

Abstract
Purpose
Although the majority of patients with HPV+ oropharyngeal cancers have a favorable prognosis, there are some patients with tumors that are resistant to aggressive chemoradiotherapy with unusual patterns of locoregional and systemic recurrence. Therefore, more effective therapies are needed. In this study, we investigated the chemosensitizing efficacy of the selective Wee-1 kinase inhibitor, AZD-1775 in HPV+ HNSCC.
Experimental Design
Clonogenic survival assays and an orthotopic mouse model of HPV+ oral cancer were used to examine the in vitro and in vivo sensitivity of HPV+ HNSCC cell lines to AZD-1775 in combination with cisplatin respectively. Cell cycle analysis, DNA damage (γH2AX), homologous recombination (HR), and apoptosis were examined to dissect molecular mechanisms.
Results
We found that AZD-1775 displays single-agent activity and enhances the response of HPV+ HNSCC cells to cisplatin both in vitro and in vivo. The sensitivity of the HPV+ HNSCC cells to AZD-1775 alone or in combination with cisplatin was associated with G2 checkpoint abrogation, persistent DNA damage, and apoptosis induction. This finding of AZD-1775 increasing the sensitivity of HPV+ HNSCC cells to cisplatin through apoptosis was not seen previously in the HPV negative HNSCC cancer cells, and is accompanied by a decreased expression of the anti-apoptotic proteins, MCl-1and XIAP, which appear to be cleaved following AZD-1775 treatment.
Conclusion
AZD-1775 selectively sensitizes HPV+ HNSCC cells and orthotopic oral xenografts to cisplatin through apoptosis and support the clinical investigation of AZD-1775 in combination with cisplatin particularly in patients with advanced and recurrent metastatic HPV+ HNSCC tumors.
Keywords: Cisplatin, HPV+ cells, Wee-1 kinase, apoptosis, unscheduled mitotic entry
Introduction
HNSCC affects over 500,000 patients worldwide annually and roughly half this number of patients will die from the disease each year (1). Over the past several decades, there has been a rapid emergence of human papillomavirus-associated squamous cell carcinoma of the oropharynx (OPSCC), with a concomitant decline in smoking related OPSCC (1,2). These distinct types of OPSCC have different clinical presentation and response patterns to the standard treatment approaches including radiation and chemoradiotherapy, and this has led to different considerations in selecting the most appropriate treatment for HPV+ and HPV- patients (3-5). Multiple studies have shown that the overall and disease-free survival for patients with HPV+ associated OPSCC is significantly better than for HPV- OPSCC patients (5,6). However, a group of HPV+ OPSCC patients' tumors remain refractory to conventional treatments with radiation given with or without chemotherapy and unusual patterns of recurrence and metastasis are emerging, particularly in patients with T4 and/or N3 staged disease (7-10). Therefore, new approaches to treatment of patients with high risk HPV+OSCC are needed. Thus, in the present study we have focused on a strategy to improve treatment of these patients by using a novel molecularly targeted agent to chemosensitize HPV+ head and neck cancers.
Wee-1 is a serine/threonine kinase that has been linked to DNA damage induced G2-M arrest, owing to its ability to inactivate the cyclin dependent kinase 1 (CDK1), also known as CDC2, through phosphorylation of the Tyr15 residue (12). Therefore, inhibition of the Wee-1 kinase activity can override a G2 cell cycle arrest and/or result in DNA damage by disrupting replication of the genome, leading to premature accumulation of cells with extensive DNA damage in mitosis and subsequent cell death through mitotic catastrophe (13, 14). Recent work with AZD-1775 (formerly known as MK-1775), a specific inhibitor of Wee-1, and siRNA-mediated depletion indicates that Wee-1 inhibition abrogates the G2 checkpoint and selectively sensitizes tumor cells defective of p53 to various DNA-damaging agents, such as gemcitabine, cisplatin, and inhibits tumor growth in in vivo models (15-18). In light of the findings from these preclinical studies, AZD-1775 has entered phase I and II clinical trials as a chemosensitizer in patients with advanced solid tumors and shows good tolerability with minimal collateral cytotoxicity. We have recently, demonstrated that MK-1775 can overcome cisplatin resistance in HPV negative HNSCC cells expressing high risk mutant p53 through mitotic arrest followed by senescence rather than apoptosis (19). Although, the status of p53 is predominantly wild type in HPV-positive HNSCC (20,21), the high-risk HPV E6 inactivates p53 through proteasomal degradation (22). Therefore, it is possible that the defective p53 status in HPV+ HNSCC cells makes these tumors more vulnerable to chemosensitization upon Wee-1 kinase inhibition. Recent in vitro data have shown that HPV+ HNSCC cells and ovarian cell lines transfected with E6 oncoprotein are sensitive to Wee-1 kinase inhibition (18,23,24). However, to our knowledge the single agent efficacy of AZD-1775 or in combination with cisplatin therapy has not been fully evaluated in vivo in high risk HPV+ HNSCC. In addition, it is not clear if AZD-1775 enhances the sensitivity of HPV+ HNSCC cells to cisplatin by mechanism(s) similar to that occurring in HPV negative HNSCC cells. In light of this information, we hypothesized that the Wee-1 kinase inhibitor, AZD-1775 will enhance the sensitivity of cisplatin both in vitro and in vivo in preclinical models of HPV+ oral cancer.
Our data show that AZD-1775 displays single-agent activity and significantly enhances the response of HPV+ HNSCC cells to cisplatin both in vitro and in vivo. Interestingly, unlike the HPV negative HNSCC cancer cells, AZD-1775 appears to increase sensitivity of HPV+ HNSCC cells to cisplatin therapy through the induction of apoptosis triggered by selective degradation (or cleavage) of the antiapoptotic, MCl-1and XIAP proteins.
Materials and Methods
Tissue culture, reagents and generation of stable cell Lines
The HPV16+ HNSCC cell lines used in this study are UMSCC47 from Dr. Thomas Carey (University of Michigan, Michigan, MI) in August 2008, HMS-001 from Dr. James Rocco (Massachusetts General Hospital, Boston, MA) in March 2011, and HB96 (transformed with HPV16 E6/E7) from Dr. Zhi-Yuan Zhang (Ninth People's Hospital School of Medicine, China, Shanghai) in November 2013. The HPV-HNSCC cell lines, HN30 (wtp53) and HN31 (mutp53) were obtained in December 2008 from the laboratory of Dr. John Ensley (Wayne State University, Detroit, MI). The cell lines (HN30, HN31, UMSCC47 and HB96) were maintained in Dulbecco's modified Eagle's medium (DMEM) and the HMS-001 in DMEM/F12 supplemented with 10% FBS, L-glutamine, sodium pyruvate, nonessential amino acids, and a vitamin solution, and incubated at 37°C in 5% CO2 and 95% Air. HPV+ HNSCC cell lines stably expressing short hairpin RNA specific for p53 (shp53) were generated according to protocol described previously (25). HPV+ HNSCC cell lines stably expressing MCl-1 were established using a pBabe-Flag h-MCl-1 vector (Addgene) as described previously (26).The identity of all cell lines was authenticated using short tandem repeat testing within 6 months of cell use. The Wee-1 inhibitor, MK-1775 was initially provided by the Merck Corporation through a collaborative agreement arrange by NCI-CTEP. The drug is currently licensed by AstraZeneca and known as AZD-1775, and its chemical structure has been described previously (15). For in vitro studies, AZD-1775 was prepared as 10 mmol/L stock solution in DMSO and stored at -20°C and diluted in culture medium (0.25 μmol/L) immediately before use. The apoptotic agent, staurosporine was purchased from Sigma (St Louis, MO) and used at 1 μmol/L final concentration. The pan-caspase inhibitor, Z-VAD-FMK was obtained from R&D System, prepared as 20 mmol/L stock in DMSO and used at 50 μmol/L final concentration.
Clonogenic survival assay
The HPV+HNSCC cells were seeded in 6-well plates at predetermined densities, concurrently exposed to different fixed-ratio combinations of cisplatin (dose range, 0.01-2 μmol/L) and AZD-1775 (dose range, 0.01-1μmol/L) for 24 hours and the clonogenic cell survival was determined as previously described (19).
Analysis of combined drug effects
Drug synergy was determined by the combination-index and isobologram analyses, which were generated according to the median-effect method of Chou and Talalay (27) using the CalcuSyn software (Biosoft, Ferguson, MO).The combination-index (CI) is a quantitative representation of the degree of drug interaction. For details, see Supplementary Materials and Methods section.
Antibodies
Antibodies used for Western blotting were phospho-γH2AX (Ser139; #2577), phospho-CDC2-Tyr15 (#9111), CDC2 (#9112), cyclin B1 (#4138), PARP-1 (#9542), MCl-1 (#4572), XIAP (#2042), Rad51 (#8875) and caspase-3 (#9661); all from Cell Signaling Technology; β-actin (#A5316; Sigma-Aldrich); p53(DO-1) (#sc-126) and MCl-1 (#sc-819) are from Santa Cruz; p21WAFI (Ab-1) (#OP64; Calbiochem); XIAP (#610716; BD Bioscience).
Western blot analysis
Cells grown on 10-cm plates were treated with clinically relevant doses of cisplatin (1.5 μmol/L), AZD-1775 (0.25 μmol/L) either alone or in combination for 16, 24 or 48 hours. For caspase cleavage detection, cells were pretreated with Z-VAD-FMK for 3 hours before addition of cisplatin and AZD-1775. Whole cell extracts were prepared and Western blot analysis was conducted with indicated antibodies as described previously (19,25).
Cell cycle analysis and apoptosis detection
Cells were seeded in 60-mm dishes, treated the next day with 1.5 μmol/L cisplatin, 0.25 μmol/L AZD-1775 either alone or in combination and then harvested at 24, 48, or 72 hours. The cell cycle analysis and apoptosis detection were performed as previously described (19).
Orthotopic mouse model of High risk HPV+ oral tongue cancer and tumor growth delay
All animal experimentation was approved by the Institutional Animal Care and use Committee (IACUC) of the University of Texas MD Anderson Cancer Center. Our orthotopic nude mouse tongue model has been previously described in the literature (28). The HPV- HNSCC (HN31) and HPV+ HNSCC (UMSCC47, HMS-001) cell lines were injected into the tongues of the male athymic nude mice and 8-10 days after injection mice were randomized into different groups. Treatment protocol and tumor growth delay measurement were conducted as previously described (19), and a detailed description of the technique is included in the Supplementary Materials and Methods.
In vivo TUNEL assay
Apoptosis was assessed in mice tissue sections with terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay with DeadEnd™ Fluorometric TUNEL System (Promega) according to the manufacturer's protocol with some modifications and a detailed description is included in the Supplementary Materials and Methods.
Immunohistochemistry
Sections were prepared from formalin-fixed paraffin embedded mice tumor tissues and subjected to immunohistochemical staining with indicated antibodies according to the protocol as described in Supplementary Materials and Methods.
Statistical analysis
The Student t and a 1-Way ANOVA tests were carried out to analyze in vitro data. For mouse studies, a 2-Way ANOVA test was used to compare tumor volumes between control and treatment groups. For immunohistocemical analyses, a chi-square test was used to compare immunostaining between control and treatment groups. All data were expressed as mean ± standard error and P values <0.05 were considered significant.
Results
AZD-1775 displays single agent activity and synergizes with cisplatin to inhibit in vitro growth of HPV+ HNSCC cells
To explore sensitivity of HPV+ HNSCC cells to cisplatin and AZD-1775 as single agents, we performed dose-response curves with each drug alone in HPV+ HNSCC cell lines (UMSCC47, HB96, HMS-001) using standard clonogenic survival assays. Compared to the relative cisplatin resistance that we previously reported in HPV negative HNSCC cells (19) (e.g., average IC50≥ 0.77 μmol/L, there was a clear trend towards increased cisplatin sensitivity in HPV+ cells with IC50 values ranging between 0.3-0.5 μmol/L. Similarly, HPV+ cells were more sensitive to AZD-1775 as a single agent (e.g., IC50 values, 0.09-0.24 μmol/L) (Fig. 1A and B) compared to IC50 values we previously reported for HPV- HNSCC cell lines which ranged from 0.25-0.375 μmol/L (19). Representative images of clonogenic survival assays following single agent AZD-1775 are shown in Fig. 1C., demonstrating the relative sensitivity of HPV+ HNSCC cells treated with various doses of AZD-1775.
Figure 1. AZD-1775 displays single agent activity and synergizes with cisplatin to inhibit in vitro growth of high risk HPV+ HNSCC cells.
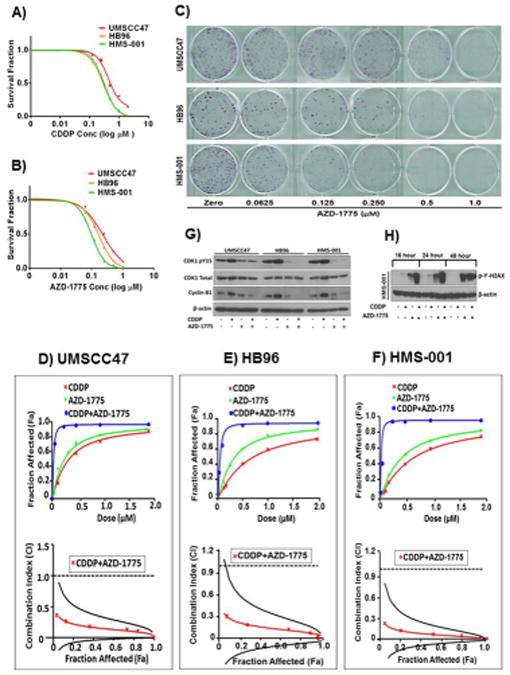
A and B, clonogenic survival curves for HPV+ HNSCC cell lines (UMSCC47, HB96, HMS-001), treated with a range of cisplatin (0.01-2 μmol/L) and/or AZD-1775 (0.01-1μmol/L) concentrations for 48 hours to determine the drugs IC50. Surviving colonies were allowed to form and counted after imaging. All treatments were performed in triplicate and each experiment was repeated at least three times. Representative images of clonogenic survival assays for Figure 1B are shown in Fig. 1C. Panel D, E and F, assessment of the degree of synergy between cisplatin and AZD-1775 in HPV+ HNSCC cell lines UMSCC47, HB96, and HMS-001, respectively using the Chou and Talalay method. Drug interactions are expressed as fraction affected (Fa) curves and combination index (CI) plots. The CI values were generated over a range of fraction affected (Fa) levels from growth inhibition percentages. Synergy was clear at relevant Fa values that are greater than 50%. A cut-off point of <0.75 was used to more rigorously define synergistic effect. Panel G and H, Western blots for HPV+ HNSCC cells treated with 1.5 μmol/L cisplatin (CDDP) alone or in combination with 0.25 μmol/L of AZD-1775 as described in Materials and Methods and analyzed for level of phospho-CDK1, CyclinB1, and phospho- γ-H2AX expression respectively. The phosphorylation of the DNA damage marker, γ-H2AX at ser139 was determined at different time points following treatment with drugs.
We next investigated whether Wee-1 kinase inhibition was synergistic with cisplatin treatment in the HPV+ HNSCC cell lines, using the combination index (CI) and fraction affected (Fa) method of Chou and Talalay (27). Addition of AZD-1775 significantly enhanced the cytotoxic effect of cisplatin in these cells and the combination effect reveals strong synergism manifested by the shift of cisplatin response curves and the CI values < 1 (Fa 0.5, ± SD) of 0.14 ± 0.09, 0.15 ± 0.13, and 0.073 ± 0.07 (Fig. 1D-1F, top plots) for UMSCC47, HB96 and HMS-001 respectively. The CI plots (Fig. 1D-1F, bottom plots) in all HPV+ HNSCC cells show a clear strong synergistic effect at the more relevant FA values (≥50%). Additionally, conservative isobologram plots of effective dose ED50, ED75, and ED90 were generated and further confirmed synergism of the drug combination in all HPV+ HNSCC cells examined (Supplementary Fig. S1A-S1C). Because HMS-001 shows greater sensitivity to AZD-1775, it was used for further experiments. The data clearly demonstrate that AZD-1775 has single agent activity and synergizes with cisplatin to inhibit in vitro tumor growth in HPV+ HNSCC.
To confirm that AZD-1775 affects its downstream target, the HPV+ HNSCC cells were treated with clinically relevant doses of the drugs as indicated previously, and phosphorylation of CDK1 was examined by Western blot (Fig.1G). Following cisplatin alone, increased phosphorylation of CDK1 was apparent in all three HPV+ HNSCC cells. AZD-1775 alone or in combination with cisplatin caused substantial suppression of CDK1 phosphorylation accompanied by decreased protein levels of its protein partner, cyclin B1 (Fig.1G), indicating an effective engagement of downstream targets and inhibition of the Wee-1 activity. Furthermore, the combination of cisplatin and AZD-1775 significantly enhanced levels of the DNA damage marker, γH2AX (phospho-ser139) in HMS001 (Fig.1H) as well as the two other HPV+ cell lines (data not shown), indicating an increase and persistence of unrepaired DNA damage in all the HPV+ HNSCC cancer cells tested. The increase in the phosphorylated γH2AX level was evident as early as 8 hours (data not shown), peaked gradually and remained elevated at 48 hours (Fig.1H). Although AZD-1775 treatment alone triggered elevated γH2AX compared to cisplatin or untreated cells by western blotting, levels were highest with the combination. Also, evident was an increase in γH2AX foci intensity in the HPV+ HNSCC cells (Supplementary Fig. S2A and S2B) 4 hours following treatment with AZD-1775 alone or in combination with cisplatin.
On the basis of our finding that inhibition of Wee-1 results in persistent cisplatin-induced DNA damage, and previous studies demonstrating a role for Wee-1 in homologous recombination (HR) (29,30), we examined alteration in the expression level of the Homologous Recombination (HR) competent marker, Rad51 in HPV+ HNSCC cells following treatment with the drugs. Significant decrease in the Rad51 protein level was observed at 16 hour and declined by 48 post treatment with AZD-1775 alone or in combination with cisplatin compared to cisplatin alone or untreated control cells (Supplementary Fig. S2C). Correspondingly, AZD-1775 treatment led to a decrease in cisplatin-induced Rad51 focus formation in HPV+ HNSCC cells (Supplementary Fig. S2D and S2E). Taken together, these data suggest that Wee-1 inhibition sensitizes HPV+ HNSCC cells to cisplatin through inhibition of Rad51-mediated HR.
AZD-1775 abrogates cisplatin-induced G2 checkpoint arrest and induces caspase-dependent apoptosis in HPV+ HNSCC cells
We have previously shown that Inhibition of Wee-1 abrogates cisplatin-induced G2 checkpoint in HNSCC cells harboring high risk mutant p53 (19). To confirm this takes place in HPV+ HNSCC cells, we performed cell cycle analysis in unsynchronized cells (UMSCC47, HMS-001) treated as previously described. In response to cisplatin alone, UMSCC47 and HMS-001 HPV+ cells were arrested at the G2/M phase (64.45%, and 49.27% respectively) 48 hours post-treatment (Fig. 2A-B and Supplementary Fig. S3A-S3B). Addition of AZD-1775 abrogated the cisplatin-induced G2 checkpoint arrest as manifested by reduction in the fraction of cells in G2 phase from 49.27% to 11.05% within 48 hours in HMS-001 cells and from 60.71% to 31.48% within 72 hours in UMSCC47 cells (Fig. 2A-B, and Supplementary Fig. S3A-S3B). Abrogation of cisplatin-induced G2 checkpoint arrest and unscheduled mitosis were confirmed by the presence of increased phospho-H3 protein levels on Western blot (data not shown). Treatment with either AZD-1775 alone or AZD-1775 plus cisplatin led to an increase in the percentage of cells with sub-G1 DNA content at 48 and 72 hours, when compared to cisplatin alone for both UMSCC47 and HMS-001 cells (Fig. 2A-B and Supplementary Fig. S3A-S3B). By 72 hours the percent of UMSCC47 with sub-G1 DNA content following treatment with AZD-1775 alone or the combination was 44% and 38%, respectively, compared to just 10% in cisplatin treated cells. In HMS-001 cells, the highest percentage of sub-G1 DNA occurred after 72 hours combination treatment (71%), compared to AZD-1775 alone (54%) or cisplatin alone (36%). No polyploidy or micronuclei were observed in these cells upon the combination treatment, indicating an absence of mitotic catastrophe (data not shown). To investigate whether cells were dying through apoptosis, HPV+ (UMSCC47, HB96, HMS-001) and HPV- (HN30, HN31) HNSCC cells were treated with the drugs as previously indicted and cell lysates examined for PARP-1 protein cleavage 48 hours later. Consistent with our previous findings (19), no PARP-1 cleavage was found in the HPV- HNSCC cell lines regardless of the drug treatments (Fig. 2C). However, in both HPV+ cell lines, AZD-1775 alone induced PARP-1 cleavage that was even greater when combined with cisplatin (Fig. 2C). The presence of apoptosis in HPV+ HNSCC cells was further confirmed with the positive APO-BrdU tunnel staining (Fig. 2D and E). In UMSCC47 the percentage of apoptotic cells was greatly enhanced by the combination treatment compared to AZD-1775 or cisplatin alone (69% versus 23% and 9.4%). However, HMS-001 cells were exquisitely sensitive to single agent AZD-1775, which induced apoptosis in greater than 90% of the population. This was consistent with high levels of PARP-1 cleavage observed in HMS-001 treated with AZD-1775 alone. Increased caspase-3/7 activity was observed at 24 hours in all HPV+ HNSCC cells following all treatments but was significantly greater in the combination of cisplatin and AZD-1775 versus single agents (Supplementary Fig. S4A-S4B). No increase in caspase-3/7 activity was observed in the HPV negative HNSCC cell lines (Supplementary Fig. S4C-S4D). Absence of caspase-dependent apoptosis in HPV-negative HNSCC cells was confirmed with western blotting (Supplementary Fig. S4E).The increased caspase-dependent activity could be blocked by the pan caspase inhibitor Z-VAD-FMK, demonstrating the specificity of the assay and the efficacy of the inhibitor. Treatment with Z-VAD-FMK profoundly inhibited the PARP-1 cleavage observed in HMS-001 exposed to AZD-1775 alone or in combination with cisplatin, confirming that PARP-1 cleavage was a consequence of drug induced caspase activation (Supplementary Fig. S4F). Collectively, these data indicate that AZD-1775 shows single agent activity and enhances cisplatin sensitivity in HPV+ HNSCC through caspase-mediated apoptosis.
Figure 2. . AZD-1775 abrogates cisplatin-induced G2 checkpoint arrest and induces caspase-dependent apoptosis in HPV+ HNSCC cells.
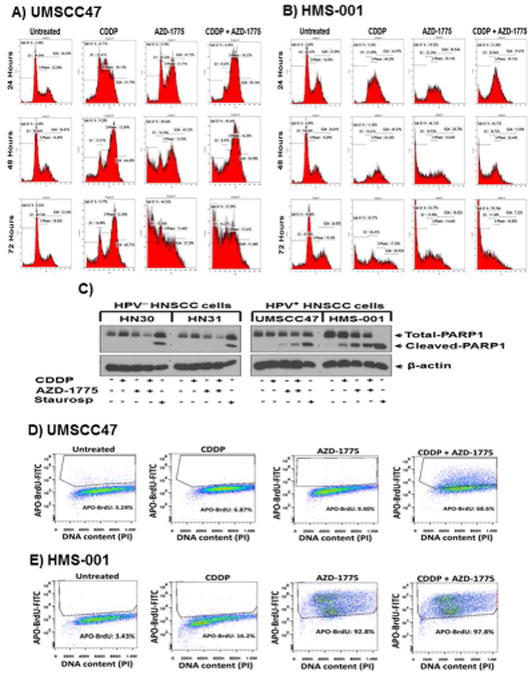
A and B, cell cycle analysis with propidium iodide (PI) staining using flow cytometry in unsynchronized cells (UMSCC47, HMS-001) treated as previously described. An abrogation of cisplatin-induced G2 checkpoint and significant increase in population of cells with sub-G1 DNA content were observed 48 hours following treatment with AZD-1775 in UMSCC47 and HMS-001 cells respectively. Data are representative of single experiment repeated at least three times. C, Western blots for HPV-negative (HN30, HN31) and HPV-positive (UMSCC47, HMS-001) HNSCC cells treated with cisplatin, AZD-1775 or in combination and analyzed for the presence of PARP-1 cleavage as indication of apoptosis. Lysates from staurosporine-treated (1 μmol/L) cells were used as positive controls for apoptosis. The β-actin serves as loading control. D and E, HPV+ HNSCC cells were subjected to FITC-APO-BrdU tunnel staining and cells stained positively for APO-BrdU were monitored by FACS analysis. Data are from a single representative experiment; two additional experiments yielded similar results.
AZD-1775 sensitizes HPV+ HNSCC cells to apoptosis in vitro accompanied by selective decrease in expression of MCl-1 and XIAP anti-apoptotic protein
Elevated expression of anti-apoptotic proteins has been reported in many tumors (31-33). In addition, several studies have shown that activation of the Wee-1 primary target, CDK1, plays a key role in mitotic arrest-induced apoptosis by decreasing stability and expression of several anti-apoptotic proteins through proteasomal degradation (34-36). Thus, we examined if the Wee-1 inhibition-induced apoptosis in HPV+ HNSCC cells (UMSCC47, HB96, HMS-001) exposed to cytotoxic treatment with cisplatin is associated with decreased levels of the anti-apoptotic proteins including Bcl-2, Bcl-xL,MCl-1, XIAP, c-IAP-1, and c-IAP-2. AZD-1775 and the combination with cisplatin selectively decreased the expression of apoptosis inhibitors MCl-1 and XIAP in HPV+ HNSCC but not in HPV negative HNSCC cells (Fig. 3A and 3B respectively). The decrease in protein expression was time dependent observed at 24 hours and remain suppressed 48 hours post-treatment. No changes in Bcl-2, Bcl-xL, c-IAP-1 or c-IAP-2 protein expression were found following all treatments (data not shown).
Figure 3. AZD-1775 sensitizes HPV+ HNSCC cells to apoptosis in vitro accompanied by selective decrease in expression of MCl-1 and XIAP anti-apoptotic protein.
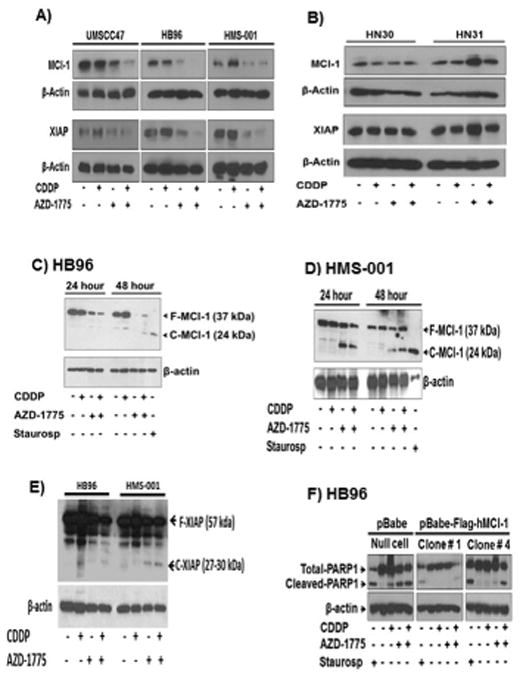
A, HPV+ HNSCC cells (UMSCC47, HB96, HMS-001) and B, HPV- HNSCC cells (HN30, HN31) grown on 10-cm plates were treated with clinically relevant doses of cisplatin (1.5 μmol/L), AZD-1775 (0.25 μmol/L) either alone expression levels of Mcl-1 and XIAP anti-apoptotic proteins were measured by Western blot analysis. C-E, detection of MCl-1 and XIAP protein cleavage in HPV+ HNSCC cells HB96 and HMS-001 respectively. F, HB96 cell clones stably expressing a retroviral pBabe-Flag-tagged human MCl-1 construct or pBabe vector alone were established, treated as indicated above, lysed and PARP1 cleavage was determined by Western blotting. Results demonstrate that forced expression of MCl-1 is sufficient for protection against AZD-1775 induced apoptosis. Data are from a single representative experiment; two additional experiments yielded similar results. F-MCl-1 and F-XIAP represent full-length protein; C-MCl-1 and C-XIAP indicate cleaved fragments.
MCl-1 and XIAP are highly regulated anti-apoptotic proteins and inactivated through cleavage by caspase activation either in spontaneous apoptosis or in drug-induced programmed cell death (37,38). To test for such possibility, the most sensitive HPV+ cells (HMS-001, HB96) were treated with cisplatin, AZD-1775 alone or in combination and MCl-1 and XIAP protein cleavage was examined by western blotting analysis. Appearance of MCl-1 and XIAP full-length and cleaved fragments was evident in these cells (Fig 3C-3E). Additionally, the decrease in both MCl-1 and XIAP protein induced by treatment with AZD-1775 or AZD-1775 plus cisplatin could be prevented in HMS-001 cells by adding Z-VAD-FMK (Supplementary Fig. 5SA), indicating that the reduction of these anti-apoptotic proteins was most likely due to their cleavage following caspase activation. Further supporting this, appearance of cleaved MCl-1 bands on western blot after exposure to AZD-1775 alone or AZD-1775 combined with cisplatin was also inhibited by the pan caspase inhibitor (Supplementary Fig. S5B). Expression of MCl-1 and XIAP can be regulated at both the transcriptional and/or translational levels (37,39,40). No changes in mRNA levels of MCl-1 and XIAP determined by qRT-PCR analysis were observed in HPV+ HNSCC cells treated with cisplatin, AZD-1775 or in combination (Supplementary Fig. S6A-S6B), suggesting that AZD-1775 enhances the ability of cisplatin to regulate anti-apoptotic proteins levels through posttranslational rather than transcriptional regulation.
To investigate the role of decreased MCl-1 in apoptosis induced by exposure to AZD-1775 or its combination with cisplatin, Flag tagged MCl-1 protein was overexpressed in HPV+ HNSCC (HB96) cells (Supplementary Fig. S6C). Overexpression of MCl-1 inhibited PARP-1 cleavage induced by either AZD-1775 or the combination with cisplatin (Fig. 3F), demonstrating that downregulation of the MCl-1 has a critical role in AZD-1775-induced apoptosis in HPV+ HNSCC tumors treated with cisplatin.
Knockdown of TP53 has no effect on the sensitivity of HPV+ HNSCC cells to cisplatin given alone or in combination with AZD-1775
A recent study reported that low levels of normally functioning wild type p53 remain in HPV+ HNSCC cells despite downregulation by the viral protein E6, and that this p53 can be activated by radiation therapy, leading to cell death through apoptosis (41).Therefore, we examined if cisplatin alone or in combination with AZD-1775 sensitizes HPV+ HNSCC cells by reactivating p53 and its immediate primary target gene p21, leading to cell death. While treatment with cisplatin alone increased p53 and p21 protein levels, addition of AZD-1775 did not further augment their protein levels in any of the HPV+ HNSCC cells tested (Fig 4A). Use of a TP53-specific shRNA, but not control shRNA, led to significant reduction in p53 expression levels in UMSCC47 and undetectable in HB96 cell lines (Fig. 4B). Therefore, the HB96 HPV+ HNSCC cell line was subsequently used for examining the role of wild type p53 on sensitivity to cisplatin in these cells. Using clonogenic survival assays, treatment of HB96 cells with cisplatin alone resulted in survival fractions that were similar between the control shRNA (IC50; 0.34 μmol/L) and shp53RNA (IC50; 0.31 μmol/L) (Fig. 4C). HPV+ HNSCC cells expressing shp53RNA showed survival fractions that were not different from those observed with control shRNA upon treatment with cisplatin and/or in combination with AZD-1775 (Fig. 4D). Finally, we examined the effect of TP53-knockdown on apoptosis by detection of PARP-1 cleavage on Western blot analysis. No difference in PARP-1 cleavage was observed in control shRNA or shp53RNA HB96 cells following cisplatin alone or in combination with AZD-1775 (Fig. 4E). These results suggest that the sensitization of HPV+ HNSCC Cells to cisplatin alone or in combination with AZD-1775 is not mediated through the p53 reactivation.
Figure 4. Knockdown of TP53 has no effect on sensitivity of HPV+ HNSCC cells to cisplatin alone or in combination with AZD-1775.
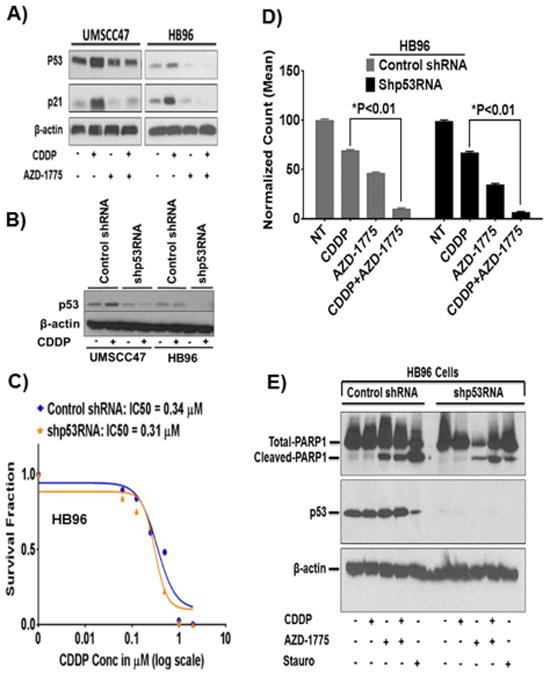
A, HPV+ HNSCC cells were treated with cisplatin and AZD-1775 as indicated previously and induction of p53 and p21 proteins were measured by Western blotting. B, stable transfection of HPV+ HNSCC cells with TP53-specific shRNA constructs produced different levels of p53 knockdown. C, clonogenic survival curves of control shRNA or shp53RNA HB96 cells treated with cisplatin alone. D, HB96 cells expressing shp53RNA showed survival fractions that were not different from those observed with control shRNA upon treatment with cisplatin and/or in combination with AZD-1775. E, HB96 cells with TP53 knockdown were treated as indicated and induction of apoptosis was examined by PARP1 cleavage on Western blot. Surviving colonies at each drug concentration were normalized to the untreated controls and plotted in the graphs. All drug treatments were carried out in triplicate and each experiment was repeated at least three times. Statistical significance (P<0.01) is indicated vs CDDP and CDDP+AZD-1775.
AZD-1775 displays single agent anti-tumor activity and synergizes with cisplatin in an in vivo orthotopic mouse model of HPV+ oral/pharyngeal cancer
To determine whether AZD-1775 can sensitize HPV+ HNSCC cells to cisplatin in vivo, UMSCC47 and HMS-001HPV+ cells were injected into the tongues of nude mice as previously described (28). Mice were treated with the drugs as described in Materials and Methods, and effects on tumor growth were examined. Cisplatin treated mice showed modest suppressive effects on tumor growth compared to untreated control groups (Fig. 5A and B). AZD-1775 alone showed significant growth inhibition compared to untreated mice (Fig. 5A and B, P < 0.001, P<0.0001 for UMSCC47 and HMS-001 respectively) but the difference from cisplatin treated mice was only significant in HMS-001 but not UMSCC47 bearing tumors (Fig. 5A and B, P<0.05). The combination of cisplatin and AZD-1775 significantly inhibited growth more than cisplatin treatment alone or control in tumors of mice bearing UMSCC47 and HMS-001HPV+ cells (Fig. 5A and B). Moreover, combination treatment showed modest effect on tumor growth inhibition over AZD-1775 alone in mice tumors carrying UMSCC47 cells (Fig. 5A, P = 0.07), but this effect was significant in mice bearing tumors with HMS-001 cells (Fig. 5B, P<0.0001). Additionally, AZD-1775 monotherapy produced greater tumor growth inhibition in mice injected subcutaneously with HOSC19 HPV-positive patient-derived tumor xenografts (PDX) (Supplementary Fig. S7A). To confirm that the enhancement of cisplatin antitumor efficacy by AZD-1775 was associated with engagement of downstream targets in vivo, phospho-CDK1 was evaluated in tissue sections obtained from tongue tumor xenografts. Consistent with earlier in vitro studies, AZD-1775 or its combination with cisplatin significantly decreased in vivo phospho-CDK1 immunostaining levels in orthotopic tongue tumors compared to cisplatin or untreated tumors (Fig. 5C-5F). A decrease in phospho-CDK1 immunostaining was also observed in PDX tumors (HOSC19) (Supplementary Fig. S7B-S7C). In addition, PCNA, a marker of proliferation, was significantly reduced in the UMSCC47 orthotopic tongue tumors from mice treated with either AZD-1775 or its combination with cisplatin compared to cisplatin alone or untreated control tumors (Supplementary Fig. S8A and S8B, Chi-square, P < 0.0001). Immunostaining with the p16 antibody confirmed that HPV+ HNSCC cells maintained HPV-positivity in vivo (Supplementary Fig S8C). Collectively, the results suggest that tumor growth inhibition is associated with decreased cellular proliferation in vivo in mice tongues bearing HPV+ HNSCC tumors treated with either AZD-1775 alone or in combination with cisplatin.
Figure 5. AZD-1775 displays single agent anti-tumor activity and synergizes with cisplatin in an in vivo orthotopic mouse model of HPV+ oral/pharyngeal cancer.
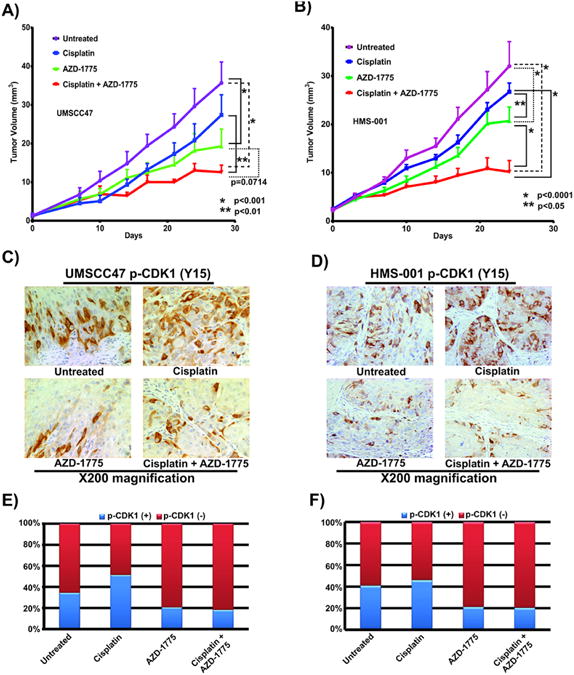
A, B, xenograft tumors made by orthotopic injection of the HPV+ HNSCC cells (UMSCC47, HMS-001) into the tongues of nude mice. The animals were treated with control, cisplatin, AZD-1775 or the combination of the two drugs once the tumors reached 5 mm in diameter according to protocol outlined in “Materials and Methods”. Tumor growth was followed for four weeks and tongue tumor size was measured with microcalipers and illustrated as tumor volume curves. C, D, evaluation of phospho-CDK1 expression by immunohistochemistry in tissue sections obtained from tongue xenografts bearing tumors with HPV+ HNSCC cells (UMSCC47, HMS-001) treated with AZD-1775 and cisplatin alone or in combination. E, F, quantification of phospho-CDK1 immunohistochemical images (percent positive and negative phospho-CDK1 tumor cells). To compare tumor growth, statistical significance (2-Way ANOVA, P < 0.001, and P < 0.01) is indicated vs control and AZD-1775 groups; CDDP and CDDP+AZD-1775 groups respectively. Statistical significance (Chi-square, P < 0.0001) is used to compare differences in Phospho-CDK1 immunostaining between AZD-1775, CDDP +AZD-1775 vs CDDP and untreated control groups. Control vehicles received only 0.5% methylcellulose solution given by oral gavage. The error bars are the standard error of the mean (SEM) tumor diameter for the group on that day and there were 8 to 9 mice in each group. Images were taken at ×200 magnification.
AZD-1775 alone or in combination with cisplatin induces apoptosis in vivo in mice bearing high risk HPV+ but not HPV- HNSCC oral/pharyngeal tumor xenografts
The occurrence of apoptosis in tongue tumor xenografts bearing HPV+ HNSCC (UMSCC47, HMS-001) cells was examined using the TUNEL assay. TUNEL-positive apoptotic cells were increased in tissue sections obtained from HPV+ tumor xenografts of mice treated with AZD-1775 alone or in combination with cisplatin compared to untreated or cisplatin treated mice (Fig. 6A and B), indicative of apoptosis and consistent with our in vitro results. Quantification of TUNEL-positive apoptotic cells shown in Fig 6A and B was presented as apoptotic index (Fig 6C and D). Furthermore, MCl-1 and XIAP expression in tumor sections of UMSCC47 oral xenograft mice significantly decreased upon therapy with combination of cisplatin and AZD-1775 or AZD-1775 alone, compared cisplatin treated and control groups (Fig. 6E and F). TUNEL positivity was also observed in the PDX tumors (HOSC19) treated with AZD-1775 (Supplementary Fig. S7D and S7E). Neither tunnel positivity nor reduction in expression of MCl-1 and XIAP was detected in mice tumors bearing HPV- HNSCC (mutp53 HN31) cells following cisplatin and AZD-1775 or AZD-1775 alone treatment, indicating minimal induction of apoptosis in these tumors (Supplementary Fig. S9A-S9C). These results suggest that AZD-1775 synergistic interaction with cisplatin therapy in vivo in HPV+ oral cancer results from apoptotic death associated with the downregulation of MCl-1 and XIAP anti-apoptotic proteins.
Figure 6. AZD-1775 alone or in combination with cisplatin induces apoptosis in vivo in mice bearing high risk HPV+ HNSCC oral/pharyngeal tumor xenografts.
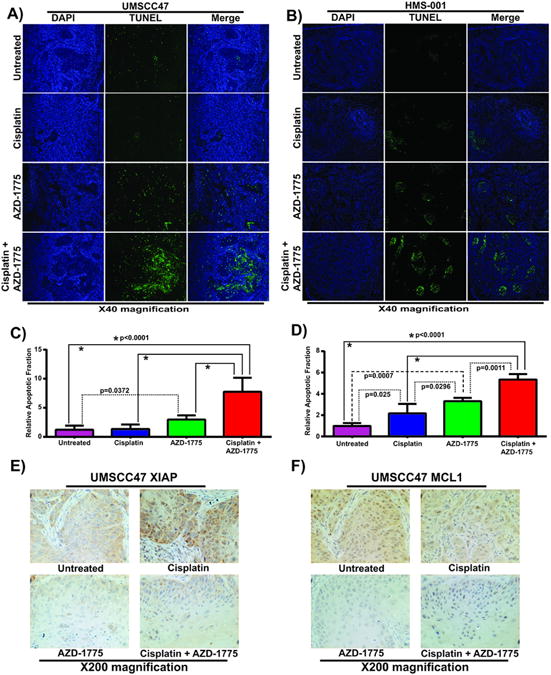
A, B, examination of the TUNEL-positive apoptotic cells (green) in tissue sections obtained from tongue xenografts bearing tumors with HPV+ HNSCC cells (UMSCC47, HMS-001) treated with AZD-1775 and cisplatin alone or in combination. The tissue samples were counterstained with DAPI (blue) and images were captured with immunofluorescence microscope at ×40 magnification. C, D, relative apoptotic fraction normalized to untreated control group (apoptotic index). E, F, immunohistochemical analyses of MCl-1 and XIAP protein expression performed in tumor sections of UMSCC47 oral xenografts mice treated as indicated above. Immunofluorescence images were taken at ×200 magnification.
Discussion
This is the first study to investigate the chemo-sensitizing effects of a highly selective inhibitor of the Wee-1 kinase, AZD-1775, in high risk HPV+ HNSCC tumor cells treated with cisplatin. This drug combination was chosen because platinum based chemotherapy plays a key role in the management of patients with HPV+ HNSCC tumors. AZD-1775 shows single agent antitumor activity synergizes with cisplatin and induces apoptosis in vitro in all HPV+ HNSCC cell lines tested. Interestingly, cisplatin arrests HPV+ HNSCC cells at the G2 phase and that addition of AZD-1775 abrogates the G2 block and pushes the cells rapidly into apoptosis. Collectively, these data suggest that AZD-1775 may be used as single agent and in combination with cisplatin therapy in HPV+ HNSCC. No apoptotic death was found in the HPV- HNSCC cells with addition of AZD-1775 or in combination with cisplatin, consistent with our recently published data, (19) in which we showed that mitotic catastrophe associated with senescence, rather than apoptosis is the major mechanism of synergistic interaction between cisplatin and AZD-1775 in high risk p53 mutant HPV- HNSCC cells. Although we did not observe apoptosis associated with AZD-1775 single agent treatment in our HPV-negative p53 mutant HNSCC cell lines, Moser and colleagues (18) have recently shown that apoptosis can occur in vitro in HPV- cells treated with higher doses of MK-1775. Most likely, the difference between their studies and ours can be attributed to different concentrations of this agent used in our versus their experiments. We used a dose of 250 nmol/L which has been shown to achieve greater than 80% reduction in phosphorylation of the Wee-1 downstream target CDK1 (15, 19, 24, 42, 43); however, we have also observed apoptosis at much higher concentrations of the drug in p53 mutant HPV- HNSCC cells.
The combination of cisplatin and AZD-1775 significantly produced high level of unrepaired and persistent DNA damage in all the HPV+ HNSCC cancer cells tested. It is possible that such high level of unrepaired DNA damage may have triggered replication stress associated with extreme lethality and unscheduled premature mitosis leading to induction of apoptosis in HPV+ tumor cells. This possibility is supported by published data showing that Wee-1 inhibition by AZD-1775 caused replication stress resulting from deregulated CDK1 activity and increased origin firing in pancreatic cancer (30). To our knowledge, this is the first report to demonstrate that the induction of apoptosis by AZD-1775 in HPV+ HNSCC tumors is accompanied by significant decrease in MCl-1 and XIAP protein levels. Because MCl-1 and XIAP are known substrates of caspase-3 and -7 (37,38), it is likely that the observed decrease in MCl-1 and XIAP was due to caspase-mediated processing after cisplatin and AZD-1775 treatment in HPV+ HNSCC. It has been previously shown that cleavage of MCl-1 and XIAP interferes with their antiapoptotic function in Jurkat and Hela cell lines (44,45). Correspondingly, we have demonstrated that both MCl-1 and XIAP are cleaved in response to treatment with AZD-1775 alone or in combination with cisplatin in HPV+ HNSCC cells. Furthermore, the downregulation of MCl-1 and its cleavage by AZD-1775 alone or in combination with cisplatin were completely attenuated by caspase inhibition, suggesting that MCl-1 and XIAP are targets of inactivation and degradation by caspase-3/7 in these cells. Additionally, caspase inhibition prevented apoptosis in these cells. We are not certain whether the degradation of Mcl-1 and XIAP we observed is a consequence of apoptosis or the cause of cell death phenotype. However, in our study, it is significant that enforced expression of MCl-1 markedly diminished AZD-1775-mediated PARP1 cleavage and apoptosis in HPV+ HNSCC (HB96) cells, suggesting that downregulation of antiapoptotic proteins is sufficient to trigger apoptosis. The mechanism by which AZD-1775 triggers the caspase-mediated apoptosis in these cells is not clear. Evidence exists that, in response to DNA damage, activation of several procaspases is required before mitochondrial permeabilization and apoptosis can take place (46,47). In light of this evidence, it is possible that Wee-1 inhibition further augments cisplatin-mediated DNA damage response, followed by casapse activation, cleavage of MCl-1 and XIAP, leading to apoptotic cell death in HPV+HNSCC. This possibility is supported by our time course experiments which revealed that the DNA damage occurred at 8 hours and preceded caspase 3/7 activation observed at 24 hours post treatment. Alternatively, the primary downstream target of Wee-1, the CDK1 may directly induce caspase activation in HPV+ HNSCC cells.
Functionally, the E6 viral oncoprotein of HPV inactivates wild type p53 and inhibits apoptosis (48). Our data revealed that complete knockdown of TP53 by shRNA did not affect clonogenic cell survival and did not block apoptosis in HPV+ HNSCC treated with cisplatin or in combination with AZD-1775. This finding indicates that reactivation of p53 is not sufficient to sensitize HPV+ HNSCC to cisplatin treatment combined with Wee-1 kinase inhibition and are not in agreement with recent published data which showed that knockdown of p53 resulted in radiation resistance in HPV+ HNSCC (41). It is possible that reactivation of p53 has different roles depending on the type of genotoxic therapy. Potentially, HPV E6 has additional effects other than inactivation of p53 that augment sensitivity to cisplatin and AZD-1775 in HPV+ HNSCC. Probably, HPV+ cells have greater deficiency repairing the DNA damage upon cytotoxic treatment, predisposing them to cell death by apoptosis. Interestingly, both E6 and E7 are involved in activation and repression of DNA damage response pathways to support viral genome maintenance and amplification in the normal viral life cycle (49). A recent study has also shown that E6 and E7 o activate caspases-3-7, and -9 upon differentiation to induce viral genome amplification (50). Understanding of how these alterations contribute to DNA damage response to cisplatin and Wee-1 kinase inhibition will warrant future investigations.
As preclinical evaluation, we demonstrated that oral administration of AZD-1775 as single-agent inhibited in vivo growth of HPV+ HNSCC oral xenografts and also synergizes with cisplatin to induce tumor regression, confirming our in vitro data. Furthermore, in vivo tumor growth inhibition was associated with an induction of apoptosis and decreased level of MCl-1 and XIAP immunostaining in the combination of cisplatin and AZD-1775 treated HPV+ HNSCC tumor xenografts compared with the control groups consistent with the in vitro results. Neither TUNEL positivity nor reduced immunostaining of MCl-1 and XIAP were detected in the HPV- HNSCC tumor sections, confirming in vitro findings and suggesting different mechanism of response to cisplatin and AZD-1775 in HPV-negative head and neck cancers as reported in our previous publication (19).
In conclusion, unlike HPV- HNSCC cells, those derived from the HPV+ HNSCC show enhanced sensitivity to cisplatin with AZD-1775 treatment through apoptosis associated with reduced expression of MCl-1 and XIAP both in vitro and in vivo. It seems that inactivation of these anti-apoptotic proteins via posttranslational cleavage induced by AZD-1775 treatment plays an important role in modulating the response of the HPV+ HNSCC tumors to chemotherapy. Our study could have implications for the use of MCl-1 and XIAP expression as surrogate end biomarkers in future clinical trials combining cisplatin and Wee-1 kinase inhibition. Additionally, our findings further suggest that MCl-1 and XIAP could be potential therapeutic targets in HPV+ HNSCC tumors. Our preclinical data on Wee-1 kinase inhibition demonstrate the susceptibility of HPV+ HNSCC tumors to deregulation of the G2-M checkpoint and support initiation of clinical trials with AZD-1775 in combination with cisplatin particularly in patients with advanced and recurrent metastatic HPV+ HNSCC tumors (18).
Supplementary Material
Translational Relevance.
Although the majority of patients with HPV+ oropharyngeal cancers have positive clinical outcomes, some patients remain resistant to chemoradiotherapy and develop regional and/or distant recurrences. Therefore, more effective therapies are urgently needed. Combinations of targeted agents, which have the potential to be efficacious and less toxic than standard radiation and/or chemotherapy, are exciting areas of investigation. In this study, we found that the addition of the Wee-1 kinase inhibitor, AZD-1775, significantly potentiates cisplatin response and produces suppressive effects on HPV+ head and neck tumor growth, illustrating the therapeutic benefit of this combination of agents. Because Wee-1 inhibition with AZD-1775 is currently under clinical investigation with cisplatin in locally advanced head and neck cancers, this study represents the preclinical foundation for initiation of clinical trials, using combinations of agents that target the G2/M checkpoint such as AZD-1775 with chemotherapy for patients with advanced and recurrent metastatic HPV+ HNSCC tumors.
Acknowledgments
Funding: This work was supported by the National Institute of Health Specialized Program of Research Excellence Grant P50CA097007 (J.N. Myers), the National Institute of Health/NIDCR R01DE024601 (J.N. Myers), the Cancer Prevention and Research Institute of Texas (CPRIT) RP12025802 (J.N. Myers), the Stiefel Oropharyngeal Cancer Program, and the UTMDACC HPV+HNSCC Moonshot Program (A.A Osman and J.N Myers).
Abbreviations
- HPV+ HNSCC
human papillomavirus positive head and neck squamous cell carcinoma
- CDDP
cisplatin
- MCl-1
(myeloid cell leukemia-1 protein)
- XIAP
(X-linked inhibitor of apoptosis)
Footnotes
Conflicts of Interest: The authors have no potential conflicts of interest to disclose
References
- 1.Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, et al. Annual Report to the Nation on the Status of Cancer, 1975–2009, Featuring the Burden and Trends in Human Papillomavirus (HPV)–Associated Cancers and HPV Vaccination Coverage Levels. J Natl Cancer Inst. 2013;105:175–201. doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturvedi AK, Goedert JJ. Human papillomavirus genotypes among women with HIV: implications for research and prevention. Aids. 2006;20:2381–3. doi: 10.1097/01.aids.0000253366.94072.b4. [DOI] [PubMed] [Google Scholar]
- 3.Mellin H, Friesland S, Lewensohn R, Dalianis T, Munck-Wikland E. Human papillomavirus (HPV) DNA in tonsillar cancer: clinical correlates, risk of relapse, and survival. Int J Cancer. 2000;89:300–4. [PubMed] [Google Scholar]
- 4.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argiris A, Li Y, Forastiere A. Prognostic factors and long-term survivorship in patients with recurrent or metastatic carcinoma of the head and neck. Cancer. 2004;101:2222–9. doi: 10.1002/cncr.20640. [DOI] [PubMed] [Google Scholar]
- 7.McBride M, Busse PM, Clark JR, Wirth LJ, Ancukiewicz M, Chan AW. Long-term survival after distant metastasis in patients with oropharyngeal cancer. Oral Oncol. 2014;50:208–12. doi: 10.1016/j.oraloncology.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Ruzevick J, Olivi A, Westra WH. Metastatic squamous cell carcinoma to the brain: an unrecognized pattern of distant spread in patients with HPV-related head and neck cancer. J Neurooncol. 2013;112:449–54. doi: 10.1007/s11060-013-1075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Sullivan B, Huang SH, Siu LL, Waldron J, Zhao H, Perez-Ordonez B, et al. Deintensification candidate subgroups in human papillomavirus-related oropharyngeal cancer according to minimal risk of distant metastasis. J Clin Oncol. 2013;31:543–50. doi: 10.1200/JCO.2012.44.0164. [DOI] [PubMed] [Google Scholar]
- 10.Gillison ML, Zhang Q, Jordan R, Xiao W, Westra WH, Trotti A, Spencer S, Harris J, Chung CH, Ang KK. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J Clin Oncol. 2012;30:2102–11. doi: 10.1200/JCO.2011.38.4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Squire CJ, Dickson JM, Ivanovic I, Baker EN. Structure and Inhibition of the Human Cell Cycle Checkpoint Kinase, Wee1A Kinase: An Atypical Tyrosine Kinase with a Key Role in CDK1 Regulation. Structure. 2005;13:541–550. doi: 10.1016/j.str.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 12.Portugal J, Mansilla S, Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des. 2010;16(1):69–78. doi: 10.2174/138161210789941801. [DOI] [PubMed] [Google Scholar]
- 13.De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–7. doi: 10.1158/1078-0432.CCR-10-2537. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther. 2004;3:305–13. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 15.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 16.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–22. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 17.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK- 1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moser R, Xu Chang, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional Kinomics Identifies Candidate Therapeutic Targets in Head and Neck Cancer. Clin Cancer Res. 2014;(20):4274–4288. doi: 10.1158/1078-0432.CCR-13-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol Cancer Ther. 2015;14(2):608–19. doi: 10.1158/1535-7163.MCT-14-0735-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balz V, Scheckenbach K, Gotte K, Bockmuhl U, Petersen I, Bier H. Is the p53 inactivationfrequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2-11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res. 2003;63:1188–1191. [PubMed] [Google Scholar]
- 22.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 Mutant Cells by PD0166285, a Novel G2 Checkpoint Abrogator. Cancer Res. 2001;(61):8211–8217. [PubMed] [Google Scholar]
- 24.Carrassa L, Chilà R, Lupi M, Ricci F, Celenza C, Mazzoletti M, et al. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;(13):2507–17. doi: 10.4161/cc.20899. [DOI] [PubMed] [Google Scholar]
- 25.Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, et al. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117(13):2926–2938. doi: 10.1002/cncr.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18(1):290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 28.Myers JN, Holsinger FC, Jasser SA, Bekele BN, Fidler IJ. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin Cancer Res. 2002;8:293–8. [PubMed] [Google Scholar]
- 29.Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Silljé HH, de Vries EG, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–3008. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- 30.Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085–96. doi: 10.1158/1078-0432.CCR-14-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Cao Z, Yan H, Wood WC. Coexistence of High Levels of Apoptotic Signaling and Inhibitor of Apoptosis Proteins in Human Tumor Cells: Implication for Cancer Specific Therapy. Cancer Res. 2003;63:6815–6824. [PubMed] [Google Scholar]
- 32.Reed JC. Dysregulation of Apoptosis in Cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 33.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4(3):267–76. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 34.Chu R, Terrano DT, Chambers TC. Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochem Pharmacol. 2012;15:199–206. doi: 10.1016/j.bcp.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–56. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1–cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosato RR, Almenara JA, Kolla SS, Maggio SC, Coe S, Gimenez MS, et al. Mechanism and functional role of XIAP and Mcl-1 down-regulation in flavopiridol/vorinostat antileukemic interactions. Mol Cancer Ther. 2007;6(2):692–702. doi: 10.1158/1535-7163.MCT-06-0562. [DOI] [PubMed] [Google Scholar]
- 38.Johnson DE, Gastman BR, Wieckowski E, Wang G, Amoscato A, Delach SM, et al. Inhibitor of apoptosis protein hILP undergoes caspase-mediated cleavage during T lymphocyte apoptosis. Cancer Res. 2000;60:1818–23. [PubMed] [Google Scholar]
- 39.Iglesias-Serret D, Pique M, Gil J, Pons G, Lopez JM. Transcriptional and translational control of Mcl-1 during apoptosis. Arch Biochem Biophys. 2003;417(2):141–152. doi: 10.1016/s0003-9861(03)00345-x. [DOI] [PubMed] [Google Scholar]
- 40.Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG, et al. Akt Phosphorylation and Stabilization of X-linked Inhibitor of Apoptosis Protein (XIAP) J Biol Chem. 2004;279:5405–5412. doi: 10.1074/jbc.M312044200. [DOI] [PubMed] [Google Scholar]
- 41.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, et al. Enhanced Radiation Sensitivity in HPV-Positive Head and Neck Cancer. Cancer Res. 2013;73:4791–800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;(17):5638–48. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarcar B, Kahali S, Prabhu AH, Shumway SD, Xu Y, Demuth T, Chinnaiyan P. Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10(12):2405–14. doi: 10.1158/1535-7163.MCT-11-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrant M, Jacquel A, Marchetti S, Belhacène N, Colosetti P, Luciano F, et al. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene. 2004;23(47):7863–73. doi: 10.1038/sj.onc.1208069. [DOI] [PubMed] [Google Scholar]
- 45.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18(19):5242–51. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122:347–57. doi: 10.1007/s10549-009-0571-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;(6):1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 49.Moody CA, Laimins LA. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009;5(10):e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. PNAS. 2007;104(49):19541–19546. doi: 10.1073/pnas.0707947104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.