGGAAGCCAATTGCCAT |
The oligonucleotide sequence for each primer sequences was obtained from the Affymetrix database using the probe set IDs. The primers were custom prepared and used as described in material and methods.
Cytokine Assays for Secreted Protein Expression
To determine cytokine secretion, media from Sum06 APM treated and untreated HAEC cultures were evaluated using a multiplex, bead-based, immunologic assay. HAEC were grown to confluence in six-well plates and then treated with control (H2O) or sum06 APM (10 μg/ml) for 3 h. After the incubation, media were collected and concentrated up to fivefold with Ultracel YM-3 Microcon centrifugal filter (Millipore, Bedford, MA). Protein concentration was determined with the bichinoic acid assay (Pierce). Cytokine assays were carried out using a commercial immuno-bead-based analytical system and human-specific assay kits (Bioplex; Bio-Rad Life Science, Hercules, CA). Cytokines assayed included interleukin-1α (IL-1α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-10 (IL-10), interleukin-13 (IL-13), GM-CSF, monocyte chemotaxtic protein-1 (MCP-1), MIP-1α, MIP-1β, vascular endothelial growth factor (VEGF), and intercellular adhesion molecule-1 (ICAM-1). The specific secreted protein expressions were expressed as pg/ml specific protein from APM-treated cells compared with those from the control-treated cells.
AhR Translocation in HAEC Exposed to Sum06 APM
The cellular localization of AhR was analyzed using endogenous expressed proteins. HAECs were grown to confluence on fibronectin-coated 12-mm round coverslips placed in 24-well medical-grade polystyrene plates (BD Falcon) and were treated with control (H2O) or sum06 APM (10 or 50 μg/ml) for 30 min, 1 h and 1.5 h. For positive control, TCDD (1 nM) and DMSO (control for TCDD) was used. After treatment, cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature and then washed 3× with PBS. Cells were then permeabilized and blocked with Superblock (Pierce, Rockford, IL) containing 0.05% saponin for 30 min. Fixed cells were incubated with rabbit polyclonal anti-AhR (1:500 dilution) and washed with 4 × 10% Superblock with 0.05% saponin to remove unbound Ab. Subsequently cells were treated with donkey-anti-rabbit antibody conjugated to Alexa Fluor 488 for 30 min, unbound material was removed as above. The nucleus was counter stained for 5 min with 4′,6-diamidino-2-phenylindole (1 μg/ml). Isotype controls were correspondingly run for all primary antibodies used in the study. Coverslips were mounted, and digital micrographs of six randomly chosen fields per coverslip obtained with an Olympus BX61 fluorescence microscope. The images shown are representative of at least four separate experiments. Photoshop CS was used to identify nuclei with enhanced AhR staining. A color range was selected using the eyedropper tool. When the bright green pixels associated with AhR-positive nuclei are selected, all nuclei containing this color range are automatically highlighted and are then counted manually. The initial selection of pixels is saved and applied to all images in the experiment. Results are expressed as % AhR positive nuclei relative to all nuclei.
Cell-Based AhR-Transcription Bioassay
Recombinant mouse hepatoma (H1L1.1c2) cells were grown and maintained in α-MEM (GIBCO/Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals) at 5% CO2 and 37°C as described (13). These cells contain a stably integrated DRE-driven firefly luciferase reporter gene plasmid whose transcriptional activation occurs in an AhR-dependent manner (13, 72). Cells were plated into white, clear-bottomed 96-well tissue culture dishes at 75,000 cells/well and allowed to attach for 24 h. Cells were incubated with carrier solvent (DMSO or water: 1% final solvent concentration), TCDD (1 nM), APM collected in summer 2006 (10 μg/ml) and winter 2007 (10 μg/ml) for 4 h at 37°C. We added 1 μl of each treatment sample to initiate the bioassay. After the incubation, sample wells were washed twice with phosphate-buffered saline, followed by addition of cell lysis buffer (Promega, Madison, WI). The plates were then shaken for 20 min at room temperature to allow cell lysis. Luciferase activity was measured using an Orion microplate luminometer (Berthold, Oak Ridge, TN) with automatic injection of Promega stabilized luciferase reagent. Luciferase activity in each well was expressed relative to that maximally induced by TCDD.
Inhibitor Studies in HAEC Exposed to Sum06 APM
To determine whether AhR signaling, oxidative stress, or bacterial endotoxin affects gene responses, HAEC were grown to confluence on six-well plates and pretreated with the AhR inhibitor α-NF (1 μM) or an inhibitor of oxidative stress, resveratrol (10 μM), or pol B (1 μg/ml), an endotoxin-binding antibiotic for 30 min, followed by treatment with 10 μg/ml sum06 APM together with inhibitor for 3 h. For α-NF and resveratrol, DMSO was used as vehicle and control. For polymyxin, H2O was used as control. After treatment, mRNA was isolated as described above in kinetic studies and specific gene expressions were analyzed by qRT-PCR.
Statistical Analysis
Data for fold-changes in gene expression obtained by qRT-PCR, Western blots, bio-plex assay, and immunofluorescence cell counts were analyzed by GraphPad PRISM 4.0 software (San Diego, CA). An unpaired student's t-test or two-way analysis of variance with repeated measures was used as appropriate for comparisons between the treatments. While qRT-PCR results were expressed as fold-change to better compare with microarray responses, statistical analysis was performed by a t-test comparing CT values for replicates of control and treated cells. In all comparisons differences with P ≤ 0.05 were considered as significant. Results are expressed as means ± SE and the P value.
RESULTS
Time-Course Studies
To determine the appropriate exposure time for transcriptional responses, HAEC were treated with H2O (control) or 10 μg/ml of sum06 APM for 30 min, 1 h, 2 h, 3 h, or 4 h, and mRNA expression of selected genes were analyzed. PAH response genes cytochrome P450 1A1 (CYP1A1) and TCDD-inducible poly (ADP-ribose) polymerase (TIPARP) were significantly increased at 1 h, 2 h, and 3 h and decreased at 4 h but still higher than control (Fig. 1). Prostaglandin-endoperoxide synthase 2 (PTGS2) was also significantly increased at 1 h, 2 h, 3 h and had decreased expression at 4 h (Fig. 1). The inflammatory response gene chemokine (C-C motif) ligand 2 (CCL-2) was significantly increased at all time points (Fig. 1).
Fig. 1.
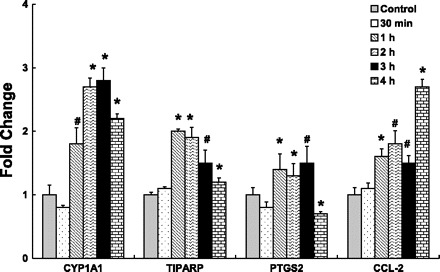
Time-course studies in human aortic endothelial cells (HAEC) exposed to summer 2006 (sum06) ambient particulate matter (APM) by quantitative real-time RT-PCR. Induction of CYP1A1, PTGS2, TIPARP, and CCL-2 gene expression in HAEC exposed to sum06 APM for 30 min, 1 h, 2 h, 3 h, and 4 h compared with control cells treated with H2O. Sum06 APM-induced gene expression was significantly increased after 1 h, 2 h, 3 h, and 4 h. n = 3, *P ≤ 0.005, #P ≤ 0.05.
Cell viability.
Using Nuclear-ID Blue Green cell viability reagent, a nonsignificant trend was observed with 10 μg/ml of sum06 or win07 APM resulting in a 15 and 9% increase in cell death, respectively, after 3 h incubation (n = 6) (data not shown).
Caspase-3/7 assay.
After 3 h exposure of HAEC to 10 μg/ml of sum06 or win07 APM, caspase-3/7 activity was measured. A modest but statistically significant increase occurred after exposure to sum06 but not win07 APM (Fig. 2) (n = 5, P ≤ 0.05).
Fig. 2.
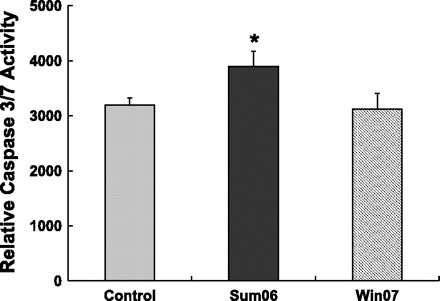
Sum06 APM increased caspase 3/7 activity. The caspase 3/7 activity was significantly increased with sum06 APM and not with winter 2007 (win07) APM compared with control H2O. n = 5, *P ≤ 0.05.
Differentially Expressed Genes
To determine APM effects, HAEC were challenged with vehicle control or APM for 3 h and cellular responses were analyzed using a high-density oligonucleotide array containing ∼22, 000 probe sets. The total number of mRNA transcripts examined in each treatment group was 22,277 probe sets with 14,500 genes. Total numbers of “present” probe sets were ∼13,000, and there was no difference in gene numbers expressed.
Differential analysis of gene expression data also showed that HAEC treated with sum06 and win07 APM affected a small percentage (0.15 and 0.04%, respectively) (Table 3) of the ∼13,000 probe set that are reliably detected: 20 genes in HAEC were upregulated by sum06 APM, four genes were upregulated, and only one gene was downregulated by Win07 APM.
Table 3.
Summary of genome-wide responses of HAEC to APM exposure
| APM in HAEC | Total Genes Affected, n | Total Genes Affected, % | Upregulated | Downregulated |
|---|---|---|---|---|
| Summer 2006 | 20 | 0.15 | 20 | 0 |
| Winter 2007 | 5 | 0.04 | 4 | 1 |
All data are based on 13,000 probe set present in human aortic endothelial cells (HAEC). The criterion for selection was ≥ ± 2-fold change as detailed in material and methods. Effects of APM on gene expression in either HAEC were obtained by comparing the entire list of genes from HAEC grown on basal media. The number differentially expressed, either summer 2006 or winter 2007 APM-sensitive genes (column 2), is also shown as the % of the total number of genes detected (column 3). The same genes may be involved in different functions.
Functional Groups Affected by APM
Functional significance of genes affected by APM was evaluated with inquiries of the PubMed Gene Database. Sum06 APM altered expression of genes involved many functional classes including AhR-responsive genes, stress-associated protein kinase/c-Jun NH2-terminal kinase (SAPK/JNK)/NF-κB pathways, proinflammatory responses, and endothelial-specific response. Win07 APM-affected genes involved AhR-responsive genes, SAPK/JNK/NF-κB pathways, and proinflammatory responses.
APM-Induced AhR and Oxidative Stress Response Genes
Global gene expression analysis identified qualitative and quantitative differences in APM treatment response genes associated with chemically induced cellular stress such as PAH metabolism and “redox-stress.” The highest gene expression for HAECs exposed to APM collected in either sum06 or win07 was that encoding cytochrome P450, family 1A1 (CYP1A1). CYP1A1 mRNA expression in sum06 and win07 was increased 3.7- and 2.3-fold, respectively (Table 4). CYP1A1 is known to be induced by environmental tobacco smoke (19). Induction of CYP1A1 genes was independently confirmed by qRT-PCR (Figs. 3A and 4).
Table 4.
Upregulated genes affected by summer 2006 and winter 2007 APM exposure in HAEC
| Fold Change |
||||
|---|---|---|---|---|
| Gene Title | Gene Symbol | Gene ID | Summer 2006 | Winter 2007 |
| AhR-responsive genes | ||||
| Cytochrome P450, family 1, subfamily A, polypeptide 1 | CYP1A1 | NM_000499 | 3.7 | 2.3 |
| Aldehyde dehydrogenase 1 family, member A3 | ALDH1A3 | NM_000693 | 2.0 | |
| Prostaglandin-endoperoxide synthase 2 | PTGS2 | NM_000963 | 2.8 | 2.0 |
| TCDD-inducible poly(ADP-ribose) polymerase | TIPARP | AL556438 | 2.0 | |
| SAPK/JNK/NF-κB pathways | ||||
| Activating transcription factor 3 | ATF3 | NM_001674 | 3.0 | |
| B-cell CLL/lymphoma 3 | BCL3 | NM_005178 | 2.0 | |
| CCAAT/enhancer binding protein (C/EBP), delta | CEBPD | M83667 | 2.3 | |
| CCAAT/enhancer binding protein (C/EBP), delta | CEBPD | AV655640 | 2.0 | |
| Proinflammatory responses | ||||
| Chemokine (C-C motif) ligand 2 | CCL2 | S69738 | 2.3 | |
| Chemokine (C-X-C motif) ligand 2 | CXCL2 | M57731 | 2.5 | |
| Selectin E | SELE | NM_000450 | 2.8 | |
| Tumor necrosis factor, alpha-induced protein 3 | TNFAIP3 | NM_006290 | 2.1 | |
| Collagen, type I, alpha 1 | COL1A1 | K01228 | 2.5 | |
| Collagen, type I, alpha 2 | COL1A2 | NM_000089 | 2.0 | |
| Suppressor of cytokine signaling 3 | SOCS3 | BG035761 | 2.6 | |
| Phospholipase A2, group IVA (cytosolic, calcium-dependent) | PLA2G4A | M68874 | 2.1 | |
| Endothelial cell-specific response | ||||
| Apolipoprotein L domain containing 1 | APOLD1 | NM_030817 | 2.8 | |
| Solute carrier family 7, member 2 | SLC7A2 | NM_003046 | 2.0 | |
AhR, arylhydrocarbon receptor.
Fig. 3.
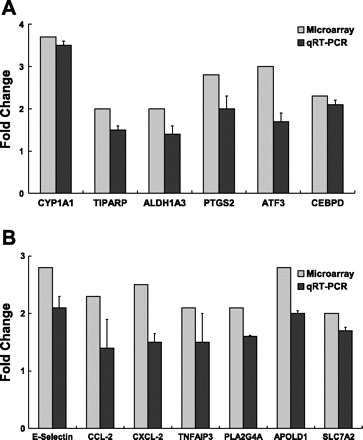
Confirmation of expression of selected genes in HAEC exposed to sum06 APM by quantitative real-time RT-PCR. A: induction of CYP1A1, TIPARP, ALDH1A3, PTGS2, ATF3, and CEBPD gene expression. B: induction of E-selectin, CCL-2, CXCL-2, TNFARP, PLA2G4A, APOLD1, and SLC7A2 gene expression in HAEC exposed to sum06 APM exposure compared with control H2O-treated group. Microarray analysis: pooled n = 3 per GeneChip. Real-time quantitative RT-PCR: n = 3, P ≤ 0.05.
Fig. 4.
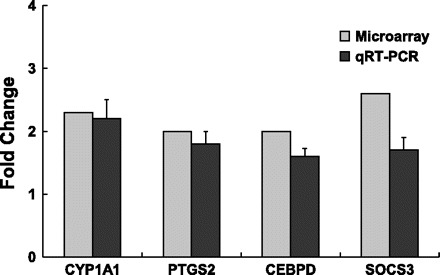
Confirmation of expression of selected genes in HAEC exposed to win07 APM by quantitative real-time RT-PCR. Induction of CYP1A1, PTGS2, CEBPD, and SOCS3 gene expression in HAEC exposed to win07 APM exposure compared with control H2O-treated group. Microarray analysis: pooled n = 4 per GeneChip. Real-time quantitative RT-PCR: n = 3, P ≤ 0.05.
Another gene associated with xenobiotic response systems, TCDD-inducible poly (ADP-ribose) polymerase (TIPARP), was also upregulated in HAEC (2.0-fold) exposed to sum06 APM (Table 4). Additional genes known to respond to AhR and oxidative stress included aldehyde dehydrogenase 1 family A3 (ALDH1A3) and prostaglandin-endoperoxide synthase 2 (PTGS2). ALDH1A3 was induced (2.0-fold) in HAEC exposed sum06. ALDH1A3 is a non-P450 aldehyde oxidizing enzyme and is a member of the aldehyde dehydrogenase superfamily (8, 37). PTGS2, also known as cyclooxygenase (COX-2) was induced 2.8-fold in HAEC treated with sum06 APM. PTGS2 is a key enzyme in prostaglandin biosynthesis and is inducible through several mechanisms including responses to active oxygen generation. Induction of ALDH1A3 and PTGS2 by APM exposure was confirmed by qRT-PCR (Figs. 3A and 4).
Inflammatory Response Genes Induced By APM
Sum06 APM.
Several biologically active proinflammatory molecules were upregulated in addition to PTGS2. Chemokine (C-C motif) ligand 2 (CCL-2), also known as MCP-1, mRNA expression was increased 2.3-fold in HAEC exposed to sum06 APM. Chemokine (C-X-C motif) ligand 2 (CXCL-2), also known as MIP-2α was also increased 2.5-fold in HAEC. Upregulated expression of the leukocyte adhesion molecule E-selectin (SELE) was induced 2.8-fold. Tumor necrosis factor, alpha-induced protein 3 (TNFAIP3), a TNF-α response protein that downregulates NF-κB responses, was increased 2.1-fold (Table 4). PLA2G4A or vascular early response gene (VERGE), apolipoprotein L domain containing 1 (APOLD1), and solute carrier family 7 member 2 (SLC7A2) were increased 2.1-, 2.8-, and 2.0-fold, respectively. Induction of CCL-2, CXCL-2, E-selectin, TNFAIP3, PLA2G4A, APOLD1, and SLC7A2 in HAEC were confirmed by qRT-PCR (Fig. 3B).
Win07 APM.
In HAEC exposed to win07 APM, suppressor of cytokine signaling 3 (SOCS3) had the highest overall expression (2.6-fold) and was the only upregulated gene related to inflammation (Table 4).
APM Activates SAPK/JNK/NF-κB Signaling Pathway
Sum06 APM.
HAEC exposed to sum06 APM had upregulation of activating transcription factor 3 (ATF3), a member of the mammalian activation transcription factor/cAMP responsive element-binding (CREB) protein family of transcription factors. ATF3 was increased 3.0-fold (Table 4) and confirmed by qRT-PCR (Fig. 3A). Other genes related to JNK/NF-κB signaling (see discussion) upregulated included the NF-κB response gene BCL3 and a JNK/NF-κB regulatory activity, CCAAT/enhancer binding protein delta (CEBPD).
Win07 APM.
HAEC exposed to win07 APM had a 2.0-fold upregulation of CEBPD (Table 4).
Secreted Protein Expression
To determine whether APM treatment resulted in secretion of proinflammatory proteins, HAEC were treated with sum06 APM for 3 h, and media were analyzed using the Bio-plex cytokine assay. Increased protein expression of IL-6 (47%) and MCP-1 (CCL-2) (55%) was found in media of HAEC treated with sum06 APM (Fig. 5). No changes in other cytokines were evident.
Fig. 5.
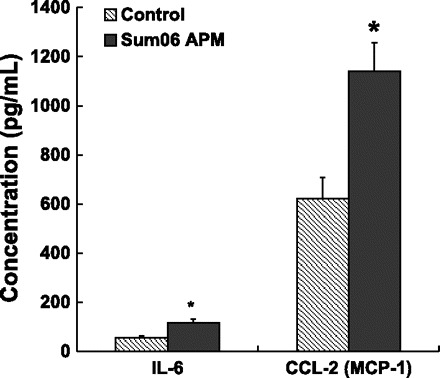
Secreted protein expression in supernatant of HAEC treated sum06 APM. IL-6 and CCL-2 (MCP-1) protein expression were significantly increased in APM treated compared with control H2O (n = 3, *P ≤ 0.05).
APM Activates AhR-Aromatic Hydrocarbon Receptor Nuclear Translocator Signaling Pathway
Induction of xenobiotic metabolizing enzymes, especially CYP1A1, is regulated through AhR (18). AhR is a cytoplasmic receptor for aromatic hydrocarbons such as TCDD present in industrial waste, byproducts of combustion and incinerator reactions, tobacco smoke, and other sources (31, 40, 58). It acts as a transcription factor for proteins involved in xenobiotic metabolism. To determine whether APM activates the AhR signaling pathway, we evaluated the time course of nuclear accumulation of AhR in response to APM. We compared the percent of nuclei exceeding a threshold intensity of immunofluorescence in response to treatment with sum06 APM compared with the AhR agonist TCDD as a positive control. Representative images of nuclear accumulation are presented for the 1 h time point in Fig. 6A. Percent positive nuclei are presented in Fig. 6B. All particle treatments had significantly increased percentage of positive nuclei. While a significant increase in positive nuclei in TCDD-treated cells occurred, cells treated with 50 μg/ml APM had the highest percentage of positive nuclei.
Fig. 6.
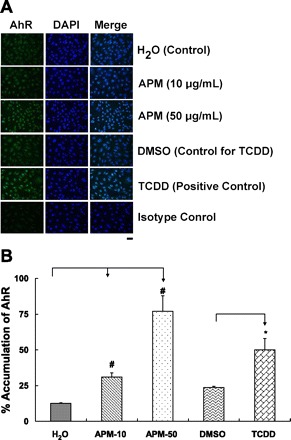
A: aryl hydrocarbon receptor (AhR) nuclear localization in HAEC treated with Sum06 APM for 1 h. Representative images of HAEC monolayers showing AhR distribution in cytosol and nucleus. Nuclear accumulation of AhR was evident after exposure to sum06 APM or tetrachlorodibenzo-p-dioxin (TCDD, positive control) compared with control cells treated with H2O or DMSO (control for TCDD) by immunofluorescence staining (n = 4). Isotype controls had no detectable staining. Bar = 40 μm. B: percent accumulation of AhR by sum06 APM. Percent positive nuclei after 1 h incubation. 31% (APM-10), 77% (APM-50), and 50% (1 nM TCDD) of nuclei stained with AhR after 1 h. APM exposure compared with control H2O and TCDD was compared with control DMSO. n = 4; *P ≤ 0.05; #P ≤ 0.005.
Cell-Based AhR-Mediated Bioassay
To confirm that activation of AhR by APM results in transcriptional activation, we used a recombinant mouse hepatoma cell line (H1L1.1c2) that contains a stably integrated firefly luciferase gene whose expression is under the control of four DREs (13). Treatment of these cells with AhR ligands (agonists) induces luciferase gene expression in a time-, dose-, and AhR-dependent manner (13, 17). Induction of luciferase following incubation of H1L1.1c2 cells for 4 h with the indicated samples is shown in Fig. 7. APM collected in sum06 and win07 induced luciferase reporter gene activity to 13 and 19% of that induced by TCDD, respectively (Fig. 7).
Fig. 7.
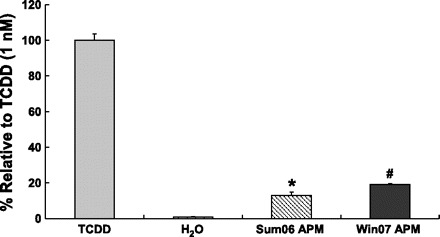
Induction of luciferase activity by APM. Induction of luciferase in H1L1.1c2 cells containing a stably integrated AhR-responsive DRE-driven firefly luciferase reporter gene plasmid. Response to a 3 h incubation with 10 μg/ml of APM collected in sum06 APM or win07 APM was compared with TCDD (100% as positive control). n = 3; *P ≤ 0.0005, #P ≤ 0.0005.
α-NF and Resveratrol Suppressed Sum06 APM-Induced Gene Expression
Our microarray data demonstrated that sum06 APM affected several functional group including xenobiotic, oxidoreductase activity, and inflammatory immune response. To determine the relative role of AhR signaling and intracellular oxidant activity, we performed a series of inhibitor studies using sum06 APM. Control cells in these studies were treated with the inhibitor vehicle DMSO. We found that 10 μg/ml Sum06 APM for 3 h induced CYP1A1, TIPARP, PTGS2, CEBPD, and CCL-2 gene expression (2.3-, 1.2-, 1.5-, 1.4-, and 2.1-fold, respectively) (Fig. 8). The AhR inhibitor α-NF significantly suppressed CYP1A1 gene expression induced by Sum06 to 1.5-fold (Fig. 8). Conversely, expression of the AhR responsive genes TIPARP, PTGS2, and CEBPD were markedly increased by cotreatment with α-NF. CCL-2 expression was not altered by α-NF. Resveratrol (3,5,4′-trihydroxy-trans-stilbene), is a potent suppressor of intracellular ROS production by inhibition of NADH/NADPH oxidase activity as well as inhibition of the production of the proinflammatory mediators NO·and PGE2 (at least in part by inhibition of NOS-2 and COX-2 gene and protein expression) (26). Resveratrol is also an AhR antagonist acting through the inhibition of recruitment of the AhR/aromatic hydrocarbon receptor nuclear translocator (ARNT) complex to xenobiotic response elements (7). Resveratrol suppressed CYP1A1, PTGS2, TIPARP, and CCL-2 gene expression to 0.3-, 1-, 1-, and 1.3-fold (Fig. 8).
Fig. 8.
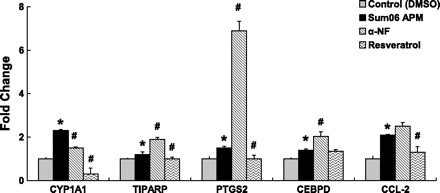
α-Napthoflavone (α-NF) and resveratrol suppressed sum06 APM-induced gene expression. RT-PCR analysis demonstrates α-NF significantly suppressed sum06 APM-induced CYP1A1 gene expression compared with control DMSO (35%). Resveratrol suppressed Sum06 APM-induced CYP1A1, TIPARP, PTGS2, and CCL-2 gene expression compared with control (87, 17, 33, 38%). n = 3; *P ≤ 0.05, Sum06 APM compared with control; #P ≤ 0.05, Sum06 APM with α-NF or resveratrol compared with Sum06 APM.
Pol B Treatment Differentially Alters HAEC Proinflammatory Responses
We also asked whether endotoxin contamination of APM activated HAEC inflammation using the endotoxin binding antibiotic pol B. Pol B had no effect on the AhR-responsive genes CYP1A1 and TIPARP. ALDH1A3 was also unaffected. Expression of PTGS2, ATF3, and E-selectin were enhanced by pol B treatment. Pol B suppressed APM-induced changes in CXCL-2 and CCL-2 (Fig. 9).
Fig. 9.
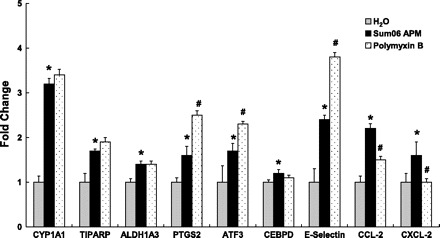
Effect of endotoxin on HAEC responses to APM. Polymyxin B, a inhibitor of bacterial endotoxin, did not suppress Sum06 APM-induced CYP1A1, TIPARP, ALDH1A3, CEBPD, and augmented PTGS2, ATF3, E-selectin expression. CCL-2 and CXCL-2 expression was significantly suppressed by polymyxin B. n = 3; *P ≤ 0.05, and Sum06 APM treatment compared with control; #P ≤ 0.05 and Sum06 APM with polymyxin compared with Sum06 APM.
DISCUSSION
For the genes surveyed, our kinetic studies demonstrated that APM-induced gene expression was maximal at 3 h. Cell viability and caspase-3/7 assays demonstrated that only the sum06 APM-treated cells had a modest increase in cell death and this was only statistically significant for the caspase-3/7 assay. Therefore, the bulk of alterations in gene expression, cytokine secretion, and alterations in AhR signaling were sufficiently adaptive or at least not detrimental to the overall survival of cultured cells. While the concentration of particles used in these experiments are higher than likely to be achieved in the systemic circulation, they more closely approximate estimates of local alveolar concentrations that would be adjacent to alveolar endothelium (23). Estimates of systemic accumulations suggest concentrations ranging from 1 to 8% of pulmonary PM may accumulate in systemic tissues (see Ref. 47 for review). Almost all studies of systemic accumulation are based on single doses, so accumulation after repetitive or continuous environmental exposure remains uncertain. Maximal absorption of ultrafine particles occurs rapidly after intratracheal instillation with a peak exposure of systemic tissues in a 4 h time course, consistent with the 3 h time point used in the majority of the current study (51).
The number of gene responses and their magnitude varied significantly between ambient particles collected in summer or winter. Samples collected in summer upregulated significantly more genes than did particles from winter. In general, the fold-changes resulting from particles collected in summer were more robust than those from winter.
While particle concentrations administered to cultures were gravimetrically equivalent, significant differences in composition likely influenced cellular responses. Particles collected in summer were higher in both elemental and organic carbon, sulfate, and reactive trace metals including vanadium, iron, lead, calcium, barium (57). Analysis of 21 PAHs spanning a range of sizes and volatility demonstrated that summer concentrations were ∼25% lower than those in winter (57). Particles collected in winter had a higher concentration of soluble ammonium nitrate, which would reduce the effective concentration of solid components in treated cultures. Conversely, elemental carbon, organic carbon, trace metals, and PAH components are generally not water soluble and thus might well have greater concentrations in individual particles and potentially greater interaction with cell cultures.
The pattern of gene responses in vascular endothelium is consistent with current hypotheses implicating pulmonary inflammation and vascular inflammation/coagulation in the mechanism of cardiovascular complications of particulate air pollution. The principal pathways implicated by gene responses in human aortic cells to collected APM include the AhR-responsive genes associated with PAH exposure and an interaction between the MAP kinase and NF-κB stress response pathways that could be activated by either oxidant- or endotoxin-mediated receptors. Several activities induced by the AhR response element in cells more traditionally associated with xenobiotic metabolism were upregulated in APM-treated HAEC. CYP1A1, PTGS2 (also known as COX-2), and TIPARP are all genes induced by the interaction of the AhR and ARNT complex with their promoters. TIPARP is a member of the poly(ADP-ribose) polymerase (PARP) family of genes that regulate protein function through adding ADP ribose units to glutamic acid residues. Induction of TIPARP in mouse liver is dependent on AhR/ARNT signaling (35). AhR agonists, PAHs, have previously been shown to increase the expression of IL-1β, IL-8, and TNF-α factors involved in adverse inflammatory responses (25, 33, 34, 68) in the following paper. N'Diaye et al. (41) have shown that benzo[a]pyrene exposure induces the production of CCL1, a known participant in inflammatory and cardiovascular disease. These studies all point to the importance and the multiplicity of mechanisms by which the AhR receptor could potentially participate in the resulting pulmonary-cardiovascular insult following exposure to PAH carrying particulate matter.
ALDH1A3 is also a xenobiotic response gene but classically is induced by phenobarbital-like P450 inducers, while a related enzyme, ALDH3A1, not seen in this study, is associated with AhR-mediated responses (52). In addition to being an AhR-responsive gene, PTGS2 is strongly induced in endothelium by IL-1β (20).
A spectrum of gene responses suggested interactions between oxidant response elements and TLR-4-related pathways that could be stimulated by endotoxin. Upregulation of the transcription factor ATF3 suggests activation of the SAPK/JNK subset of MAP kinase signaling (16). CEBPD is a third transcription factor that participates in the interactions between these two counterbalancing regulators of endothelial stress and inflammatory responses (29).
The SAPK/JNK pathway responds to a variety of cellular stressors including endotoxin (9), UV radiation, and oxidants (1). Second messengers in this pathway act through JNK to phosphorylate cJun, which is translocated to the nucleus where it interacts with ATF3 to form the transcriptional complex known as AP-1. The ATF3 subunit undergoes auto-stimulatory upregulation in response to this transcriptional activation. Other AP-1 responses in endothelial cells include E-selectin and IL-8 (28, 32).
A key pathway activating NF-κB in monocytes is mediated by endotoxin stimulation of the TLR-4 receptor. A complex interaction between NF-κB and inhibitory proteins including IκB regulates NF-κB stability and its accumulation in the nucleus as a transcription factor. BCL3 is an NF-κB response element that acts in an autoregulatory way to modulate NF-κB signaling. A multitude of cellular responses characterize NF-κB activation including transcription of IL-6, CXCL-2, and cellular survival signals.
A complex interplay between SAPK/JNK and NF-κB signaling directs a balance between the spectrum of proinflammatory responses and survival/apoptosis signals in leukocytes. The CEBPD transcription factor participates in this signaling interface as an amplifier of NF-κB-mediated stimulus of IL-6 transcription. IL-6 has previously been shown to be elevated in PM-exposed mice (71). ATF3 attenuates this response. The balance between ATF3 attenuation and CEBPD enhancement acts through epigenetic modulation of transcription and is theorized to discriminate between short and persistent LPS signals (29). SAPK/JNK and NF-κB signaling are also important in endothelial cell biology. In particular, regulation of SAPK/JNK activity directs site-specific apoptotic signals in relation to flow and turnover of endothelium at atheroprone sites in the arterial tree (9).
CCL-2 upregulation in HAEC occurs through LPS- and TNF-α-mediated NF-κB pathways. HAEC also upregulate CCL-2 (MCP-1), E-selectin, and IL-8 in response to LPS (61). However, both NF-κB and AP-1 pathways upregulate CCL-2 (70). Regulation of CCL-2 secretion by endothelium through NF-κB- and AP-1-mediated pathways is affected by such diverse stimuli as LPS, TNF-α, anthrax toxin (70), and activated platelet interactions (14). Our data suggest a similar synergistic effect on CCL-2 secretion in APM-treated endothelial cells. This could have important implications for systemic disease associated with APM exposure as CCL-2 plays a key role in the vascular and renal consequences of the metabolic syndrome (15, 36, 54). Finally, CEBPD has been reported to enhance CCL-2 transcription separate from NF-κB activity in vascular smooth muscle cells (60).
CXCL-2 (also known as MIP-2α) acts primarily as a neutrophil chemokine. CXCL-2 is sequestered in the heparin sulfate surface lining of activated endothelial cells and directs neutrophil migration along the consequent chemotactic gradient (38). There appears to be a connection between AhR signaling in endothelial cells, p38 and JNK subsets of MAP kinase signaling, and the induction of CXCL-2 in response to aromatic hydrocarbons (22). Induction of CYP1A1 appears to be caveolin dependent, and overexpression of CYP1A1 results in oxidative stress that likely activates the JNK pathway and subsequent CXCL-2 induction.
The last group of genes identified as responsive to PM treatment include endothelial cell-specific activation pathways including PLA2G4A, APOLD1 [also known as vascular early response gene (VERGE)], and SLC7A2. PLA2G4A or phospholipase 2 catabolizes membrane lipids to release arachidonic acid, thus initiating eicosanoid and other lipid mediator signaling pathways. Its activation occurs through a plethora of signals including reactive oxygen species. Similarly, transcription of PLA2G4A is induced through multiple cytokine pathways including TNF-α, IL-1β, and IFN-γ as well as subsequent to endotoxin (64).
APOLD1 or VERGE is highly expressed in developmental angiogenesis and transiently expressed in adult CNS vasculature in response to seizures or in vitro in response to hypoxia or fibroblast growth factor (56). It has also been shown to upregulate in response to exercise (63). VERGE protein localizes to endothelial cell borders and is speculated to interact with PKC-mediated induction of microvascular leak (56).
SLC7A2 is a solute transporter specific for uptake of l-arginine, the substrate for nitric oxide synthase (NOS) (45). In addition to vaso-relaxation, NO acts to decrease endothelial cell proinflammatory responses by downregulating JNK with subsequent decrease in IL-8 secretion (43). Interestingly, the induction of inducible NOS in endothelial cells by IL-1β acts through a CEBPD promoter site (21).
A distinctive pattern of upregulation of genes associated with xenobiotic metabolism was present in response to particulates collected in both summer and winter. CYP1A1 was induced in endothelial cells, and it is classically recognized as an enzyme that metabolizes exogenous many different compounds, enhancing their elimination (11, 12). Halogenated aromatic hydrocarbons and PAHs have been shown to be strong exogenous inducers of CYP1A1 genes by virtue of their ability to bind to and activate the AhR, a ligand-dependent transcription factor (2, 12, 58). The AhR acts in a well-characterized signaling cascade involving occupancy by an AhR agonist (of which there are a large number of structurally diverse chemicals) (12, 53, 58), which leads to nuclear translocation and its association with the ARNT, which converts the AhR into its high-affinity DNA binding form (12). In addition to stimulating transcription of CYP-related phase I enzymes, the AhR/ARNT complex also induces expression of a variety of phase II enzymes, including specific forms of glutathione S-transferase, quinone reductase, UDP-glucronosyl transferase, and aldehyde dehydrogenase (12, 35). CYP1A1 also has endogenous activities that more directly relate to endothelial cell function, particularly its metabolism of arachidonic acid to a variety of eicosanoids that modulate inflammation and vaso-reactivity (44).
The induction of a strong AhR-like response in endothelial cells was somewhat unexpected. We sought to confirm AhR signal transduction through evaluating nuclear translocation and demonstrating transcriptional activation of AhR-responsive genes in an established hepatocyte luciferase reporter assay. Our findings demonstrate AhR nuclear translocation by immunofluorescence in HAEC exposed to summer source APM. Similarly, APM from both summer and winter sources were positive in the AhR-dependent reporter gene assay. Activation of AhR response pathways also potentially affects stress response pathways through interaction with REL-B, a member of the NF-κB family (69).
The interlocking networks of cytokine and stress response proteins upregulated in APM-treated HAEC led us to examine whether inhibition of specific PM components would alter the responses or delineate their relative importance. We compared the effects of inhibition of the AhR pathway, intracellular antioxidant supplementation, and endotoxin binding by pol B on AhR, SAPK/JNK, and NF-κB-related gene transcription. α-NF, a selective inhibitor of AhR, predictably inhibited transcription of CYP1A1 but had no effect on the inflammatory gene CCL-2. Surprisingly, AhR-responsive genes TIPARP and PTGS2 were increased by α-NF treatment, suggesting these genes have a more complex relationship with other signaling pathways. We found resveratrol suppressed CYP1A1, TIPARP, PTGS2, and CCL-2 gene expression. While the broad spectrum of inhibition would not be unexpected given the dual role of resveratrol as an AhR antagonist (7), as well as inhibitor of Nox-based reactive oxygen synthesis, the suppression of inflammatory cytokines suggests a role for active oxygen in signaling their expression. Inhibition of endotoxin by pol B had no effect on AhR-responsive genes or on endothelial stress response genes but did inhibit both inflammatory cytokine gene responses. This suggests a selective role for endotoxin carried by APM in the induction of inflammation and recapitulates a similar selective proinflammatory effect our laboratory previously demonstrated in isolated human monocytes (10).
In summary, gene response patterns to collected APM suggest a complex relationship between PAH responses, oxidant-associated stress responses, and synthesis of proinflammatory mediators by vascular endothelium. Results of this study and others in the recent literature support a developing hypothesis that connects PAH exposure, AhR responses, and oxidative stress with proinflammatory consequences. A schematic diagram of AhR signaling pathway activation by APM exposure in HAEC is presented in Fig. 10.
Fig. 10.
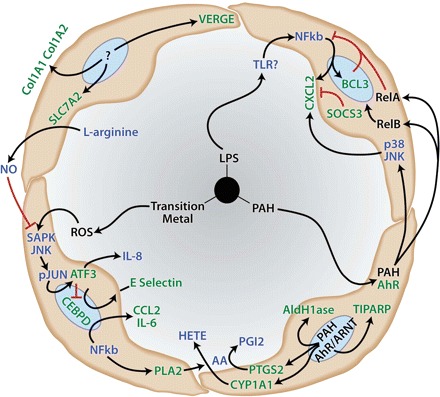
Current hypotheses of endothelial cell responses to environmental particulate matter (PM). PM components including polycyclic aromatic hydrocarbons (PAH), endotoxin (LPS), and transition metal-induced reactive oxygen species (ROS) activate endogenous stress response and xenobiotic metabolism pathways leading to a proinflammatory endothelial cell phenotype characterized by upregulation of adhesion molecules and secretion of cytokines and arachidonic acid (AA)-derived lipid mediators of inflammation such as hydroxyeicosatetraenoic acids (HETE). Activities in green found in the present study. Processes in blue represent connecting endothelial signal-response pathways derived from current literature. Red connectors illustrate inhibitory pathways.
GRANTS
Although the research described in the article has been funded wholly or in part by the United States Environmental Protection Agency through grant RD-83241401-0 to the University of California, Davis, it has not been subject to the Agency's required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1. Abe T, Oue N, Yasui W, Ryoji M. Rapid and preferential induction of ATF3 transcription in response to low doses of UVA light. Biochem Biophys Res Commun 310: 1168–1174, 2003. [DOI] [PubMed] [Google Scholar]
- 2. Arrieta DE, Ontiveros CC, Li WW, Garcia JH, Denison MS, McDonald JD, Burchiel SW, Washburn BS. Aryl hydrocarbon receptor-mediated activity of particulate organic matter from the Paso del Norte airshed along the US-Mexico border. Environ Health Perspect 111: 1299–1305, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aung HH, Vasu VT, Valacchi G, Corbacho AM, Kota RS, Lim Y, Obermueller-Jevic UC, Packer L, Cross CE, Gohil K. Effects of dietary carotenoids on mouse lung genomic profiles and their modulatory effects on short-term cigarette smoke exposures. Genes Nutr 4: 23–39, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aust AE, Ball JC, Hu AA, Lighty JS, Smith KR, Straccia AM, Veranth JM, Young WC. Particle characteristics responsible for effects on human lung epithelial cells. Res Rep Health Eff Inst: 1–65; discussion 67–76, 2002. [PubMed] [Google Scholar]
- 5. Becker S, Dailey LA, Soukup JM, Grambow SC, Devlin RB, Huang YC. Seasonal variations in air pollution particle-induced inflammatory mediator release and oxidative stress. Environ Health Perspect 113: 1032–1038, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Becker S, Mundandhara S, Devlin RB, Madden M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies. Toxicol Appl Pharmacol 207: 269–275, 2005. [DOI] [PubMed] [Google Scholar]
- 7. Beedanagari SR, Bebenek I, Bui P, Hankinson O. Resveratrol inhibits dioxin-induced expression of human CYP1A1 and CYP1B1 by inhibiting recruitment of the aryl hydrocarbon receptor complex and RNA polymerase II to the regulatory regions of the corresponding genes. Toxicol Sci 110: 61–67, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beedham C. The role of non-P450 enzymes in drug oxidation. Pharm World Sci 19: 255–263, 1997. [DOI] [PubMed] [Google Scholar]
- 9. Chaudhury H, Zakkar M, Boyle J, Cuhlmann S, van der Heiden K, Luong le A, Davis J, Platt A, Mason JC, Krams R, Haskard DO, Clark AR, Evans PC. c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler Thromb Vasc Biol 30: 546–553, 2010. [DOI] [PubMed] [Google Scholar]
- 10. den Hartigh LJ, Lame MW, Ham W, Kleeman MJ, Tablin F, Wilson DW. Endotoxin and polycyclic aromatic hydrocarbons in ambient fine particulate matter from Fresno, California initiate human monocyte inflammatory responses mediated by reactive oxygen species. Toxicol In Vitro 24: 1993–2002, 2010. [DOI] [PubMed] [Google Scholar]
- 11. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43: 309–334, 2003. [DOI] [PubMed] [Google Scholar]
- 12. Denison MS, Phelen D, Elferink CJ. The Ah receptor signal transduction pathway. In: Xenobiotics, Receptors and Gene Expression, edited by Denison MS, Helferich WG. Philadelphia, PA: Taylor and Francis, 1998, p. 3–33. [Google Scholar]
- 13. Garrison PM, Tullis K, Aarts JM, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam Appl Toxicol 30: 194–203, 1996. [DOI] [PubMed] [Google Scholar]
- 14. Gawaz M, Page S, Massberg S, Nothdurfter C, Weber M, Fisher C, Ungerer M, Brand K. Transient platelet interaction induces MCP-1 production by endothelial cells via I kappa B kinase complex activation. Thromb Haemost 88: 307–314, 2002. [PubMed] [Google Scholar]
- 15. Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Jr, Luster AD, Luscinskas FW, Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 398: 718–723, 1999. [DOI] [PubMed] [Google Scholar]
- 16. Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA 88: 3720–3724, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han D, Nagy SR, Denison MS. Comparison of recombinant cell bioassays for the detection of Ah receptor agonists. Biofactors 20: 11–22, 2004. [DOI] [PubMed] [Google Scholar]
- 18. Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol 71: 1475–1486, 2007. [DOI] [PubMed] [Google Scholar]
- 19. Kim JH, Sherman ME, Curriero FC, Guengerich FP, Strickland PT, Sutter TR. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol Appl Pharmacol 199: 210–219, 2004. [DOI] [PubMed] [Google Scholar]
- 20. Kirtikara K, Raghow R, Laulederkind SJ, Goorha S, Kanekura T, Ballou LR. Transcriptional regulation of cyclooxygenase-2 in the human microvascular endothelial cell line, HMEC-1: control by the combinatorial actions of AP2, NF-IL-6 and CRE elements. Mol Cell Biochem 203: 41–51, 2000. [DOI] [PubMed] [Google Scholar]
- 21. Kolyada AY, Madias NE. Transcriptional regulation of the human iNOS gene by IL-1beta in endothelial cells. Mol Med 7: 329–343, 2001. [PMC free article] [PubMed] [Google Scholar]
- 22. Kopf PG, Huwe JK, Walker MK. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc Toxicol 8: 181–193, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kreyling WG, Blanchard JD, Godleski JJ, Haeussermann S, Heyder J, Hutzler P, Schulz H, Sweeney TD, Takenaka S, Ziesenis A. Anatomic localization of 24- and 96-h particle retention in canine airways. J Appl Physiol 87: 269–284, 1999. [DOI] [PubMed] [Google Scholar]
- 24. Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdorster G, Ziesenis A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A 65: 1513–1530, 2002. [DOI] [PubMed] [Google Scholar]
- 25. Lecureur V, Ferrec EL, N'Diaye M, Vee ML, Gardyn C, Gilot D, Fardel O. ERK-dependent induction of TNFalpha expression by the environmental contaminant benzo(a)pyrene in primary human macrophages. FEBS Lett 579: 1904–1910, 2005. [DOI] [PubMed] [Google Scholar]
- 26. Leiro J, Alvarez E, Arranz JA, Laguna R, Uriarte E, Orallo F. Effects of cis-resveratrol on inflammatory murine macrophages: antioxidant activity and down-regulation of inflammatory genes. J Leukoc Biol 75: 1156–1165, 2004. [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Stonehuerner J, Devlin RB, Huang YC. Discrimination of vanadium from zinc using gene profiling in human bronchial epithelial cells. Environ Health Perspect 113: 1747–1754, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Libby P. Inflammation in atherosclerosis. Nature 420: 868–874, 2002. [DOI] [PubMed] [Google Scholar]
- 29. Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol 10: 437–443, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta CT) method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 31. Lofroth G, Rannug A. Ah receptor ligands in tobacco smoke. Toxicol Lett 42: 131–136, 1988. [DOI] [PubMed] [Google Scholar]
- 32. Lusis AJ. Atherosclerosis. Nature 407: 233–241, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lyte M. Regulation of interleukin-1 production in murine macrophages and human monocytes by a normal physiological constituent. Life Sci 38: 1163–1170, 1986. [DOI] [PubMed] [Google Scholar]
- 34. Lyte M, Bick PH. Modulation of interleukin-1 production by macrophages following benzo(a)pyrene exposure. Int J Immunopharmacol 8: 377–381, 1986. [DOI] [PubMed] [Google Scholar]
- 35. Ma Q. Induction and superinduction of 2,3,7,8-tetrachlorodibenzo-rho-dioxin-inducible poly(ADP-ribose) polymerase: role of the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator transcription activation domains and a labile transcription repressor. Arch Biochem Biophys 404: 309–316, 2002. [DOI] [PubMed] [Google Scholar]
- 36. Majkova Z, Smart E, Toborek M, Hennig B. Up-regulation of endothelial monocyte chemoattractant protein-1 by coplanar PCB77 is caveolin-1-dependent. Toxicol Appl Pharmacol 237: 1–7, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol 4: 697–720, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 116: 1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, Chandel NS, Dean DA, Sznajder JI, Budinger GR. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest 117: 2952–2961, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health 44: 171–174, 1989. [DOI] [PubMed] [Google Scholar]
- 41. N'Diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, Monteiro P, Rauch C, Galibert MD, Fardel O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem 281: 19906–19915, 2006. [DOI] [PubMed] [Google Scholar]
- 42. Nadadur SS, Haykal-Coates N, Mudipalli A, Costa DL. Endothelial effects of emission source particles: acute toxic response gene expression profiles. Toxicol In Vitro 23: 67–77, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Natarajan R, Gupta S, Fisher BJ, Ghosh S, Fowler AA., 3rd Nitric oxide suppresses IL-8 transcription by inhibiting c-Jun N-terminal kinase-induced AP-1 activation. Exp Cell Res 266: 203–212, 2001. [DOI] [PubMed] [Google Scholar]
- 44. Nebert DW, Karp CL. Endogenous functions of the aryl hydrocarbon receptor (AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem 283: 36061–36065, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nelin LD, Nash HE, Chicoine LG. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 281: L1232–L1239, 2001. [DOI] [PubMed] [Google Scholar]
- 46. Nemmar A, Hoet PH, Dinsdale D, Vermylen J, Hoylaerts MF, Nemery B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation 107: 1202–1208, 2003. [DOI] [PubMed] [Google Scholar]
- 47. Nemmar A, Hoylaerts MF, Hoet PH, Dinsdale D, Smith T, Xu H, Vermylen J, Nemery B. Ultrafine particles affect experimental thrombosis in an in vivo hamster model. Am J Respir Crit Care Med 166: 998–1004, 2002. [DOI] [PubMed] [Google Scholar]
- 48. Nemmar A, Hoylaerts MF, Hoet PH, Vermylen J, Nemery B. Size effect of intratracheally instilled particles on pulmonary inflammation and vascular thrombosis. Toxicol Appl Pharmacol 186: 38–45, 2003. [DOI] [PubMed] [Google Scholar]
- 49. Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet PH, Verbruggen A, Nemery B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am J Respir Crit Care Med 164: 1665–1668, 2001. [DOI] [PubMed] [Google Scholar]
- 50. Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Lunts A, Kreyling W, Cox C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J Toxicol Environ Health A 65: 1531–1543, 2002. [DOI] [PubMed] [Google Scholar]
- 51. Palko HA, Fung JY, Louie AY. Positron emission tomography: a novel technique for investigating the biodistribution and transport of nanoparticles. Inhal Toxicol 22: 657–688, 2010. [DOI] [PubMed] [Google Scholar]
- 52. Pappas P, Sotiropoulou M, Karamanakos P, Kostoula A, Levidiotou S, Marselos M. Acute-phase response to benzo[a]pyrene and induction of rat ALDH3A1. Chem Biol Interact 143–144: 55–62, 2003. [DOI] [PubMed] [Google Scholar]
- 53. Petkov PI, Rowlands JC, Budinsky R, Zhao B, Denison MS, Mekenyan O. Mechanism-based common reactivity pattern (COREPA) modelling of aryl hydrocarbon receptor binding affinity. SAR QSAR Environ Res 21: 187–214, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Piemonti L, Calori G, Lattuada G, Mercalli A, Ragogna F, Garancini MP, Ruotolo G, Luzi L, Perseghin G. Association between plasma monocyte chemoattractant protein-1 concentration and cardiovascular disease mortality in middle-aged diabetic and nondiabetic individuals. Diabetes Care 32: 2105–2110, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pope CA, 3rd, Verrier RL, Lovett EG, Larson AC, Raizenne ME, Kanner RE, Schwartz J, Villegas GM, Gold DR, Dockery DW. Heart rate variability associated with particulate air pollution. Am Heart J 138: 890–899, 1999. [DOI] [PubMed] [Google Scholar]
- 56. Regard JB, Scheek S, Borbiev T, Lanahan AA, Schneider A, Demetriades AM, Hiemisch H, Barnes CA, Verin AD, Worley PF. Verge: a novel vascular early response gene. J Neurosci 24: 4092–4103, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ruehl CR, Ham WA, Kleeman MJ. Temperature-induced volatility of molecular markers in ambient airborne particulate matter. Atmos Atmos Chem Phys 11: 67–76, 2011. [Google Scholar]
- 58. Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs). Crit Rev Toxicol 21: 51–88, 1990. [DOI] [PubMed] [Google Scholar]
- 59. Seaton A, MacNee W, Donaldson K, Godden D. Particulate air pollution and acute health effects. Lancet 345: 176–178, 1995. [DOI] [PubMed] [Google Scholar]
- 60. Sekine O, Nishio Y, Egawa K, Nakamura T, Maegawa H, Kashiwagi A. Insulin activates CCAAT/enhancer binding proteins and proinflammatory gene expression through the phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. J Biol Chem 277: 36631–36639, 2002. [DOI] [PubMed] [Google Scholar]
- 61. Shi Q, Cox LA, Glenn J, Tejero ME, Hondara V, Vandeberg JL, Wang XL. Molecular pathways mediating differential responses to lipopolysaccharide between human and baboon arterial endothelial cells. Clin Exp Pharmacol Physiol 37: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shimada A, Kawamura N, Okajima M, Kaewamatawong T, Inoue H, Morita T. Translocation pathway of the intratracheally instilled ultrafine particles from the lung into the blood circulation in the mouse. Toxicol Pathol 34: 949–957, 2006. [DOI] [PubMed] [Google Scholar]
- 63. Simonsen ML, Alessio HM, White P, Newsom DL, Hagerman AE. Acute physical activity effects on cardiac gene expression. Exp Physiol 95: 1071–1080, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem 103: 1–16, 2007. [DOI] [PubMed] [Google Scholar]
- 65. Takenaka S, Karg E, Roth C, Schulz H, Ziesenis A, Heinzmann U, Schramel P, Heyder J. Pulmonary and systemic distribution of inhaled ultrafine silver particles in rats. Environ Health Perspect 109, Suppl 4: 547–551, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Valavanidis A, Fiotakis K, Vlachogianni T. Airborne particulate matter and human health: toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 26: 339–362, 2008. [DOI] [PubMed] [Google Scholar]
- 67. van Eeden SF, Tan WC, Suwa T, Mukae H, Terashima T, Fujii T, Qui D, Vincent R, Hogg JC. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM10). Am J Respir Crit Care Med 164: 826–830, 2001. [DOI] [PubMed] [Google Scholar]
- 68. Vandebriel RJ, Meredith C, Scott MP, Roholl PJ, Van Loveren H. Effects of in vivo exposure to bis(tri-n-butyltin)oxide, hexachlorobenzene, and benzo(a)pyrene on cytokine (receptor) mRNA levels in cultured rat splenocytes and on IL-2 receptor protein levels. Toxicol Appl Pharmacol 148: 126–136, 1998. [DOI] [PubMed] [Google Scholar]
- 69. Vogel CF, Matsumura F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem Pharmacol 77: 734–745, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Warfel JM, D'Agnillo F. Anthrax lethal toxin enhances IkappaB kinase activation and differentially regulates pro-inflammatory genes in human endothelium. J Biol Chem 284: 25761–25771, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wilson DW, Aung HH, Lame MW, Plummer L, Pinkerton KE, Ham W, Kleeman M, Norris JW, Tablin F. Exposure of mice to concentrated ambient particulate matter results in platelet and systemic cytokine activation. Inhal Toxicol 22: 267–276, 2010. [DOI] [PubMed] [Google Scholar]
- 72. Ziccardi MH, Gardner IA, Mazet JA, Denison MS. Application of the luciferase cell culture bioassay for the detection of refined petroleum products. Mar Pollut Bull 44: 983–991, 2002. [DOI] [PubMed] [Google Scholar]