

Abstract
Objective
Exposure to particulate matter air pollution may be an independent risk factor for cardiovascular morbidity and mortality; however, the biological mechanisms are unclear. We hypothesize that exposure to diesel exhaust (DE), an important source of traffic-related particulate air pollution, promotes changes of atherosclerotic plaque component that may lead to plaque vulnerability.
Methods and results
30-week old ApoE knockout mice fed with regular chow inhaled DE (at 200 μg/m3 of particulate) or filtered-air (control) for 7 weeks (6 h/day, 5 days/week) (12 mice/group). Total number of alveolar macrophages (p < 0.01) and alveolar macrophages positive for particles (p < 0.0001) were more than 8-fold higher after DE inhalation than the control. DE inhalation caused 1.5 to 3-fold increases in plaque lipid content (p<0.02), cellularity (p<0.02), foam cell formation (p<0.04), and smooth muscle cell content (p<0.05). The expression of oxidative stress markers, iNOS, CD36, and nitrotyrosine was significantly increased by 1.5 to 2-fold in plaques, with enhanced systemic lipid and DNA oxidation (p<0.02). Increased foam cells and the expression of iNOS (R2 = 0.72, p = 0.0081) and CD36 (R2 = 0.49, p = 0.015) in plaques were positively correlated with the magnitude of DE exposure.
Conclusions
Exposure to DE promotes changes in atherosclerotic plaques characteristic of unstable vulnerable plaques. Increased systemic and plaque oxidative stress markers suggest that these changes in plaques could be due to DE-induced oxidative stress.
Keywords: Diesel exhaust, Atherosclerotic plaque, ApoE knockout mice
1. Introduction
Epidemiological studies conducted over the past 20 years showed that exposure to ambient particulate matter air pollution with aerodynamic diameter less than 2.5 μm (PM2.5) may be an independent risk factor for increased cardiovascular morbidity and mortality [1,2], and recent studies suggest that reducing ambient particles resulted in declined cardiovascular morbidity and deaths [3,4]. Evidence from other and our own laboratories suggested that deposition of particles in the lung causes alveolar inflammation resulting in a systemic inflammatory response that impacts the blood vessels [5–8], which could be responsible for the downstream adverse cardiovascular effects [1]. This hypothesis is supported by several animal studies [6–8] and human studies showing that elevations of 10μg/m3 and 20μg/m3 in PM2.5 were associated with 5.9% and 12.1% increases in the development of atherosclerosis, respectively [9].
Inflammation plays a central role in all stages of atherosclerosis development [10]. Exposure to particulate matter air pollution results in excessive production of reactive oxidative species (ROS) [11]. These ROS contribute to many cardiovascular complications via exacerbating interactions with lipids, proteins, and DNA. An in vitro study showed that diesel exhaust particle (DEP)-induced ROS exerted lipid peroxidation to produce oxidized low-density-lipoprotein (oxLDL), which can serve as a stimulus for monocyte migration into the sub-endothelial space representing an key step in atherogenesis [10,12]. These monocyte-derived macrophages phagocytose oxLDL, contributing to foam cell formation, and the development of fatty streaks, a hallmark of early atherosclerotic lesion [10]. Macrophage CD36 is the major scavenger receptor for oxLDL, hence promoting the progression of atherosclerosis [13]. Active unstable atherosclerotic plaques are characterized by increased lipid accumulation, foam cell formation, smooth muscle migration from the sub-endothelial layer and a thin fibrous cap [14]. Plaque disruption and atherothrombosis are the underlying causes of approximately 60–80% of all sudden cardiac deaths [15].
Diesel exhaust particulate is a major component of urban PM2.5, accounting for up to 90% of the fine particulate mass in ambient air of many major cities, such as London [16]. Diesel Exhaust (DE) is a mixture of fine particles and gases, and represents a useful model of traffic-related air pollutants. The particulate from DE consists of a central carbon core nucleus onto which an estimated 18,000 combustion products are absorbed, including organic chemicals, such as polycyclic aromatic hydrocarbons, and transition metals [17,18].
Recent studies have shown an association between the progression of atherosclerosis and people living near major roads, and traffic-related air pollution exposure and acute coronary events [19]. The association between DE exposure and cardiovascular diseases is compelling; however, the impact of DE exposure on atherogenesis is poorly understood. In this study we exposed ApoE knockout mice to DE via whole-body inhalation for 7 weeks, and measured morphological alteration of atherosclerotic lesion to test the hypothesis that DE exposure causes changes in atherosclerotic plaque characteristic of unstable plaque or plaque vulnerable to cause acute vascular events. We also explored the impact of the magnitude of DE exposure and downstream oxidative stress on the changes in plaque morphology.
2. Material and methods
2.1. Exposure protocol and experimental animals
Characteristics of the exposure system have been described elsewhere in detail [20] (See online supplementary “Material and methods” for more details).
Male ApoE knockout mice fed with regular chow, at the age of 30-week, were exposed for 7 weeks (5 days/week, 6 h/day) to DE controlled at the concentration of 200 μ/m3 PM2.5. Mice exposed to filtered air were the control (12mice/group). Animal procedures were approved by the Animal Care and Use Committee of the University of Washington.
2.2. Sample processing and image acquisition
After exposure, sodium pentobarbital (100mg/kg, Abbott Laboratories) and heparin sulfate (500U/kg) were administered intraperitoneally. Plasma, thoracic aorta, aortic root, lung and urine were collected and kept in appropriate conditions until assay. For morphometric analysis, images were captured by a spot digital camera (Microspot, Nikon), coded and analyzed using Image Pro Plus software. 10–12 fields per view of each animal were randomly chosen.
2.3. Plasma cholesterol and triglyceride
Plasma cholesterol and triglyceride levels were measured using commercially available kits (Cayman Chemical).
2.4. Lung tissue analysis
Lungs were inflated and fixed with 10% neutral formalin for 24 h, cut into 4 pieces longitudinal slices, embedded in paraffin blocks, sectioned at 10 μm and stained with hematoxylin and eosin (H and E). Random images were captured to represent the whole lung and analyzed using point counting to determine the volume fraction (V/v%) of alveolar macrophages. The V/v% of alveolar macrophages positive for particulate matter was measured as an indicator for the magnitude of lung exposure.
2.5. Histo- and immunohistochemical analysis of atherosclerotic plaque
Aortic root was used to quantify the changes in plaque. Aortic root tissues were fixed with 10% neutral formalin for 24 h, embedded in paraffin, sectioned at 5 μm and stained for Movat pentachrome. Point counting was used to determine the V/v% of plaques, total cell counts (plaque cellularity) and foam cells. The nuclei of cells in each lesion were enumerated and normalized to the plaque size.
Picro-sirius red (Sigma) staining was used to identify collagen content and collagen was quantified (V/v%) in plaque tissues.
The expression of iNOS, CD36, nitrotyrosine and α-actin was quantified after immunohistochemical staining for aortic root tissues. Sections were incubated with antibodies F4/80, a maker for mature macrophages (1:50, AbD Serotec); inducible nitric oxide synthase (iNOS) (1:100); CD36 (1:50); nitrotyrosine (NT) (1:400), and α-actin (1:600) at 4°C overnight. Subsequently, sections were incubated with biotinylated goat anti rabbit IgG (1:800, Vector Laboratories), followed by avidin- biotin conjugated alkaline phosphatase and Vector red (Vector Laboratories) to detect the antigen-antibody complexes. The area of positive staining was recognized, and the volume fraction (V/v%) of iNOS, CD36 and NT was determined.
2.6. Lipids in plaque
Frozen aortic root was embedded in OCT (Sakura Finetek), sectioned at 5 μm, stained with Oil-Red-O and V/v% of positive staining was determined.
2.7. Systemic oxidative stress
Urine samples were purified using 8-isoprostane affinity sorbent, and 15-F2t-isoprostane (15-F2t-IsoP), a marker of lipid oxidation, was measured using a 8-Isoprostane EIA kit (Cayman Chemical). As a marker of DNA oxidation, 8-hydroxy-2-deoxy guanosine (8-OH-dG) concentration was measured using a 8-OH-dG kit (Cayman Chemical). Creatinine levels were measured by a creatinine assay kit (Cayman Chemical). The plasma myeloperoxidase (MPO) levels were also measured using an ELISA kit (Invitrogen).
2.8. Statistical analysis
Data are shown as mean ± SEM. The statistical significance was evaluated using the unpaired Student’s t-test for simple comparison between two values. Linear regression modeling was used to assess the relationship between exposure markers (alveolar macrophages with particles) and changes in plaques and oxidative stress markers. In all experiments, n equals the number of mice from which samples were obtained. p < 0.05 was considered to be significant.
3. Results
3.1. Alveolar macrophages in lung tissue
DE exposure significantly increased the number of alveolar macrophages positive for particles (3.5 ±1.2% in Filtered air vs. 85.7 ± 1.7% in DE; p < 0.0001) (Fig. 1A). The total number of alveolar macrophages was also increased in DE exposure group, compared with filtered air exposure (1.3±0.2% vs. 10.2±1.1%; Filtered air vs. DE; p <0.01) (Fig. 1B).
Fig. 1.
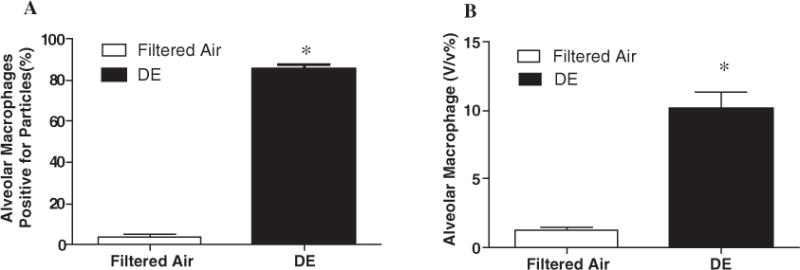
Exposure effects. (A) Exposure to DE increased the number of alveolar macrophages positive for particles, n=10, *p < 0.0001; (B) Total alveolar macrophages were also increased after DE exposure, n=6, *p < 0.01. Values are mean±SE.
3.2. No changes in plasma cholesterol and triglyceride
To determine whether DE increased circulating lipids and thereby lipids in atherosclerotic plaques, we measured plasma cholesterol and triglyceride levels. Exposure to DE did not affect plasma lipid levels (Supplementary Table 1).
3.3. Changes in atherosclerotic plaque composition
Fig. 2A, C and E shows different morphological features of plaques on Movat stains. There was no difference in the volume fraction of the size of atherosclerotic lesions between DE and filtered air exposed mice (Fig. 2A and B). Histological characterization of plaque morphology reveals that DE exposure increased plaque cellularity (70.0±4.0 vs. 100.0±9.8 cells/100 μm of lesion area; Filtered air vs. DE; p < 0.02) (Fig. 2C and D), foam cell formation (12.2±1.7% vs. 27.3±5.4%; Filtered air vs. DE; p < 0.04) (Fig. 2E and F), increased lipid accumulation (16.1±2.1% vs. 31.9±4.7%; Filtered air vs. DE; p < 0.02) (Fig. 3A–C), and smooth muscle cell content (7.5±2.0% vs. 25.5±9.1%; Filtered air vs. DE; p < 0.05) (Fig. 3D–F). We observed a non-significant decrease in collagen content of plaques (90.2±9.6% vs. 69.4±9.1%; Filtered air vs. DE; p > 0.05) (Fig. 3G–I). The proteoglycan content of plaques was similar between DE and Filtered air exposure groups (data not shown).
Fig. 2.
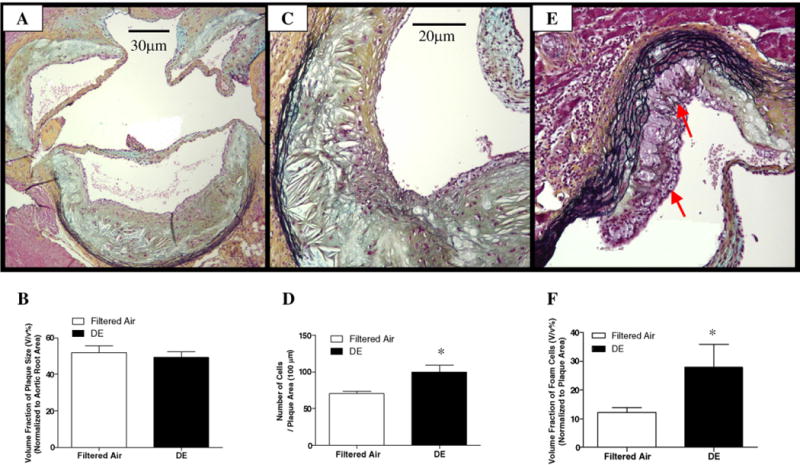
Movat staining analysis of atherosclerotic lesion size, cellularity and the number of foam cells in aortic root. (A) Representative photomicrographs of Movat staining for plaque size analysis (100×); (B) no changes in volume fraction of plaques were observed, n = 10, p > 0.05; (C) movat stainingfor total cell count (200×); (D) the cellularity in atherosclerotic lesion was significantly increased after DE exposure, n = 9, *p < 0.02; (E) movat staining for foam cell count (200×); (F) DE exposure significantly increased foam cells formation, n = 12, *p < 0.04. Values are mean±SE.
Fig. 3.
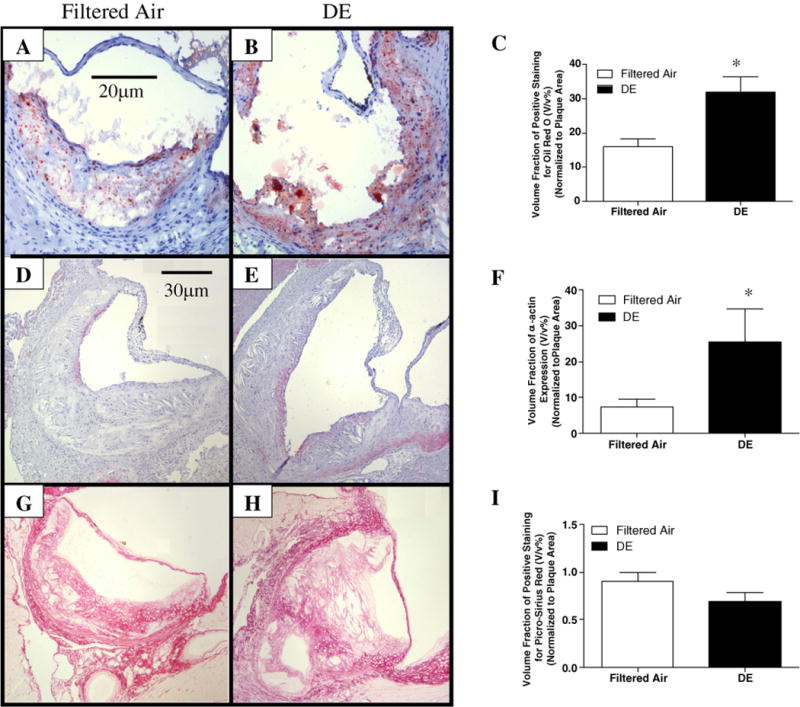
Histochemical analysis of the components of atherosclerotic plaque in aortic root. ((A) and (B)) Representative photomicrographs of oil red O staining for lipid content (200×); (C) Exposure to DE increased lipid content occupied in atherosclerotic lesion, n = 6, *p < 0.02; ((D) and (E)) Staining of α-actin for smooth muscle cells (100×); (F)The smooth muscle cell content was significantly increased after DE exposure, n =6, *p < 0.05; ((G) and (H)) picro-sirius red staining for collagen content (100×); (I) DE exposure induced non-significant reduction for collagen content, n =7, p > 0.05. Values are mean±SE.
3.4. Increased oxidative stress in atherosclerotic plaque
The expression of oxidative stress markers: iNOS, CD36, and nitrotyrosine in plaques was increased after DE exposure; iNOS (18.7±5.5% vs. 50.8±12.2%; Filtered air vs. DE;p <0.05) (Fig. 4A–C), CD36 (36.1±5.6% vs. 66.3±6.9%; Filtered air vs. DE; p < 0.005) (Fig. 4D–F), and nitrotyrosine (43.4±8.9% vs. 75.6±8.6%; Filtered air vs. DE; p < 0.02) (Fig. 4G–I), respectively. To further examine the relationship between the magnitude of DE exposure and down stream effect, we examined the association between the number of alveolar macrophages positive for particles and the numberoffoam cells, the expression of CD36 and iNOS in plaque, respectively. The number of alveolar macrophages positive for particles in filtered air group was significantly lower than that in DE exposure group (3.5±1.2% in Filtered air vs. 85.7±1.7% in DE); therefore, we used data from just DE exposure animals. We found that there were positive correlations between alveolar macrophages positive for particles and plaque foam cell formation (R2 = 0.35,p <0.027), CD36 expression (R2 = 0.49, p =0.015) and iNOS expression (R2 = 0.72, p = 0.0081), respectively (Fig. 5).
Fig. 4.
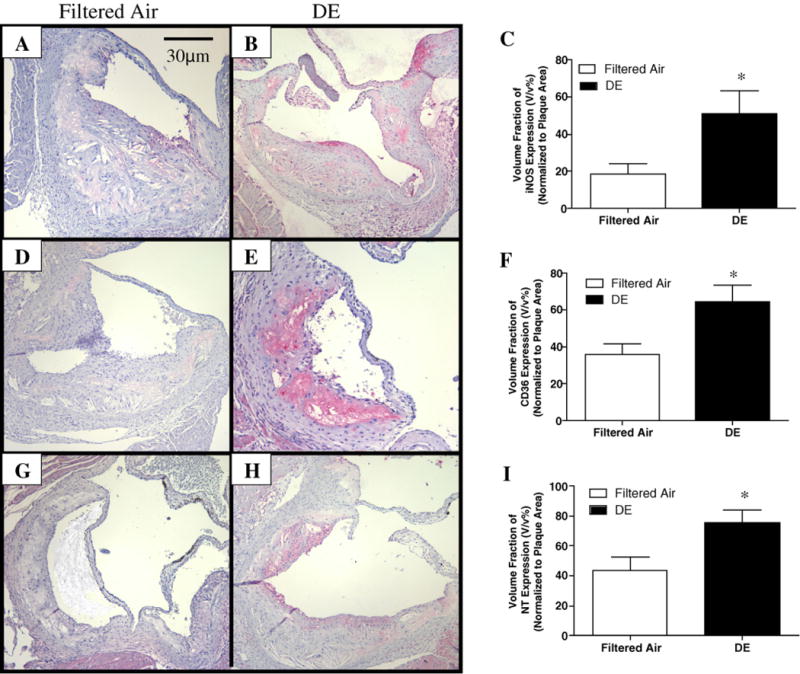
Immunohistochemical analysisofthe generationofoxidative stressinatherosclerotic plaqueinaortic root. ((A) and (B)) Representative photomicrographsofimmuno-histochemical staining for iNOS expressioninaortic root (100×); (C) exposuretoDEincreased iNOS expression,n = 8, *p < 0.05; ((D) and (E)) immunohistochemical staining for CD36 expression(100×); (F) CD36 expression was significantly increased after DE exposure, n = 12, *p < 0.005; ((G) and (H)) immunohistochemical staining for nitrotyrosine (NT) expression (100×); (I) DE exposure significantly enhanced nitrotyrosine expression, n = 9, *p < 0.04. Values are mean±SE.
Fig. 5.
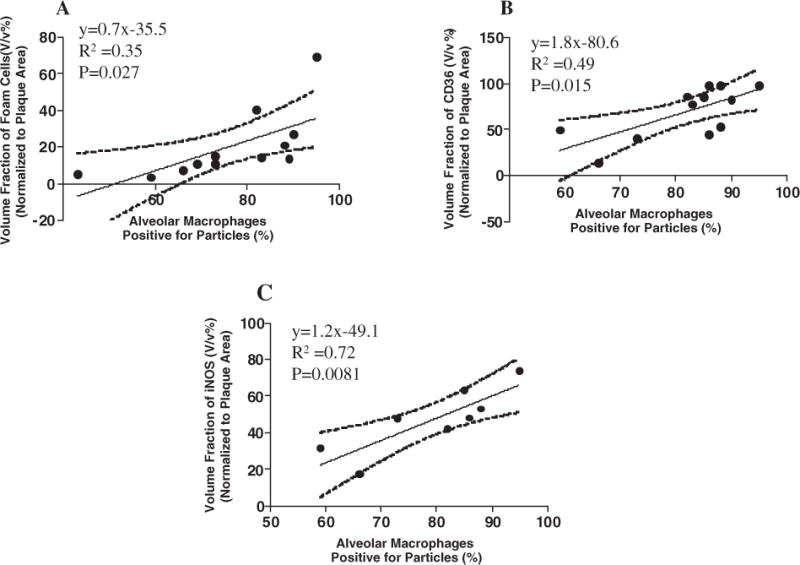
Association forDE exposure, plaque foam cell formation and oxidative stress. (A) Positive correlation between increased foam cell formation and alveolar macrophages positive for particles (R2 = 0.35,p = 0.027); (B) positive correlation between enhanced CD36 expression in atherosclerotic lesion in aortic root and alveolar macrophages positive for particles (R2 = 0.49, p = 0.015); (C) positive correlation between iNOS expression in aortic root and alveolar macrophages positive for particles (R2 = 0.72, p =0.0081).
3.5. Systemic oxidative stress
The urine levels of both 15-F2t-isoprostane (11.7±3.4 vs. 27.7±4.1 ng/creatinine(mmol); Filtered air vs. DE; p < 0.02) and 8-hydroxy-2-deoxy guanosine (1422±106 vs. 5110±1452 ng/creatinine(mmol); Filtered air vs. DE; p < 0.02) were increased by DE exposure (Supplementary Fig. 1). The plasma MPO levels were similar between DE and filtered air exposure groups (Supplementary Fig. 2).
4. Discussion
In this study, we demonstrate that DE exposure induced changes in atherosclerostic plaque that are characteristic of unstable plaques associated with vascular events [14,15]. We show that exposure to DE increased plaque lipids, plaque cellularity, foam cell formation and smooth muscle cell migration into plaque (Figs. 2 and 3). The positive correlation between the increased plaque foam cell formation and particle positive alveolar macrophages (Fig. 5A), links DE exposure to these morphological changes in atherosclerotic lesions. We also show an increase in oxidative stress markers both in the circulation and in the plaques themselves (Fig. 4 and Supplementary Fig. 1), and the positive association between particles in the lung and increased expression of iNOS and CD36 in aortic root (Fig. 5B and C) suggests that oxidative stress contributes to these DE-induced plaque changes.
With the rapid industrialization inboth developed and developing world, DE has become one of the most important components of urban particulate matter [15]. This is the first study to quantify the morphological changes in atherosclerotic plaque induced by exposure to DE and linked these changes to oxidative stress.
In this study, we exposed ApoE knockout mice to DE for 7 weeks using a well-controlled whole-body inhalation system that mimics real-world exposure near roadways. The average calculated exposure throughout a 24-hour period in our study was less than 35 μg/m3, which is within the range of the National Ambient Air Quality Standard for PM2.5 [21].
We demonstrate that exposure to DE resulted in an increase in alveolar macrophages that phagocytosed particles as well as the total number of alveolar macrophages (Fig. 1), indicating lung inflammation. Alveolar macrophages are key in processing inhaled particulate matter, and this study confirms previous findings from our laboratory using a different exposure model (EHC93 instillation), that the majority of particulate in the lungs were found in alveolar macrophages [8,22]. Alveolar macrophages are also powerful producers of mediators (such as IL-1β, IL-6, IL-8, and GM-CSF) that fuel the systemic inflammatory response when processing inhaled ambient particulate matter [8]. These pro-inflammatory mediators are known to activate the endothelium and up-regulate VCAM-1, ICAM-1, selectins, which are key steps for monocyte migration into the subendotheliumto engorge lipid hence promoting atherogenesis [10]. We have recently shown that mediators produced in the lung translocate to the circulating blood stream following instilled ambient particulate [5] underscoring the importance of the lung inflammatory response induced by PM exposure to the downstream vascular responses [1,10]. These findings support the concept that thesystemic inflammatory response is responsible for the downstream adverse vascular effects following exposure to particulate matter air pollution. The association between lung alveolar macrophages phagocytosed particles and foam cell formation in plaques (Fig. 5A) supports the link between the magnitude of exposure to PM and vascular disease.
4.1. Morphological changes in atherosclerotic plaques
Instability and rupture of an atherosclerotic plaque are implicated in the majority of acute ischemic syndromes, including myocardial infarction and stroke [15]. Histopathological features of these vulnerable plaques in humans include a large necrotic core (≥40% of plaque area), increased inflammatory cells (including macrophages/foam cells) with less smooth muscle cells and a thin fibrous cap (<65 μm) accompanied by reduced collagen and proteoglycans [14]. Although the features of vulnerable plaque in animal models are controversial [23], abundant evidence has suggested that the features in murine atherosclerotic lesions, including a large lipid core, increased inflammatory cells (e.g., macrophages) can be considered as histological markers for plaque progression and vulnerability [14].
Our study shows that exposure to DE promoted changes in plaques, such as an increase in lipid accumulation, higher cellularity, more foam cell formation and smooth muscle cell content (Figs. 2 and 3). Studies from other [7,8] and our own laboratories [6] have suggested that exposure to ambient PM is associated with the development and progression of athereosclerosis. Using Watanabe heritable hyperlipidemic rabbits, and instillation of PM into the lung,we previously demonstrated that exposure toPM collected over a major North American city (EHC93) not only caused an increase in the extent of atherosclerosis [6], but also induced changes in plaques (such as increase cellularity and lipid accumulation) similar to what we found in this study using an inhalation model. In subsequent studies using the rabbit model, we showed the increased adhesion molecule expression in the endothelium overlying plaques, and an acceleration of monocyte migration into atherosclerotic lesions [6]. Using a high fat diet mouse model, Sun and colleagues demonstrated that 6-month inhalation exposure to concentrated ambient PM2.5 particles enlarged plaque area and increased lipid content of atherosclerosis [8]. Similar to the method of Sun’s study, we also used ApoE knockout mice and inhalation exposure, but we fed these mice with regular chow and our exposure period was shorter (7 weeks vs. 6 months). Albeit the shorter exposure period, we found more extensive modifications of atherosclerotic lesions (Figs. 2 and 3), and enhanced oxidative stress in plaques (Fig. 4), suggesting that DE may be more active in modifying atherosclerotic plaques [24] and particle composition may play an important role in PM-induced adverse cardiovascular effects [25].
In the study, we did not observe significant differences of body weight, food consumption (data not shown) and lipid levels (Supplementary Table 1) between two groups. Using aortic root as a representative of large vessel atherosclerosis, we show that the volume fraction of atherosclerosis in aortic root was not different between DE and filtered air exposed mice (Fig. 2A and B). The results support findings from a recently published study using an even longer DE exposure model [25]. Studies in humans showed that the majority of acute vascular events originate in active plaques with more lipid accumulation and inflammatory cells (e.g., macrophages) [26] underlining the importanceof plaque composition rather than plaque size as a risk for vascular events. We show that DE exposure caused increased foam cell formation, cells originate from circulating monocytes [6], which differentiate into macrophages that specialize in phagocytosis of modified LDLs via their scavenger receptors, such as CD36, thereby transforming into foam cells [10]. Lipid uptake by macrophages stimulates the release of various cytokines, which support foam cell formation and smooth muscle recruitment from the media into the intima. Pro-inflammatory mediators, such as IL-1, IL-6, MIP-1 and TNF, promote smooth muscle cell migration, whereby contributing to the evolvement of smooth muscle cells from a “contractile” phenotype state to active “synthetic” state. Synthetic smooth muscle cells synthesize pro-inflammatory cytokines and release matrix metalloproteinases that can digest the components of extracellular matrix (e.g., collagen and proteoglycan) leading to weakening the integrity or stability of plaques [27]. Our findings of increased plaque lipid content, increased cellularity with more foam cells and smooth muscle cells, support the notion that DE exposure causes a persistent inflammatory milieu in plaques, all hallmarks of unstable vulnerable plaques. As mentioned, human studies have shown that plaque activity reflected by plaque composition rather than plaque size, is a stronger indicator for downstream vascular events [24]. We postulate that DE-induced compositional changes in atherosclerotic plaque could explain atleast part of the epidemiological associations between traffic pollution and cardiovascular events [2,15].
4.2. Oxidative stress and compositional changes in atherosclerotic plaque
Our results suggest that DE exposure induces oxidative stress that contributes to the compositional changes observed in atherosclerotic plaque. This notion is supported by the increased expression of iNOS, CD36 and nitrotyrosine in atheroslclerotic lesions (Fig. 4), enhanced systemic lipid and DNA oxidation, and the association between alveolar macrophages that phagocytosed particles (magnitude of exposure) and the increased expression of oxidative stress markers (e.g., iNOS and CD36) (Fig. 5B and C). ROS generation can arise directly from the surface of ambient particles, and soluble compounds (e.g., transition metals and organic compounds) [22]. Airway alveolar macrophages are the first cells to come in contact with inhaled particles. These cells release a large number ofpro-inflammatory mediators upon DE stimulation, at the same time they produce ROS at the sites of inflammation, wherebypromoting thelocaland systemicinflammatoryresponses [22]. Nitric oxide (NO) is an important biological mediator in living organisms that is synthesized from L-arginine using NADPH and molecular oxygen. The overproduction of NO catalyzed by iNOS is cytotoxic. iNOS is not expressed under physiology conditions, but up-regulated by pro-inflammatory mediators. iNOS cannot only catalyze NO production, but also generate superoxide in macrophages [28]. The concomitant NO production with increased levels of superoxide leads to the generation of a strong NO-derived oxidant, peroxynitrite, which is implicated in the pathogenesis of a number of cardiovascular diseases, including atherosclerosis [29].
Oxidative modification of LDL accelerates lipoprotein-uptake by macrophages hence promoting the formation of foam cells, which is the landmark of the development of atherosclerosis. This oxLDL uptake is mainly mediated by a macrophage scavenger receptor CD36, also known as oxLDL receptor [13]. CD36 is regulated by proatherogenic cytokines and lipids. We suspect that the increased CD36 expression in plaques promotes the increased foam cell formation and lipid accumulation as we observed (Figs. 2F and 3C). Collectively, the increase in both systemic oxidative stress markers and the markers of oxidative stress in plaque tissues support our hypothesis that exposure to DE induces ROS, which promote the changes we observed in the atherosclerotic lesion.
5. Conclusion
In summary, we demonstrate that DE exposure results in specific changes in atherosclerotic plaques that are associated with plaque activity and vulnerability. We also show a strong association between these changes in plaques and the particle deposition in the lung. The association between traffic related air pollution exposure and vascular events is increasingly well established [1] and our study supports the notion that changes in atherosclerotic plaques induced by traffic related pollutants (e.g., DE) contribute to these vascular events.
Supplementary Material
Acknowledgments
This work was supported by the NIH grants R01ES13434 (MER), K24ES013195 (JDK), P50ES015915 (MER,SVE,JDK), The Heart and Stroke Foundation of Canada (SVE, NB), and Michael Smith Foundation for Health Research (NB). SVE is a Senior Scholar with the Michael Smith Foundation for Health Research and CIHR/GSK professor in Chronic Obstructive Pulmonary Disease.
Abbreviations
- DE
diesel exhaust
- ELISA
enzyme-linked immunosorbent assay
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- H and E
hematoxylin and eosin
- HPRT1
hypoxanthine phosphoribosyltransferase-1
- ICAM
intercellular adhesion molecule
- iNOS
inducible nitric oxide synthase
- IL-1
interleukin-1
- MIP
macrophage inflammatory protein
- MPO
myeloperoxidase
- NF-κB
nuclear factor-kappa B
- NT
nitrotyrosine
- OxLDL
oxidized low-density-lipoprotein
- PM2.5
particulate matter air pollution with aerodynamic diameter less than 2.5 μm
- RT-PCR
reverse transcription polymerase chain reaction
- ROS
reactive oxidative species
- TNF
tumor necrosis factor
- VCAM-1
vascular cell adhesion molecule-1
- 8-OH-dG
8-hydroxy-2-Deoxy guanosine
- 15-F2t-IsoP
15-F2t-isoprostane
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.atherosclerosis.2011.02.019.
References
- 1.Brook RD, Rajagopalan S, Pope CA, III, et al. Particulate matter air pollution and cardiovascular disease:an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–78. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 2.Peters A, von KS, Heier M, et al. Exposure to traffic and the onset of myocardial infarction. N Engl J Med. 2004;351:1721–30. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- 3.Clancy L, Goodman P, Sinclair H, Dockery DW. Effectof air-pollution control on death rates inDublin, Ireland: anintervention study. Lancet. 2002;360:1210–4. doi: 10.1016/S0140-6736(02)11281-5. [DOI] [PubMed] [Google Scholar]
- 4.Laden F, Schwartz J, Speizer FE, Dockery DW. Reduction in fine particulate air pollution and mortality: extended follow-up of the Harvard Six Cities study. Am J Respir Crit Care Med. 2006;173:667–72. doi: 10.1164/rccm.200503-443OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kido T, Tamagawa E, Bai N, et al. Particulate matter induces IL-6 translocation from the lung to the systemic circulation. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0427OC. [DOI] [PubMed] [Google Scholar]
- 6.Yatera K, Hsieh J, Hogg JC, et al. Particulate matter air pollution exposure promotes recruitment of monocytes into atherosclerotic plaques. Am J Physiol Heart Circ Physiol. 2008;294:H944–53. doi: 10.1152/ajpheart.00406.2007. [DOI] [PubMed] [Google Scholar]
- 7.Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF. Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol. 2002;39:935–42. doi: 10.1016/s0735-1097(02)01715-1. [DOI] [PubMed] [Google Scholar]
- 8.van Eeden SF, Tan WC, Suwa T, et al. Cytokines involved inthe systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)) Am J Respir Crit Care Med. 2001;164:826–30. doi: 10.1164/ajrccm.164.5.2010160. [DOI] [PubMed] [Google Scholar]
- 9.Kunzli N, Jerrett M, Mack WJ, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113:201–6. doi: 10.1289/ehp.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 11.Li XY, Gilmour PS, Donaldson K, MacNee W. Free radical activity and pro-inflammatory effects of particulate air pollution (PM10) in vivo and in vitro. Thorax. 1996;51:1216–22. doi: 10.1136/thx.51.12.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda M, Shitashige M, Yamasaki H, Sagai M, Tomita T. Oxidative modification of low density lipoprotein by diesel exhaust particles. Biol Pharm Bull. 1995;18:866–71. doi: 10.1248/bpb.18.866. [DOI] [PubMed] [Google Scholar]
- 13.Podrez EA, Febbraio M, Sheibani N, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ni M, Chen WQ, Zhang Y. Animal models and potential mechanisms of plaque destabilisation and disruption. Heart. 2009;95:1393–8. doi: 10.1136/hrt.2008.143461. [DOI] [PubMed] [Google Scholar]
- 15.Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–82. doi: 10.1056/NEJM199705013361802. [DOI] [PubMed] [Google Scholar]
- 16.Guo X, Liu ES, Ko JK, et al. Protective role of cyclooxygenase inhibitors in the adverse action of passive cigarette smoking on the initiation of experimental colitis in rats. Eur J Pharmacol. 2001;411:193–203. doi: 10.1016/s0014-2999(00)00914-6. [DOI] [PubMed] [Google Scholar]
- 17.Schuetzle D, Lee FS, Prater TJ. The identification of polynuclear aromatic hydrocarbon (PAH) derivatives in mutagenic fractions of diesel particulate extracts. Int J Environ Anal Chem. 1981;9:93–144. doi: 10.1080/03067318108071903. [DOI] [PubMed] [Google Scholar]
- 18.Weisenberger B. Health effect of diesel emissions: a current status report. J Soc Occup Med. 1981;31:4–8. doi: 10.1093/occmed/31.1.4. [DOI] [PubMed] [Google Scholar]
- 19.Hoek G, Brunekreef B, Goldbohm S, Fischer P, van den Brandt PA. Association between mortality and indicators of traffic-related air pollution in the Netherlands: a cohort study. Lancet. 2002;360:1203–9. doi: 10.1016/S0140-6736(02)11280-3. [DOI] [PubMed] [Google Scholar]
- 20.Gould T, Larson T, Stewart J, Kaufman JD, Slater D, McEwen N. A controlled inhalation diesel exhaust exposure facility with dynamic feedback control of PM concentration. Inhal Toxicol. 2008;20:49–52. doi: 10.1080/08958370701758478. [DOI] [PubMed] [Google Scholar]
- 21.Brook RD, Franklin B, Cascio W, et al. Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation. 2004;109:2655–71. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 22.Bai N, Khazaei M, van Eeden SF, Laher I. The pharmacology of particulate matter air pollution-induced cardiovascular dysfunction. Pharmacol Ther. 2007;113:16–29. doi: 10.1016/j.pharmthera.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–13. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 24.Hirano S, Furuyama A, Koike E, Kobayashi T. Oxidative-stress potency of organic extracts of diesel exhaust and urban fine particles in rat heart microvessel endothelial cells. Toxicology. 2003;187:161–70. doi: 10.1016/s0300-483x(03)00053-2. [DOI] [PubMed] [Google Scholar]
- 25.Quan C, Sun Q, Lippmann M, Chen LC. Comparative effects of inhaled diesel exhaust and ambient fine particles on inflammation, atherosclerosis, and vascular dysfunction. Inhal Toxicol. 2010;22:738–53. doi: 10.3109/08958371003728057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation. 1994;90:775–8. doi: 10.1161/01.cir.90.2.775. [DOI] [PubMed] [Google Scholar]
- 27.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–52. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA. 1997;94:6954–8. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cromheeke KM, Kockx MM, De Meyer GR, et al. Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res. 1999;43:744–54. doi: 10.1016/s0008-6363(99)00148-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.