

Abstract
Malignant rhabdoid tumors arise in several anatomic locations and are associated with poor outcomes. In the brain, these tumors are known as atypical teratoid/rhabdoid tumors (AT/RT). While genetically engineered models for malignant rhabdoid tumors exist, none of them recapitulate AT/RT for which preclinical models remain lacking. In the majority of AT/RT, loss of heterozygosity occurs at the genetic locus SNF5 (Ini1/BAF47/Smarcb1), which functions as a subunit of the SWI/SNF chromatin-remodeling complex and a tumor suppressor in familial and sporadic malignant rhabdoid tumors. Therefore, we generated mice in which Snf5 was ablated specifically in nestin-positive and/or glial fibrillary acid protein (GFAP)-positive progenitor cells of the developing central nervous system (CNS). Snf5 ablation in nestin-positive cells resulted in early lethality that could not be rescued by loss of p53. However, Snf5 ablation in GFAP-positive cells caused a neurodegenerative phenotype exacerabated by p53 loss. Notably, these double mutants exhibited AT/RT development, associated with an earlier failure in granule neuron migration in the cerebellum, reduced neuronal projections in the hippocampus, degeneration of the corpus callosum and ataxia and seizures. Gene expression analysis confirmed that the tumors which arose in Snf5/p53 mutant mice were distinct from other neural tumors and most closely resembled human AT/RT. Our findings uncover a novel role for Snf5 in oligodendrocyte generation and survival, and they offer evidence of the first genetically engineered mouse model for AT/RT in the CNS.
Keywords: Smarcb1, pediatric brain tumors, neurodevelopment, genetic mouse model, neurodegeneration
Introduction
Malignant rhabdoid tumors (MRT) are aggressive, poorly differentiated pediatric cancers, characterized by the presence of germline and/or somatic mutations in Snf5/Ini1/Baf47/Smarcb1 which functions as a component of the SWI/SNF core chromatin-remodeling complex (1). Tumors arise frequently in kidney and soft tissues, and less frequently in the central nervous system (CNS). In the CNS, neoplastic lesions, referred to as Atypical Teratoid/Rhabdoid Tumors (AT/RT), occur primarily in early childhood with a median age of onset of 11 months (2). Most children die from disease within 1 year of diagnosis, despite use of intensive therapy.
Snf5/Ini1 is a nuclear protein that is constitutively expressed in normal cells but absent in AT/RT (3). The majority of rhabdoid tumors exhibit loss of heterozygosity (LOH) at the Snf5/Ini1 locus and harbor recurrent biallelic alterations (deletions and point mutations) irrespective of the tissue of origin (2,4). While several studies demonstrate a role for Snf5 in the regulation of cyclin D1, p16Ink4a and pRb, through the ATP-dependent chromatin-remodeling SWI/SNF complex, the mechanism responsible for oncogenesis remains unclear (5). Ink4 and Arf signaling have been suggested to be disrupted in AT/RT(6), and reintroduction of Snf5/Ini1 into rhabdoid tumor cells causes G0/G1 arrest and senescence by direct repression of cyclin D1 and activation of p16Ink4a (7–9). It has been proposed that Snf5/Ini1 activates the mitotic spindle checkpoint through the p16-cyclinD1/Cdk4-pRb-E2F pathway (10). The interferon and hedgehog (Hh) signaling pathways have also been proposed to be affected by Snf5/Ini1 (11,12) and it has been suggested that oncogenesis in the absence of Snf5 requires residual activity of BRG1-containing SWI/SNF complexes (13).
Alterations in Snf5/Ini1 have also been reported in familial and sporadic schwannomatosis (14,15) and in gastrointestinal stromal tumors (GIST) (16). Furthermore, loss of Snf5/Ini1 has been documented in renal medullary carcinoma, possibly as a consequence of epigenetic silencing (17). Taken together, these data imply that Snf5 is a tumor suppressor gene involved in oncogenic transformation of cells present in a broad range of developing tissues.
In mice, homozygous deletion of Snf5/Ini1 results in early embryonic lethality, whereas heterozygous loss, similar to the situation in humans, predisposes to development of aggressive sarcomas (18–20). Conditional inactivation of Snf5 in mice results in profound cancer susceptibility, with all animals developing tumors at a median age of 11 weeks (21). These lesions exhibit many features of rhabdoid tumors, including the complete absence of Snf5/Ini1 expression. Homozygous or heterozygous deletion of p53 in Snf5 heterozygous mice accelerates the appearance of MRTs (22,23). However, to date, no brain tumors have been reported in mice carrying mutations in Snf5.
To investigate the role of Snf5 in the developing CNS, we targeted Snf5 and p53 mutations to neuronal progenitor cells using Cre-lox technology. Ablation of Snf5 in nestin-positive neural progenitor cells resulted in embryonic lethality, which was not rescued by the absence of p53. In contrast, initially only modest neuroanatomic defects were observed in Snf5-Flox/GFAP-Cre mice indicating a strong lineage-dependent effect of Snf5 ablation. However, in the absence of p53, Snf5-Flox/GFAP-Cre mice exhibited neurodegeneration, defects in granule neuron migration, ataxia and seizure activity. These phenotypes appear to arise as a consequence of the loss of oligodendrocytes throughout the developing brain. In addition, all adult Snf5Flox/Flox/p53lox/lox/GFAP-Cre mice examined, after the age of four weeks, exhibited highly aggressive brain tumors displaying many hallmarks of CNS AT/RT, including loss of Snf5 expression.
Materials and Methods
Transgenic mice
GFAP-Cre (GFAP, glial fibrillary acidic protein) transgenic mice, a gift from David H. Gutmann (Washington University School of Medicine, St. Louis, MO), have been described previously (24,25) to drive expression of cre recombinase specifically in the developing nervous system of the mouse. To obtain Snf5 conditional loss in the nervous system of the mouse, Snf5Flox/Flox mice (21) provided by Charles Roberts (Dana-Farber Cancer Institute and Children's Hospital, Harvard Medical School, Boston, MA) were crossed with GFAP-Cre mice for two generations to generate Snf5Flox/Flox/GFAP-cre mice, as well as littermate controls. Inactivation of p53 was achieved using p53lox/lox mice, obtained from the National Cancer Institute (Bethesda, MD), to generate Snf5Flox/Flox/p53lox/lox/GFAP-Cre animals or also referred to as Snf5F/F/p53L/L/GFAP-Cre mice. Genotyping of Snf5 flox, p53 lox and Cre are described in Supplemental data. Ptc1+/− mice obtained from Dr. Mathew Scott (Stanford University, CA, USA) were maintained on a mixed C57Bl/6 and 129Sv strain background and were crossed with p53+/− mice (Jackson Laboratory, USA) to generate Ptc1+/− p53+/− and Ptc+/− p53-/− mice. Genotyping of Ptc1 and p53, was performed as previously described (26,27)
Histology and immunohistochemistry
Animals were transcardially perfused with 1x phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. For some experiments, mice were sacrificed by cervical dislocation after intraperitoneal (IP) injection with ketamine (100–300 mg/kg) and xylazine (16–48mg/kg). Following dissection, brain tissues were transferred to PBS and subsequently embedded in paraffin and sectioned at a thickness of 5 µm. Every tenth section was stained with hematoxylin and eosin (H&E), and sections from similar anatomical planes were chosen for histology and immunohistochemical (IHC) analyses. Histopathological analysis was performed in a blinded manner. Immunohistochemistry and double-label immunofluorescence (IF) staining were performed on formalin fixed paraffin embedded (FFPE) brain sections. Procedure details and antibodies are described in Supplemental data.
EEG analysis
All EEG studies were performed on Snf5/p53/GFAP-Cre mice. Mice carrying the Cre alleles were used as controls. The methods for implantation and recording have been previously published (28,29) and details are presented in Supplemental data. EEG tracings were visually reviewed for the presence of electrographic seizures. If a seizure was suspected, the video was reviewed to ensure the electrographic change was associated with a seizure and not associated with movements, such as scratching, which could produce an artifact. The EEG was also qualitatively reviewed to characterize the background activity, including the amplitude, frequencies, and the presence of normal transients (e.g. theta in hippocampal electrodes during exploration), and interictal epilpetiform abnormalities such as spikes. These patterns were compared in all groups monitored.
Microarray gene expression analyses
Total RNA was isolated from mouse AT/RT derived from Snf5F/F p53L/L GFAP-Cre mice (n = 5) and from medulloblastoma derived from Ptc1+/− p53+/− (n = 2) and Ptc1+/− p53-/− (n = 3) mice using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen). 200 ng of total RNA was amplified and converted to biotinylated cRNA using the MessageAmp Premier Ampification Kit (Revision G, Life Technologies, Carlsbad, CA). 10 ug of fragmented cRNA were used for each Affymetrix Mouse Genome 430A 2.0 microarray, with hybridization, washing, staining, and scanning performed according to the standard protocol (Expression Analysis Technical Manual, Revision 3, Affymetrix, Santa Clara, CA). Raw data from the GeneChip 3000 7G scanner, converted to CEL intensity files using Affymetrix Command Console version 4.1.2, were used for subsequent analyses. Detailed quantitative Real-time PCR (qRT-PCR) and gene expression of human and mouse data sets are described in Supplemental data. All data have been deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=mropwgcsphczhat&acc=GSE68627).
Results
Snf5 and p53 loss in neural stem/progenitor cells of the mouse CNS
The nestin-Cre transgene (30) was introduced into mice to ablate Snf5 and p53, individually and together, from the developing nervous system. Loss of Snf5 alone in neuronal progenitor cells resulted in embryonic lethality, as no live pups homozygous for Snf5 loss (0/79) were obtained from heterozygous intercrosses of Snf5+/F NestinCre mice. In contrast, heterozygous Snf5+/F Nestin-Cre mice (n = 85) were grossly normal and fertile, and did not exhibit any signs of disease up to one and half years of age. Very small litter sizes (average of 5) were obtained from these crosses indicating that most embryos died in utero. To rescue the suspected lethality, we crossed Snf5+/F Nestin-Cre mice with p53 Lox mice, however, even in the absence of p53, we did not obtain any live Snf5F/F Nestin-Cre pups (0/151). Thus, Snf5 expression in nestin-expressing neural progenitor cells is essential for completion of embryonic development.
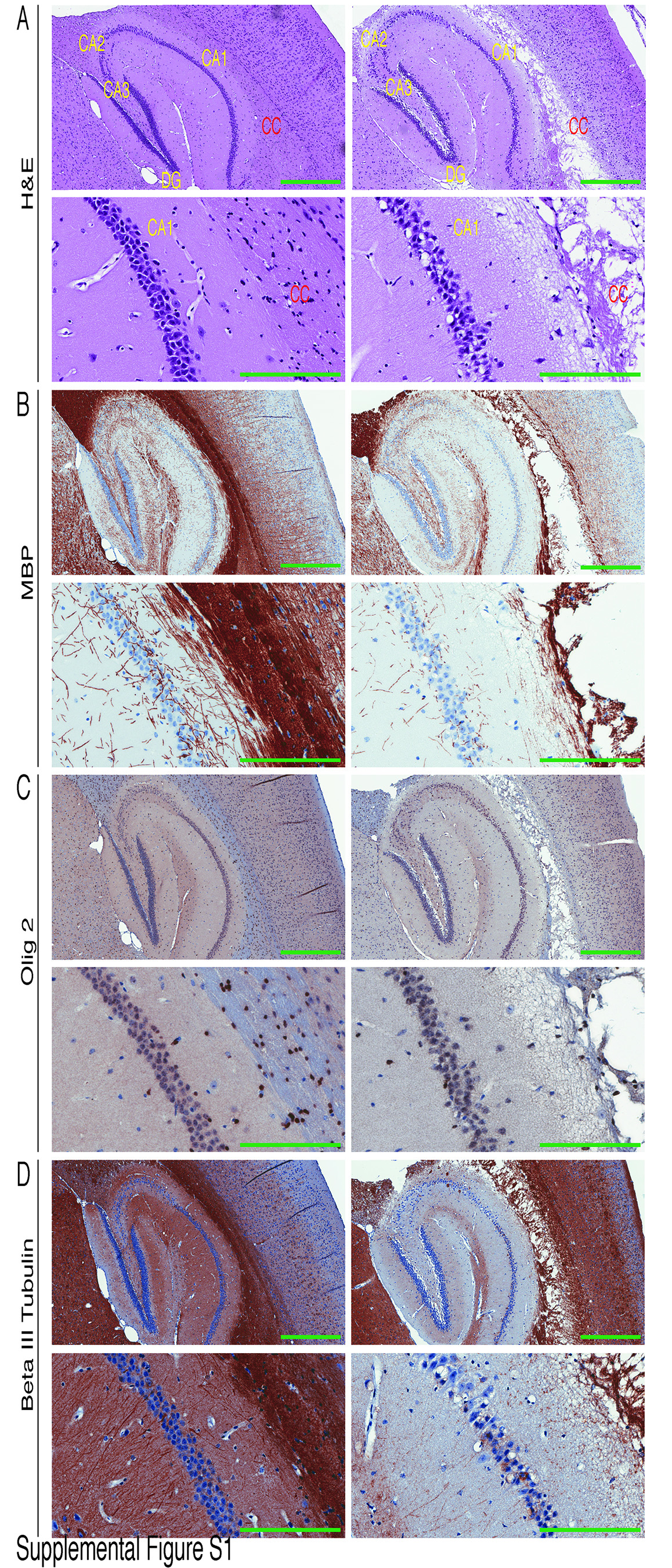
To target additional mouse neural stem/progenitor cell populations, we used the human glial fibrillary acidic protein (GFAP) promoter to program Cre expression (GFAP-Cre) (24,25) in Snf5-Flox mice. This promoter drives Cre expression in neural progenitor cells and a small subset of astrocytes (31). Snf5+/F GFAP-Cre and Snf5F/F GFAP-Cre mice were grossly normal and fertile. However, histological analysis of brain sections revealed evidence of a degenerative phenotype in all Sn5F/F GFAP-Cre mice examined (Suppl. Fig. S1). The extent of the damage was variable and it progressed with age. The lesions were prominent in the white matter of the corpus callosum with vacuolization, spongy changes, cystic-like breakdown and destruction of tissue. The posterior portion of the corpus callosum (splenium) was more affected than the anterior portion (genu). Loss of axons and myelin were also detected in the mutant brains that were stained with myelin basic protein (MBP) and proteolipid protein (PLP) antibodies while both proteins were abundant in control animals. However, the cerebral cortex, although thinner, and the pyramidal cells of the hippocampus, were mostly well preserved. The cerebellum appeared grossly normal, although there was a variable, mild defect in granule neuron migration and disorganization of the Purkinje cell layer. We also found slightly reduced levels of cells expressing oligodendrocyte lineage transcription factor 2 (Olig 2) in the hippocampal region of the mutant brain (Suppl. Fig. S1C). In the control brain, we counted 388 cells in total of which 229 cells were positive for Olig 2, whereas in the mutant brain 174 total cells were counted of which 89 cells were positive for Olig 2. This indicates that on a unit basis there is a reduction in the total number of cells from 194 (control) to 58 (mutant) whereas the number of Olig 2 positive cells drops from 150 (control) to 30 (mutant). These data suggest that there is a 80% reduction in the percentage of Olig 2 positive cells in the area examined in the mutant brain. Additionally, there was decreased expression of the specific neuronal marker beta-III-tubulin in the mutant brains (Suppl. Fig. S1D).
Over the course of more than one year, no brain tumors developed in any Snf5+/F GFAP-Cre or Snf5F/F GFAP-Cre mice. We performed histological analyses of a total of thirty brains from Snf5+/F GFAP-Cre and Snf5F/F GFAP-Cre mice, from ages 2 months to 1 year, and did not detect any evidence of neoplastic growth. Since p53 loss or reduced p53 expression have been reported to accelerate peripheral tumors in mice heterozygous for Snf5 (19,22,23) we investigated whether p53 ablation would promote brain tumor formation by crossing Snf5F/F GFAP-Cre mice with p53 Lox mice.
Mice heterozygous for Snf5 (Snf5+/F p53+/L GFAP-Cre, n = 54 and Snf5+/F p53L/L GFAP-Cre, n = 110) appeared grossly normal and fertile, although they displayed excessive scratching behavior, resulting in dermatitis around the neck, earlobes and the flank region. In contrast, Snf5F/F p53+/L GFAP-Cre mice showed similar phenotypic and pathohistological features to those seen in Snf5F/F GFAP-Cre animals, including, extreme lesions in the white matter of the corpus callosum and cingulum, reduced levels of expression of MBP, PLP, Olig 2, and beta-III-tubulin. However, there was no evidence of neoplastic growth. Double homozygous mice (Snf5F/F p53L/L GFAP-Cre, n = 240) were much smaller than their heterozygous littermates (Fig. 1A, left), with bodyweight reduced by approximately 40% (Fig. 1A, right), and they required water gel for survival. In addition, they displayed severe ataxia with dystonia and seizures. Many mice also appeared quite lethargic with hind limb paralysis. Furthermore, they exhibited a more severe version of the scratching behavior seen in Snf5+/F p53L/L GFAP-Cre and Snf5F/F p53+/L GFAP-Cre mice.
Figure 1. Snf5 is required for a normal growth and behavior development.

(A) Left panel: A 3-week old control mouse (agouti, 14.3 gram) and its smaller Snf5F/F p53L/L GFAPCre (black, 7.2 gram) littermate. Right panel: Growth curve of Snf5 p53 GFAPCre mice: Snf5+/+ p53L/L GFAPCre (n = 6), blue) and Snf5F/F p53L/L GFAPCre (n = 6), green). The average body weights (gram) were followed in time over a period of 4 weeks.
(B) Footprint patterns of 4 months old control (left) and Snf5F/F/p53L/L/GFAP-Cre (right) mice. The front paws are blue and the back paws are red. Scale bar represents 1 cm.
Neurological abnormalities in Snf5F/F/p53L/L/GFAP-Cre mice
Snf5F/F/p53L/L/GFAP-Cre mice exhibited a strong neurological phenotype, tumbling repeatedly during attempted locomotion. Motor coordination problems, manifest as tremors, dystonia and abnormal posture of the hind limbs, became evident from around 3 weeks of age to adulthood. We used a foot-painting assay to trace the walking gait of adult mice. Two colors of nontoxic water-based paint were used to mark the bottom of the forepaws (blue) and hindpaws (red), and the mice were placed onto white paper to trace their footprints. Wildtype animals displayed steady, evenly measured strides, with hind- and forepaw positioning resulting in overlapping footprints and a uniform gait width (Fig. 1B, left). In contrast, Snf5F/F/p53L/L/GFAP-Cre mice had a non-uniform gait and exhibited circling behavior. The traces indicated uneven stride length and width, with smeared non-overlapping footprints, indicating dragging rather than precise positioning of the hindpaws and an inability to keep the hindquarters upright (Fig. 1B, right). Also, Snf5F/F/p53L/L/GFAP-Cre mice frequently fell over, and they repeatedly lifted and replaced the same paw in various positions during each stride. Ataxia, combined with dystonia and tremors, persisted into adulthood. Footprint tracings from single mutant floxed (Snf5F/F/GFAP-Cre) or double heterozygous mutant floxed mice (Snf5+/F/p53+/L/GFAP-Cre Snf5+/F/p53L/L/GFAP-Cre) appeared quite normal, indicating a straight path with regular alternating strides, and they were never observed to lose balance during locomotion. These results demonstrate that loss of Snf5 results in ataxia and they imply that Snf5 is required for normal functioning of the cerebellum.
Loss of Snf5 results in seizures
Observations of Snf5F/F/p53L/L/GFAP-Cre mice suggested possible seizure activity. To investigate this possibility, we performed long-term video electroencephalography (EEG) analysis of Snf5F/F/p53L/L/GFAP-Cre and wild type mice at 3 months of age. EEG measures electrical activity in the brain. The three mutant mice examined showed lower voltage and slower background EEG activity compared to wild type mice (Fig. 2; Note the tracings are displayed at the same gain (mV/mm) making the control look higher than the mutant). The low voltage tracing included intermittent runs of sharp waves (bracketed arrows in A- lower tracings) that sometimes evolved into clear seizures (bracketed arrows in B- upper tracings). The Snf5F/F/p53L/L/GFAP-Cre mice also exhibited intermittent electrographic seizures (Fig. 2B) associated either with forelimb clonus, evolving into body jerking, and wild running (stage 5 seizure) (32), or more commonly arrest of activity during the ictal EEG discharges. All three of the recorded mice developed spontaneous seizures. The EEG changes were consistently comprised of a build up of rhythmic spiking in both the cortex and hippocampus (arrows in Fig. 2B).
Figure 2. Loss of Snf5 results in electrographic seizures.

(A) Background EEG findings of 3 months old control and Snf5F/F/p53L/L/GFAP-Cre mice. For all tracings a cortical electrode (Cx, upper tracing) and hippocampal electrode (Hp, lower tracing) are presented. Representative EEG of adult C57/Bl6 (control, upper panel) and two Snf5F/F/p53L/L/GFAP-Cre (middle and lower panels) mice are shown. Compared with the control mouse, the Snf5F/F/p53L/L/GFAP-Cre mice showed lower voltage and slow background EEG activity with intermittent runs of sharp waves (bracketed arrows in lower tracings).
(B) Ictal EEG of three Snf5F/F/p53L/L/GFAP-Cre mice. The EEG changes consistently comprised of a build up of rhythmic spiking in both the cortex and hippocampus electrodes (arrows indicate time of seizure onset). The intermittent runs of sharp waves (bracketed arrows in figure A, lower tracings) sometimes evolved into clear seizures (bracketed arrows in figure B, upper tracings).
Snf5F/F/p53L/L/GFAP-Cre mice were also observed to have intermittent episodes of tail extension and dystonic posturing of the body, frequently in response to handling. To determine if these movements were seizures, during the EEG recordings, the mice were manipulated to induce the tail extension and dystonic posturing. For all three mutant mice recorded, we observed these behaviors after the mice were manipulated and while the animals were spontaneously walking around the recording cage. At no time, was there an associated seizure like electrographic discharge, strongly suggesting that these movements were not related to cortical or hippocampal seizure activity. Together, these data indicate that the Snf5F/F/p53L/L/GFAP-Cre mice had both spontaneous electrographic seizures as well as paroxysmal events of odd positioning and abnormal gait that did not have an electrographic correlate.
Disruption of hippocampus, cortex, cerebellum in Snf5/p53 GFAP-Cre mice
Histological analysis of adult Snf5F/F p53L/L GFAP-Cre mouse brain revealed profound defects in the white matter, corpus callosum, hippocampus and cerebellum compared with Snf5F/F GFAP-Cre animals (Fig. 3). Immunostaining using Ini1/Baf47 antibody demonstrated normal, nuclear expression in most cell types in control brain sections throughout the cortex, hippocampus and cerebellum (Fig. 3B and 3D). In contrast, loss of Snf5 expression was evident throughout Snf5F/F p53L/L GFAP-Cre brains in several specific cell populations. For example, loss of Snf5 expression was observed in approximately 20% of cells in the CA1 hippocampal region of mutant mice (52/280) compared to control mice (0/511) (Fig. 3B). Snf5 loss was evident in rare scattered cells throughout the thinner cerebral cortex of mutant mice (Fig. 3B). However, the most prevalent site of Snf5 loss was in the cerebellum in both white matter and some populations of cells in the disrupted granule cell layer (Fig. 3D, red arrowheads). Thus, Snf5F/F p53L/L GFAP-Cre mice showed Snf5 inactivation in several subsets of brain cells. Cell counting also revealed reduced total cell numbers in specific regions of the mutant mouse brains. The amount of cell loss increased over time and, ultimately, there was complete loss of tissue in regions of white matter.
Figure 3. Snf5 is essential for a normal brain architecture.

(A–B) H&E (A), Baf47 (B) of sagittal brain sections of the hippocampus of an adult control mouse (left: top 40x magnification; bottom 200x magnification) and Snf5F/F p53L/L GFAP-Cre mouse (right: top 40x magnification; bottom 200x magnification). In comparison to the control (A and B left), the empty space above the hippocampus represents the completely disrupted corpus callosum that is transformed to a cystic-like space in the Snf5F/F p53L/L GFAP-Cre mouse (A and B right). Scale bars represent 600 µm (top) and 200 µm (bottom). (C–D) H&E (C), Baf47 (D) of sagittal brain sections of the cerebellum of an adult control mouse (left: top 40x magnification; bottom 200x magnification) and Snf5F/F p53L/L GFAP-Cre mouse (right: top 40x magnification; bottom 200x magnification). Blue arrows indicate a group of malignant cells that are Snf5 negative in the subpial area of the molecular layer and red arrowheads mark individual Baf47/Snf5 negative cells (C and D right). Scale bars represent 600 µm (top) and 300 µm (bottom).
Inactivation of Snf5 leads to loss of oligodendrocytes
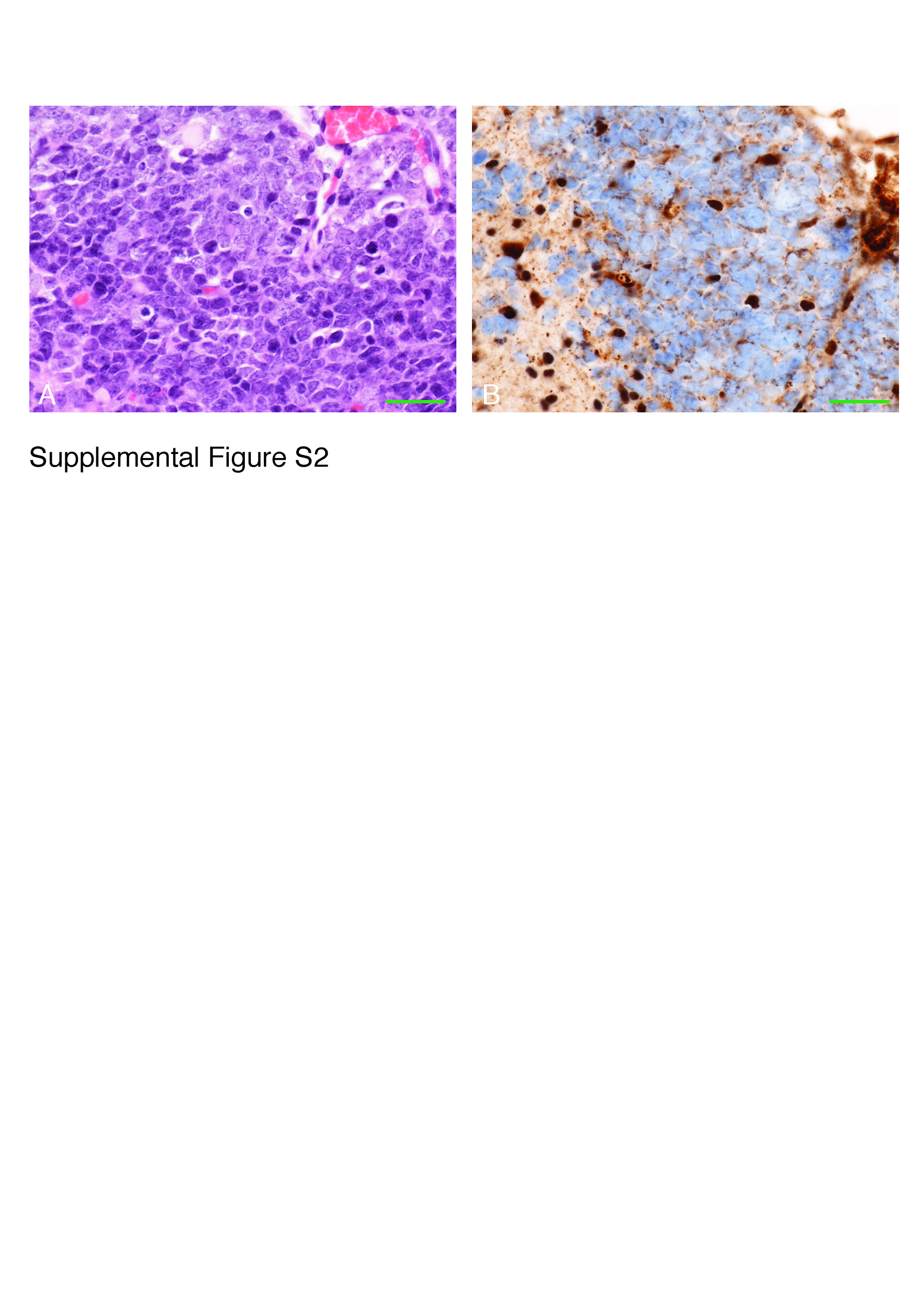
Analysis of the cerebellar organization of Snf5F/F p53L/L GFAP-Cre mice revealed defects in cytoarchitecture and altered granule neuron migration (Fig. 3C and 3D). Some granule neurons failed to migrate out of the external germinal layer, others remained trapped in the molecular layer and the boundary between the internal granule layer and the molecular layer remained diffuse. Remarkably, Snf5 was expressed in the majority of ectopic granule neurons suggesting that the defect in migration may be non-cell autonomous. In addition, there was a small patch of neoplastic, proliferating cells involving the subpial molecular region, located superficially at the interface of two cerebellar folia, representing an early neoplastic lesion. As expected, these neoplastic cells demonstrated a loss of nuclear expression of Ini1/Baf47 (Fig. 3D blue arrows). At higher magnification, large atypical cells, some with rhabdoid features and many mitoses, supporting a highly proliferative activity, are also present (Suppl. Fig. S2).
Compared with control cerebellum, white matter fiber tracks appeared to be much reduced in the mutant mice. Many of the cell nuclei present in these regions were negative for Snf5. To determine if this was a developmental phenotype, we examined the brains of three-week old mice (Fig. 4). The defects observed in adult mice, including lesions in the corpus callosum, reduced cerebral cortex, loss of white matter, altered cerebellar granule neuron migration (Fig. 4A and 4B, yellow arrowheads), were all present at three weeks of age. However, in this case there were many more cells present in the cerebellar white matter, and in the most superficial aspect of the cerebellar folia, that were negative for Snf5 staining (Fig. 4B, red arrows). Staining with GFAP antibodies revealed high levels of gliosis in the fiber tracks and in the molecular layer as well as in the remnants of the aberrant external germinal layer (Fig. 4C, red arrow and arrowhead, respectively). These findings imply that there is a progressive loss of cells and neuronal projections in the cerebellum of Snf5F/F p53L/L GFAP-Cre mice.
Fig 4. Snf5 loss causes early onset of granule neuron migration.

(A–B) H&E (A), Baf47 (B) of sagittal brain sections of a 3-week-old control mouse (left: top (whole brain) 8x magnification; middle (cerebellum) 40x magnification, bottom (cerebellum) 100x magnification) and Snf5F/F p53L/L GFAP-Cre mouse (right: top (whole brain) 8x magnification; middle (cerebellum) 40x magnification, bottom (cerebellum) 100x magnification). Yellow arrowheads indicate altered cerebellar granule neuron migration and red arrows mark individual Baf47/Snf5 negative cells. Scale bars represent 3mm (top) and 600 µm (middle) and 300 µm (bottom).
(C) GFAP of sagittal sections of the cerebellum of a 3-week-old control mouse (left 100x magnification) and Snf5F/F p53L/L GFAP-Cre mouse (right: 100x magnification). Red arrow and arrowhead indicate high levels of gliosis in the fiber tracks and in the molecular layer as well as in the remnants of the aberrant external germinal layer, respectively. Scales bar represent 300 µm.
Since the most prevalent site of loss of Snf5 expression in the cerebellum was observed in fiber tracks, we hypothesized that these cells may be oligodendrocytes. Immunofluorescent histochemical analysis revealed a profound loss of cells positive for the oligodendrocyte markers MBP, PLP, and Olig 2, in Snf5F/F p53L/L GFAP-Cre mice compared with control mice (Fig. 5). The prevalence of Snf5 negative oligodendrocytes dropped dramatically as the animals aged indicating that these cells are lost. Loss of oligodendrocytes is associated with neurodegeneration and seizure activity (33,34). In fact, ablation of mouse oligodendrocytes during the first 3 postnatal weeks causes a developmental phenotype, including abnormal cerebellar foliation, impaired neuronal migration, seizures, tremors and retarded growth, that is remarkably similar to that of Snf5F/F p53L/L GFAP-Cre mice (33,34). In contrast, the levels of NG2+ oligodendrocyte precursor cells were comparable in three-week old mutant mouse cerebellum and hippocampus compared with control mice (Supl Fig. S3A). Similar levels of NG2+ oligodendrocyte precursor cells were also seen in adult mutant mouse cerebellum and hippocampus compared with control mice (Suppl. Fig. S3B). In adult mutant mice, very few Olig2-positive/Baf47-negative remain in the cerebellum (Suppl. Fig. S4). These results imply that Snf5 plays a fundamental role in the generation and maintenance of oligodendrocytes after the NG2+ stage.
Figure 5. Snf5 Inactivation leads to loss of oligodendrocytes.

(A–C) Brains sections of the cerebellum of a 3-week-old control mouse (left) and Snf5F/F p53L/L GFAP-Cre mouse (right) with (A) double immunofluorescent labeling of DAPI (blue), Baf47 (green) and MBP (red), (B) double immunofluorescent labeling of DAPI (blue), Baf47 (green) and PLP (red), and (C) double immunofluorescent labeling of DAPI (blue), Baf47 (green) and Olig2 (red).
Scale bars represent 100 µm.
Snf5F/F p53L/L GFAP-Cre mice develop CNS AT/RT
Several Snf5F/F p53L/L GFAP-Cre adult mice of various ages died suddenly, independently of the severity of their ataxic or seizure phenotypes. Histological examination of brain tissues from these mice revealed the presence of high grade, aggressive tumors, that infiltrated and obliterated the surrounding cerebellar folia (Fig. 6). Subpial and leptomeningeal spread as well as infiltration into the mesencephalon was also seen in some mice, however all tumors appeared to arise from the cerebellum. Mice were examined when they showed the first signs of disease. However, the age of onset, was distributed across a 12-month period. Histologically, the tumors displayed many of the hallmarks of AT/RT. The classical rhabdoid cells, with eccentrically placed nuclei, prominent nucleoli and eosinophilic inclusion-like cytoplasm (Fig. 6B and 6C, arrows and arrowheads) were intermingled with primitive neuroectodermal tumor (PNET)-like small cells. In many cases, this PNET-like appearance was the predominant pattern. This is similar to human AT/RT in which only a minority of neoplastic cells demonstrate typical rhabdoid features with the majority represented by primitive PNET-like small cells. In keeping with the immunohistochemical profile of AT/RT, all tumors examined (n = 36), demonstrated loss of nuclear Ini1/Baf47 staining. Positive staining was seen only in stromal endothelial cells (Fig. 6D) (3). Strong reactivity for cytokeratin was seen in groups of cells, or single elements, with a maximum of up to 30%-40% of the tumor cells staining positive, depending on the specific case (CK, Fig. 6E). The same variability, with patchy and focal staining, was observed with synaptophysin (SYN, Fig. 6F) and glial fibrillary acidic protein (GFAP, Fig. 6G). Some tumors were negative for SYN and the scattered cells reactive for GFAP were identified as reactive astrocytes rather than tumor cells. Neither human AT/RT (n = 17) or our mouse tumors stained positive for NG2 (data not shown). In addition, Ki67, a nuclear marker of the cell cycle, demonstrated high reactivity (50%-60% and in some cases even up to 70–80%) (Fig. 6H), and the apoptotic marker Caspase-3 was also observed (not shown). The other widely-used markers for AT/RT, smooth muscle actin (SMA) and epithelial membrane antigen (EMA), do not work on mouse tissues.
Figure 6. Snf5F/Fp53L/LGFAP-Cre mice develop CNS AT/RT.

(A–C) H&E stained sagittal section of a 5 months old mouse Snf5F/Fp53L/LGFAP-Cre brain, demonstrates a tumor infiltrating the cerebellum with obliteration of the folia (A). The tumor high grade, poorly differentiated tumor (B) characterized by the presence of cells with eccentrically placed nuclei, prominent nucleoli (arrowhead) and eosinophilic inclusion-like cytoplasm (arrow), resembling rhabdoid cells (C) in a background of non-descriptive primitive neuroectodermal cells.
(D–H) Ini1/Baf47 expression (D) is loss in the nuclei of all tumor cells, but is retained in the vascular endothelial elements (brown). Focal expression (brown) of cytokeratin (E), synaptophysin (F), and GFAP (G) and high nuclear expression of Ki67 (H) was also observed in the tumor cells. Scale bars represent 800 µm (A), 40 µm (B, D-H) and 27 µm (C).
Snf5F/F p53L/L GFAP-Cre mice developed brain tumors starting at around 1 month of age (n=8) and they were observed in all adult mice examined (n = 58). Taking together the histologic appearance, immunohistochemical profile, and loss of Snf5 expression in tumors cells, we conclude that the tumors arising in Snf5F/Fp53L/LGFAP-Cre mice should be considered as AT/RT.
Snf5F/Fp53L/LGFAP-Cre tumors and human ATRT exhibit similar gene expression profiles
Human medulloblastoma (MB) can be segregated into four major categories based on gene expression patterns: Hedgehog (HH) subtype, Wnt subtype, Group 3, and Group 4 (35,36). However, they all exhibit strikingly different gene expression profiles from AT/RT (37). We re-analyzed published gene expression microarray dataset of pediatric brain tumors to identify a gene expression profile that clearly distinguished human AT/RT from human MB (37). We used this profile, choosing only genes that have clear murine orthologs, to compare tumors from Snf5F/Fp53L/LGFAP-Cre mice with MB from Ptc1+/− mice. As illustrated in Fig. 7A–C, Snf5F/Fp53L/LGFAP-Cre tumors are clearly very different from mouse MB and the markers that distinguish human AT/RT from human MB also distinguish the mouse tumors. We confirmed this gene expression array finding using a selection of specific marker genes in a quantitative real-time PCR (qRT-PCR) assay (Fig. 7D). The set of genes include 3 controls, 5 genes more highly expressed in human AT/RT and 5 genes more highly expressed in human MB. The data are very consistent with analysis of human brain tumors and clearly show that our mouse tumors closely resemble human AT/RT. Details of the analysis, including gene set enrichment analysis (GSEA) are presented in Suppl. Fig. S5).
Figure 7. Snf5F/Fp53L/LGFAP-Cre tumors and human AT/RT show similar gene expression signatures.

(A–D) Differential expression analysis of 4,473 genes, which are included in the published Pomeroy human tumor (AT/RT and MB) dataset that have clearly identifiable mouse orthologs, with Snf5F/Fp53L/LGFAP-Cre tumors and mouse MB from Ptch1+/− mice (A). The plot shows the global similarity of AT/RT-MB differential expression between the human and mouse data sets. Each point represents the log2-ratio of mean expression measurements of a single gene. These 2 sets of values are positively correlated. (Pearson’s r = 0.22, p < 10e-16). A set of genes was selected based on this plot to represent AT/RT-specific markers (FGFR1, FGFR2, ID3, PMP22, and PRCP; red dots) and MB-specific markers (NEUROD1, NEUROD2, NTRK3, RELN, SNAP25; blue dots). Comparison on these markers in bar graphs of human data (B) and mouse data (C) indicate they clearly distinguish the two sets of tumors. To validate these results, we used specific probes for qRT-PCR analysis of RNA from Snf5F/Fp53L/LGFAP-Cre tumors and MB from Ptch+/− mice (D). In addition, 3 control genes (Atad3a, Creb3, and Prdx5) that were expressed at the same levels in each tumor type were also examined. The color represents no change (grey bars), AT/RT > medulloblastoma (red bars), and AT/RT < medulloblastoma (blue bars). The error bars represent 95% confidence interval of group difference. The delta cycle threshold (CT) method was used to compute the expression levels of individual genes after data normalization as described below.
Discussion
Genetically-engineered mouse (GEM) models have allowed significant advances to be made in the understanding of human cancer and, in the case of Hedgehog pathway inhibitors, they allowed critical proof of concept studies that accurately predicted subsequent clinical responses (38,39). Here we report a new GEM, Snf5F/Fp53L/LGFAP-Cre that provides a model for CNS AT/RT. While no mouse model can encompass the diversity of human cancer, Snf5F/Fp53L/LGFAP-Cre mice recapitulate the histopathology, invasive phenotype and aggressive growth of AT/RT brain tumors. In addition, gene expression analysis indicated that our mouse tumors share a gene expression profile with human AT/RT that distinguishes them from MB. We hope that these mice, and tumor cells derived from the mice, propagated in culture or in allograft tumor transplants, can be used to develop new approaches for the treatment of this devastating pediatric disease.
Ablation of Snf5 using a nestin-Cre expression construct resulted in prenatal lethality. The cause of the lethality was not determined, but it was not rescued by the absence of p53. Complete ablation of Snf5 results in embryonic lethality around the time of implantation (18,20) and targeted ablation at birth, using the inducible MX-Cre construct to achieve widespread excision in many tissues, with the exception of the brain, results in hematopoietic failure leading to death of most mice at 1–3 weeks of age (40). Thus, Snf5 seems to be essential for survival of neuronal lineages as it is in the majority of cell types.
In the case of the GFAP-Cre construct, mice did survive, but they exhibited an unexpected progressive neurological phenotype. Although the age of onset and severity of the phenotype varied, all mice examined displayed some histopathological evidence of white matter degeneration, neuronal loss and mild defects in cerebellar granule neuron migration. The defects in granule neuron migration appear to be non-cell autonomous since Snf5 was present in ectopic granule neurons. The human GFAP-Cre construct (Gfa2) used for these studies has been widely studied and shown to direct Cre-mediated recombination in astrocytes, oligodendroglia, ependymal cells and a subset of neurons (24,41,42). Initial analysis indicated there was some loss of oligodendrocytes and neuronal projections.
Whole exome sequencing of 32 human MRTs, including 20 CNS AT/RT revealed only one p53 mutation that was predicted not to be detrimental, although this was not demonstrated (43). However, previous mutational analysis identified p53 mutations in 3/19 CNS AT/RTs nucleophosmin (6). In addition, the majority of CNS AT/RT express p53 and it was proposed that p53 is inhibited in these tumors by binding to (44,45). Thus, there is support for the notion that deregulation of p53 plays a role in AT/RT. Furthermore, loss of p53 accelerates tumor formation in mice with heterozygous mutations in Snf5 (19,23) and it has been argued that reduced expression of p53 cooperates with loss of Snf5 to decrease the latency of tumors arising in sites observed infrequently in Snf5+/− mice, because Snf5 regulates a subset of p53 target genes (22,46). Thus, there is strong evidence of cooperation between these two tumor suppressor pathways. In the case of the GFAP-Cre GEM, we observed a very high degree of cooperativity, since no tumors were observed in the presence of p53 and 100% of Snf5F/Fp53L/LGFAP-Cre mice examined after 3 weeks of age harbored brain tumors with many hallmarks of AT/RT. Cooperativity extended to the neurological phenotype observed in Snf5F/FGFAP-Cre mice which was much more severe in Snf5F/Fp53L/LGFAP-Cre mice. We did not detect any tumors or enhancement of the neurological phenotype in Snf5F/Fp53+/LGFAP-Cre mice, therefore, the basis of the cooperativity we observed may be different than that proposed to explain acceleration of peripheral MRTs (46). In general, p53 loss during tumor initiation has been suggested to promote tumorigenesis by blocking apoptosis. However, there was increased cell death in the oligodendrocyte lineage, and in some neuronal populations, in Snf5F/Fp53L/LGFAP-Cre mice. While we do not yet know the cell of origin of AT/RT, these results imply that there are additional biological consequences of the interactions between loss of Snf5 and p53 that may be cell-type dependent.
The profound neurodegeneration phenotype observed in Snf5F/Fp53L/LGFAP-Cre mice was quite unexpected. The phenotype seems to be initiated by a dramatic loss of oligodendrocytes, as it is very similar to the phenotype described in mice lacking oligodendrocytes (33,34). At three weeks of age in Snf5F/Fp53L/LGFAP-Cre mice, there are many oligodendrocytes that are negative for Snf5 immunohistochemistry. Subsequently, these cells were lost leading to a progressive degeneration of nerve fibers and a host of ancillary phenotypes including seizure activity. Since some myelin positive cells can be detected initially, it seems that Snf5 may be more critical for the maintenance rather than the generation of oligodendrocytes. Children with AT/RT suffer a broad range of neurological deficits many of which have been ascribed to the invasive nature of the tumor, or the side effects of therapy (47). However, it is also possible that germline, or somatic, alterations in Smarcb1, such as those seen in patients with Coffin-Siris syndrome, could also contribute to the neurodevelopmental phenotypes seen in these patients (48). Our studies imply that some of these effects may be a consequence of loss of oligodendrocytes.
The earliest tumors observed in mutant mice appeared to arise in the most superficial aspects of the cerebellar folia. The tumors are invasive and highly aggressive destroying surrounding cerebellar tissues as they grow. The co-morbidity arising from the loss of oligodendrocytes begs the question of whether AT/RT are derived from progenitor cells in this lineage. Interestingly, germline or somatic loss of Snf5 is prevalent in schwannoma (15,49). Furthermore, nPDGFA/NG2 expressing glial progenitor/stem cells can produce both oligodendrocytes and Schwann cells during repair of chemical CNS demyelination (50). Thus, it is not out of the question that rhabdoid tumors are derived from cells in these allied lineages of the central and peripheral nervous systems.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Grant Support
This work was supported by grants from the Children’s Brain Tumor Foundation, the Brain Tumor Society and the National Institutes of Health (NIH) (CA 096832). E. Marsh was supported by grants from the NINDS/NIH (R01 NS082761-01, 1-KO2-NS065975).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
5 supplemental figures and 1 supplemental table
Supplementary materials and methods
Highlighted manuscript (for review only)
Authors’ Contributions
Conception and design: J. M. Y. Ng, T. Curran
Development of methodology: J. M. Y. Ng, T. Curran, D. Martinez, E. D. Marsh, Z. Zhang, E. Rappaport
Acquisition of data (provided animals, acquired and managed patients provided facilities, etc.): J. M. Y. Ng, T. Curran.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. M. Y. Ng, T. Curran, D. Martinez, E. D. Marsh, M. Santi, Z. Zhang, E. Rappaport
Writing, review, and or revision of the manuscript: J. M. Y. Ng, T. Curran, D. Martinez, E. D. Marsh, M. Santi, Z. Zhang, E. Rappaport
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. M. Y. Ng, D. Martinez, E. D. Marsh, Z. Zhang, E. Rappaport
Study supervision: J. M. Y. Ng, T. Curran
References
- 1.Sevenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D, et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet. 1999;8(13):2359–2368. doi: 10.1093/hmg/8.13.2359. [DOI] [PubMed] [Google Scholar]
- 2.Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 3.Judkins AR, Burger PC, Hamilton RL, Kleinschmidt-DeMasters B, Perry A, Pomeroy SL, et al. INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. Journal of neuropathology and experimental neurology. 2005;64(5):391–397. doi: 10.1093/jnen/64.5.391. [DOI] [PubMed] [Google Scholar]
- 4.Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus. 2006;20(1):E11. doi: 10.3171/foc.2006.20.1.12. [DOI] [PubMed] [Google Scholar]
- 5.Sansam CG, Roberts CW. Epigenetics and cancer: altered chromatin remodeling via Snf5 loss leads to aberrant cell cycle regulation. Cell cycle. 2006;5(6):621–624. doi: 10.4161/cc.5.6.2579. [DOI] [PubMed] [Google Scholar]
- 6.Venneti S, Le P, Martinez D, Eaton KW, Shyam N, Jordan-Sciutto KL, et al. p16INK4A and p14ARF tumor suppressor pathways are deregulated in malignant rhabdoid tumors. Journal of neuropathology and experimental neurology. 2011;70(7):596–609. doi: 10.1097/NEN.0b013e31822146ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21(34):5193–5203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- 8.Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21(42):6403–6412. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- 9.Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D, et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol. 2002;22(16):5975–5988. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vries RG, Bezrookove V, Zuijderduijn LM, Kia SK, Houweling A, Oruetxebarria I, et al. Cancer-associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes Dev. 2005;19(6):665–670. doi: 10.1101/gad.335805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–1433. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morozov A, Lee SJ, Zhang ZK, Cimica V, Zagzag D, Kalpana GV. INI1 induces interferon signaling and spindle checkpoint in rhabdoid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(16):4721–4730. doi: 10.1158/1078-0432.CCR-07-0054. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Sansam CG, Thom CS, Metzger D, Evans JA, Nguyen PT, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69(20):8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyd C, Smith MJ, Kluwe L, Balogh A, Maccollin M, Plotkin SR. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet. 2008;74(4):358–366. doi: 10.1111/j.1399-0004.2008.01060.x. [DOI] [PubMed] [Google Scholar]
- 15.Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. American journal of human genetics. 2007;80(4):805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto H, Kohashi K, Tsuneyoshi M, Oda Y. Heterozygosity loss at 22q and lack of INI1 gene mutation in gastrointestinal stromal tumor. Pathobiology. 2011;78(3):132–139. doi: 10.1159/000323564. [DOI] [PubMed] [Google Scholar]
- 17.Cheng JX, Tretiakova M, Gong C, Mandal S, Krausz T, Taxy JB. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21(6):647–652. doi: 10.1038/modpathol.2008.44. [DOI] [PubMed] [Google Scholar]
- 18.Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21(10):3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1(6):500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(25):13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer cell. 2002;2(5):415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- 22.DelBove J, Kuwahara Y, Mora-Blanco EL, Godfrey V, Funkhouser WK, Fletcher CD, et al. Inactivation of SNF5 cooperates with p53 loss to accelerate tumor formation in Snf5(+/−);p53(+/−) mice. Mol Carcinog. 2009;48(12):1139–1148. doi: 10.1002/mc.20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(49):17745–17750. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22(14):5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1994;14(3 Pt 1):1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 27.Wetmore C, Eberhart D, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Research. 2001;61(2):513–516. [PubMed] [Google Scholar]
- 28.Marsh ED, Peltzer B, Brown MW, 3rd, Wusthoff C, Storm PB, Jr, Litt B, et al. Interictal EEG spikes identify the region of electrographic seizure onset in some, but not all, pediatric epilepsy patients. Epilepsia. 2010;51(4):592–601. doi: 10.1111/j.1528-1167.2009.02306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain : a journal of neurology. 2009;132(Pt 6):1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16(20):2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP:12 V-Ha-Ras mouse astrocytoma model. The American journal of pathology. 2005;167(3):859–867. doi: 10.1016/S0002-9440(10)62057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and clinical neurophysiology. 1972;32(3):281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 33.Collin L, Usiello A, Erbs E, Mathis C, Borrelli E. Motor training compensates for cerebellar dysfunctions caused by oligodendrocyte ablation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(1):325–330. doi: 10.1073/pnas.0305994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathis C, Hindelang C, LeMeur M, Borrelli E. A transgenic mouse model for inducible and reversible dysmyelination. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20(20):7698–7705. doi: 10.1523/JNEUROSCI.20-20-07698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular subgroups of medulloblastoma. Expert Rev Neurother. 2012;12(7):871–884. doi: 10.1586/ern.12.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(12):1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 37.Pomeroy S, Tamayo P, Gaasenbeek M, Sturla L, Angelo M, McLaughlin M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 38.Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer cell. 2004;6(3):229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 39.Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(22):6305–6312. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, et al. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. 2002;62(1):323–328. [PubMed] [Google Scholar]
- 41.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132(24):5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of clinical investigation. 2012;122(8):2983–2988. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eberhart CG, Chaudhry A, Daniel RW, Khaki L, Shah KV, Gravitt PE. Increased p53 immunopositivity in anaplastic medulloblastoma and supratentorial PNET is not caused by JC virus. BMC cancer. 2005;5:19. doi: 10.1186/1471-2407-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dufour C, Beaugrand A, Le Deley MC, Bourdeaut F, Andre N, Leblond P, et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer. 2012;118(15):3812–3821. doi: 10.1002/cncr.26684. [DOI] [PubMed] [Google Scholar]
- 46.Kuwahara Y, Wei D, Durand J, Weissman BE. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Molecular cancer research : MCR. 2013;11(3):251–260. doi: 10.1158/1541-7786.MCR-12-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Squire SE, Chan MD, Marcus KJ. Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol. 2007;81(1):97–111. doi: 10.1007/s11060-006-9196-z. [DOI] [PubMed] [Google Scholar]
- 48.Kosho T, Miyake N, Carey JC. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet. 2014;166C(3):241–251. doi: 10.1002/ajmg.c.31415. [DOI] [PubMed] [Google Scholar]
- 49.Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatric blood & cancer. 2011;56(1):7–15. doi: 10.1002/pbc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R, Jamen F, et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell stem cell. 2010;6(6):578–590. doi: 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.