

Abstract
Identifying genetic alterations that prime a cancer cell to respond to a particular therapeutic agent can facilitate the development of precision cancer medicines. Cancer cell-line (CCL) profiling of small-molecule sensitivity has emerged as an unbiased method to assess the relationships between genetic or cellular features of CCLs and small-molecule response. Here, we developed annotated cluster multidimensional enrichment analysis to explore the associations between groups of small molecules and groups of CCLs in a new, quantitative sensitivity dataset. This analysis reveals insights into small-molecule mechanisms of action, and genomic features that associate with CCL response to small-molecule treatment. We are able to recapitulate known relationships between FDA-approved therapies and cancer dependencies and to uncover new relationships, including for KRAS-mutant cancers and neuroblastoma. To enable the cancer community to explore these data, and to generate novel hypotheses, we created an updated version of the Cancer Therapeutic Response Portal (CTRP v2).
INTRODUCTION
Patient response to cancer medicines is increasingly tied to genetic or lineage features of cancer cells (1). Identifying the small-molecule targets that exploit the context-specific vulnerabilities of cancer cells can be critical for the development of novel precision medicines. Furthermore, given the heterogeneous response of patients to cancer therapies and the frequent development of drug resistance, it is imperative to identify the molecular settings in which small molecules or combinations of small molecules lead to durable patient responses (2, 3).
Cancer cell lines (CCL) have long served as models for therapeutics discovery, and profiling the small-molecule sensitivity of CCL panels has emerged as a systematic method to relate the genetic or cellular features of CCLs to patterns of small-molecule response (4). The first high-throughput CCL sensitivity profiling effort, the NCI-60, evaluated the response of 59 CCLs to >105 small-molecule probes to identify instances of differential responses to small-molecule treatment among CCLs from various lineages (5). Unlike with cytotoxic chemotherapies, patients respond at much lower rates and in a more context-restricted manner to targeted therapies (4). In order to capture these clinically relevant, context-specific events in profiling experiments, larger panels of CCLs that faithfully represent the diversity of patient tumors are needed. Several recent studies have demonstrated the feasibility of profiling the small-molecule sensitivity of much greater numbers of CCLs that are genetically and cellularly characterized, with the goal of identifying genomic markers of CCL response (6–9).
The ability of CCL sensitivity profiling to inform development of new patient-matched therapies, particularly those targeting proteins other than oncogenes, remains to be proven. Furthermore, optimal methods to design, execute, and analyze such experiments are still being determined. For example, one report recently highlighted inconsistencies in drug-response measurements within and between two reported sensitivity profiling studies (10). Despite these open questions, many clinical genomic predictors of cancer-cell response to small-molecule treatment can be recapitulated in CCL models (6–9). Furthermore, recent efforts have demonstrated that CCLs derived from patients who developed resistance to targeted therapies can be used in vitro to suggest combinations for overcoming resistance (11). As such, we believe that the use of prudently selected small-molecule and CCL collections in well-controlled sensitivity profiling studies, coupled with effective analysis methods, has the potential to reveal context-specific dependencies of cancer cells.
With this goal in mind, we measured the viability of 860 genetically characterized CCLs individually against members of an “Informer Set” of 481 compounds, in 16 concentrations in duplicate. The Informer Set comprises FDA-approved drugs, clinical candidates, and small-molecule probes, resulting in the largest quantitative CCL sensitivity dataset available to date. The small molecules were carefully selected to target a diverse array of protein targets; the CCL panel encompasses 25 lineages, and comprehensive genetic characterization is publicly available (8). To support the discovery of predictors of small-molecule response, we developed a new annotated cluster multidimensional enrichment (ACME) analysis. Compared with previously described methods, which aim to identify response markers for each compound individually (6–9), the new method simultaneously integrates information from multiple compounds and CCLs. ACME analysis detects small molecules sharing a common target that perform similarly in the profiling experiment to robustly identify protein targets upon which CCLs are dependent. Concurrently, the common genetic and cellular information of CCLs that share patterns of response is found, thereby pinpointing the likely CCL vulnerabilities that result in small-molecule sensitivity from the large number of cataloged CCL genetic alterations (e.g., passenger mutations). Using ACME analysis, we identified mechanisms of action previously unreported for compounds in preclinical development, as well as candidate dependencies in the context of neuroblastoma and KRAS mutation. A new resource containing associations based on our method, and an exploratory visualization tool for browsing clusters, is publicly available in an updated version of the Cancer Therapeutics Response Portal (CTRP v2; ref. 9), enabling the cancer biology community to evaluate associations between the genetic and cellular features of CCLs and small-molecule sensitivity profiles.
RESULTS
Generation of a New Small-Molecule Sensitivity Resource
The small-molecule Informer Set contains 70 FDA-approved agents, 100 clinical candidates, and 311 small-molecule probes, enabling us to find clinically relevant genomic predictors of drug response and potential opportunities for drug development or repurposing (Supplementary Fig. S1A). To aid in the interpretation of the profiling experiments, we curated known protein targets, creating a set of high-quality annotations (Fig. 1; ref. 12). Around 115 of the probe compounds have no confirmed protein targets, including a number of novel mechanism-of-action (nMoA) compounds selected for their ability to effect unique gene-expression changes (Supplementary Fig. S1B; ref. 13). Overall, this Informer Set targets more than 250 distinct proteins, encompassing a broad range of cell circuitry relevant to CCL growth and survival. Moreover, we included small molecules with distinct structures targeting the same protein targets, or with varying selectivities for overlapping protein targets, to assess shared sensitivity patterns (Supplementary Fig. S1C and S1D).
Figure 1.
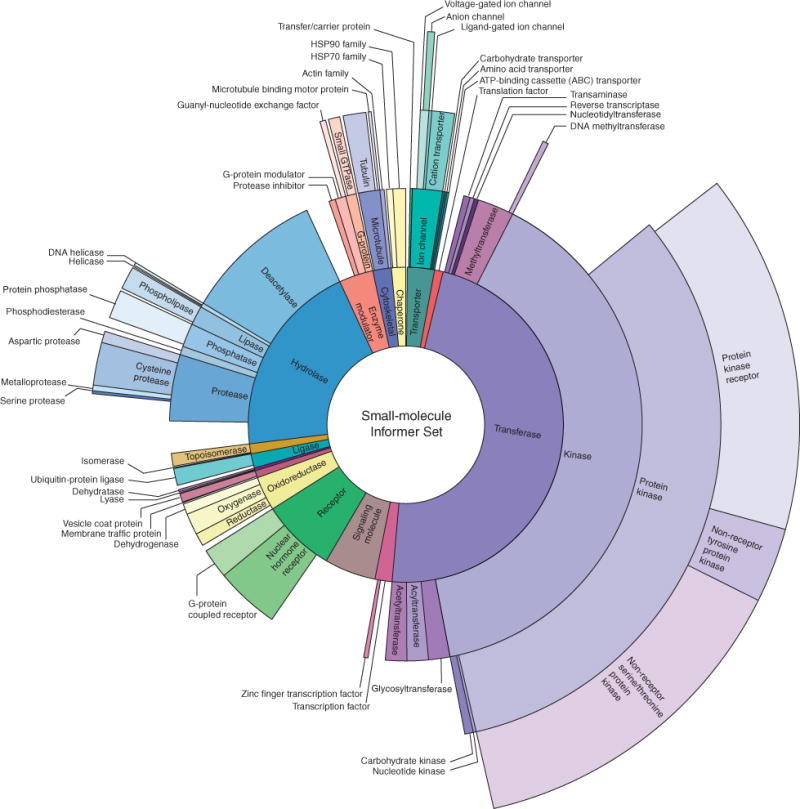
The Informer Set comprises 481 small-molecule compounds targeting a wide range of proteins involved in cell growth and survival. Sunburst visualization of validated protein targets of the small molecules in the Informer Set using protein family hierarchy from the Panther database (12). Approximately 115 compounds within the Informer Set do not have validated protein targets (nMoA). These compounds were included because they are known to affect a specific process or pathway, or to elicit gene-expression responses not seen in compounds having known mechanisms of action; however, they are not represented in this visualization.
Similarly, each CCL has been deeply characterized with respect to its genetic and cellular features as part of the Cancer Cell Line Encyclopedia (CCLE) effort (8). In the profiling experiment, CCLs were individually treated with each small molecule at 16 concentrations in duplicate. Percent-viability curves were fit and an area-under-concentration-response curve (AUC) was calculated for each compound–CCL pair, as a measure of CCL sensitivity to small-molecule treatment. Additional quality-control (QC) heuristics to ensure reliable data were also implemented (Supplementary Methods). In analyses relating CCL features to compound response, we included (i) classifications of primary site, histology, and histologic subtype (Supplementary Figs. S1E and S2), and (ii) mutations and indels in 1,651 genes, determined by targeted massively parallel sequencing (8). We further classified mutations as follows: (i) nonsynonymous mutations in the coding region of a given gene; (ii) mutations at a specific amino acid location in a gene; and (iii) specific amino acid changes at a given location (e.g., associations to BRAF, BRAFV600, BRAFV600E).
ACME Analysis
Initially, we performed unsupervised, hierarchical clustering of AUC data independently for each of compounds and 664 CCLs to examine the overall patterns of sensitivity present in the profiling experiment (Fig. 2A; refs. 14, 15). We restricted the analysis to adherent CCLs due to the greater sensitivity of suspension CCLs in this assay format (9), and further excluded 3 CCLs lacking genetic or cellular characterization (Supplementary Table S2). In order to account for the varying number of shared CCLs between small-molecule or CCL pairs, the Pearson correlation coefficient for each pair was normalized to the number of overlapping AUCs by Fisher z-transformation (Supplementary Fig. S3A; ref. 16). The resulting z-score was then reconverted to a distance using a monotonic double-sigmoid transformation that preserves the overall ranking of distances and emphasizes linkage variation in the relevant part of the dendrogram (Supplementary Fig. S3B).
Figure 2.
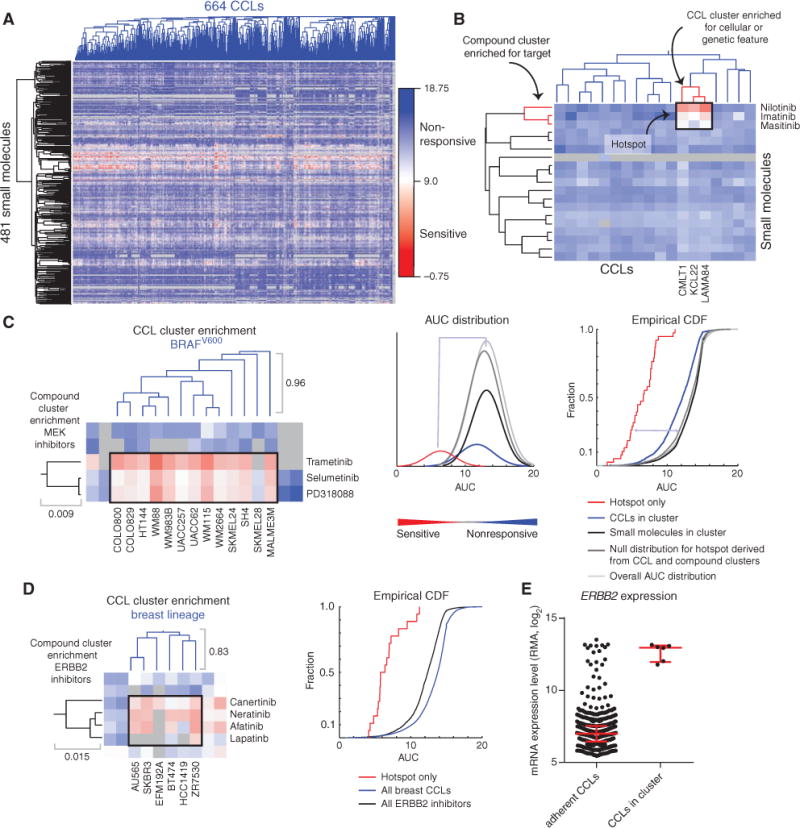
ACME analysis identifies hotspots that link genetic features of CCLs to patterns of small-molecule response. A, clustered AUC matrix for 481 small molecules and 664 adherent, genomically characterized CCLs, and the corresponding dendrograms (blue, CCLs; black, small molecules). Gray represents AUC values that were not measured or did not pass QC metrics. B, visualization of ACME analysis, which requires enrichment of small-molecule annotation, CCL annotation, and sensitivity of CCLs to the small molecules. Depicted are data for the association of sensitivity of chronic myeloid leukemia cell lines to treatment with inhibitors of ABL1. Both compound and CCL clusters (red) and the area of sensitivity in the AUC matrix (black) are depicted. C, hotspot and corresponding dendrograms for MEK inhibitors and BRAFV600 CCLs (left), including AUCs corresponding to the intersection of the two clusters (black box). The distributions of the AUCs for compound clusters, as well as the null distributions, can be visualized either with a fitted density curve (middle; bin number selected to scale with the number of CCLs), or empirical cumulative distribution function (CDF) plot (right). Each dendrogram segment is marked with the corresponding maximum height. The purity of both the compound cluster and the CCL cluster is 1. D, association of CCLs from the breast lineage with response to ERBB2 inhibitors. Visualization of the hotspot (black box) in the AUC matrix with the corresponding dendrograms (left), and the corresponding empirical CDF plot (right). The purity of both the compound cluster and the CCL cluster is 1. E, ERBB2 expression from CCLE for available adherent CCLs and the CCLs in the breast CCL-enriched cluster.
Visual inspection of the resulting clustered response matrix revealed horizontal and vertical “stripes” of high sensitivity (low AUCs; Fig. 2A). These stripes likely correspond to broadly (vs. narrowly) bioactive small molecules or “fragile,” frequently sensitive, CCLs (9). Importantly, we observed sensitivity “hotspots,” restricted areas of sensitivity in the clustered matrix, for small groups of compounds and CCLs. We then reasoned that certain sensitivity hotspots, those which occur at the intersection of a cluster of CCLs sharing genetic or cellular features and a cluster of small molecules with overlapping targets, could reveal vulnerabilities of CCLs that impart sensitivity to small molecules (Fig. 2B).
To identify sensitivity hotspots automatically, we developed ACME analysis, in which we applied three statistical tests to the clustered matrix. First, we located regions of sensitivity in the clustered matrix that correspond to the intersection of one compound cluster and one CCL cluster (Supplementary Fig. S3C). Second, to determine if the corresponding compound or CCL clusters were statistically enriched for a feature annotation (protein targets for compound clusters; mutation or lineage for CCL clusters), we probed each node of the row or column dendrogram for annotation enrichment. We also used purity (fraction of cluster members sharing the annotation) and confidence (fraction of total members sharing the annotation present in the cluster) to identify nodes with a coherent biologic signal (17). We retained sensitivity hotspots that simultaneously connected to an enriched cluster of compounds and an enriched cluster of CCLs. The resulting associations represent a new resource that can be mined to uncover relationships between small-molecule targets and genetic or lineage features of CCLs. Insights resulting from sensitivity hotspots revealed by ACME analysis are highlighted below, as well as potential use cases of the new resource now in CTRP.
Querying the Resource: Validating Known Vulnerabilities
First, we were able to recover clinically relevant relationships between the genetic vulnerabilities of cancer cells sharing a cellular feature and FDA-approved therapeutics. For example, a hotspot was observed for a cluster of proto-oncogene BRAFV600-mutant CCLs and MEK/MAP2K1 inhibitors, such as selumetinib (18), and BRAF inhibitors, such as PLX-4720 (Fig. 2C; Supplementary Fig. S4A). This particular group of CCLs is restricted to the skin lineage, a finding that is also observed in patients (19). We also found that ERBB2/HER2 inhibitors target a cluster of CCLs from the breast lineage (Fig. 2D). Investigation of copy-number variation and basal gene expression of this cluster reveals that these CCLs are ERBB2 amplified, resulting in ERBB2 overexpression, thereby providing a mechanistic link to the observed association (Fig. 2E; Supplementary Fig. S4B; ref. 20). ACME analysis associates neuroblastoma cell lines with sensitivity to Aurora kinase (AURK) inhibitors. Several studies have noted that models derived from childhood cancers with high MYCN levels respond to AURKB inhibitors (Supplementary Fig. S4C; ref. 21). Intriguingly, we observed that EGFR inhibitors target squamous cell carcinoma CCLs and upper aerodigestive tract CCLs (Supplementary Fig. S4D). Sensitivity to EGFR inhibitors is often linked to EGFR mutation status, and two CCLs harboring the EGFR746del mutation, PC14 and HCC827, cluster together and respond dramatically to these EGFR inhibitors (9). The CCLs in the two clusters above, however, are EGFR wild-type with high EGFR expression (Supplementary Fig. S4E). This finding suggests two contexts where CCL response to EGFR inhibitors may be dependent on high EGFR expression, an observation that is borne out in clinical findings (22, 23). Thus, the clinically relevant response of specific patient populations to small molecules can be captured by automated analysis of the profiling data.
Insights into Small-Molecule Mechanism of Action
ACME analysis inherently depends on knowledge of the small-molecule targets. The analysis can also be used to assess the accuracy of reported targets by examining the small-molecule clusters. We found clusters of small molecules enriched for BRAF, MTOR, MDM2, or NAMPT modulators, where these targets represent the sole target annotation of the compound. This observation indicates that CCL sensitivity measurements can capture a variety of small-molecule modes of action, including protein–protein interaction inhibitors and protein kinase inhibitors (Fig. 3A). These clusters have high purity and confidence with respect to these protein target annotations, indicating high quality of the small-molecule probes and suggesting high reproducibility within the profiling experiment (Supplementary Fig. S4F). Furthermore, inhibitors targeting similar cell circuitry often cluster together. For example, the AKT1 inhibitor MK-2206 clusters with other inhibitors of the PI3K pathway (e.g., inhibitors of PIK3CA). Because activation of AKT1 occurs through a PI3K-dependent mechanism, clustering of these compounds is probably mechanistically linked (Fig. 3A). For compounds with multiple reported targets, clustering can reveal likely mechanisms of action by which the compounds affect cell viability in the profiling experiment. KW-2449 is reported to inhibit FLT3, ABL1, and AURKA (Supplementary Fig. S5A; ref. 24). This small molecule clusters with multikinase inhibitors that modulate FLT3 and members of the VEGFR, FGFR, and PDGFR families, but not with other ABL1 or AURKA inhibitors in the Informer Set, suggesting that the inhibition of FLT3 or related kinases by KW-2449 is likely responsible for cell death across the CCL panel (Supplementary Fig. S5B).
Figure 3.
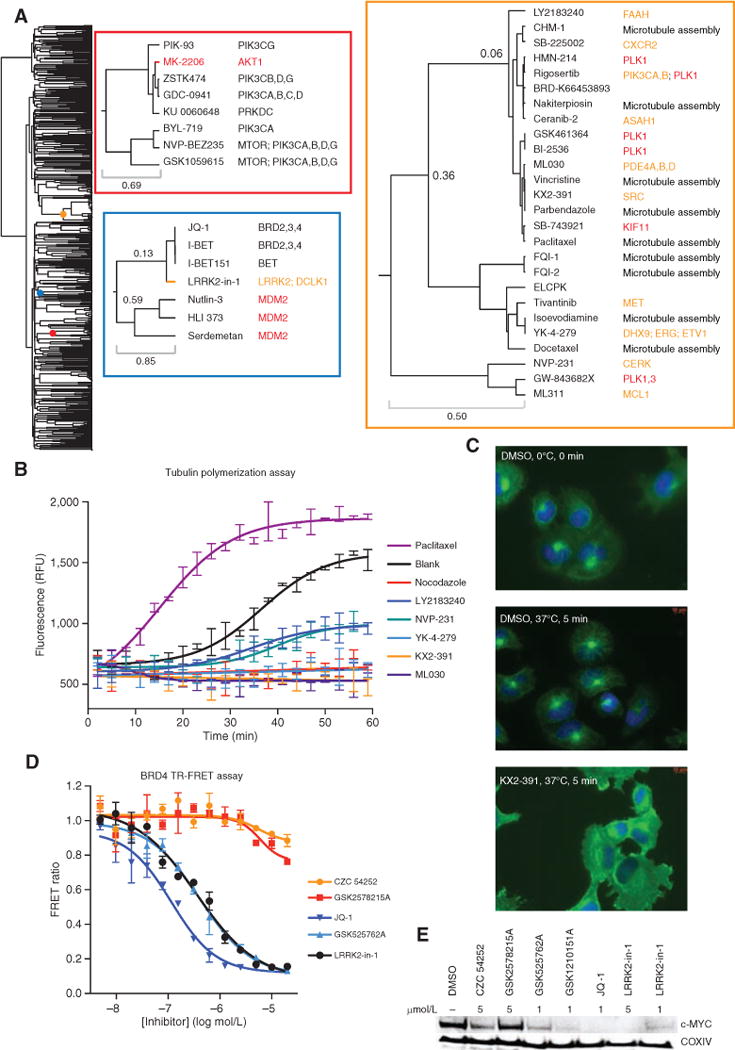
ACME analysis sheds light on small-molecule mechanism of action. A, dendrogram of the small-molecule Informer Set. Colored dots on the dendrogram denote the location of the enlarged dendrogram segments displayed to the right (color-coded to boxes). The top left inset (red box) is a “zoom in” of the cluster enriched for PI3K signaling, and the bottom left inset (blue box) contains one cluster enriched for MDM2 and one enriched for bromodomain inhibitors. The right inset (orange box) is the antimitotic cluster discussed in the text. For this inset, protein targets are colored by class: inhibitors of microtubule assembly (black), antimitotic kinase inhibitors with targets other than tubulin (red), and compounds with nominal protein targets unrelated to microtubule assembly or mitotic kinases (orange). B, recombinant tubulin polymerization assay. Every third data point is displayed. Each compound was run in duplicate (paclitaxel, 3 μmol/L; nocodazole, 3 μmol/L; KX2-391, 10 μmol/L; YK-4-279, 10 μmol/L; NVP-231, 10 μmol/L; LY2183240, 10 μmol/L). RFU, relative fluorescence units. C, microtubule regrowth assay in NCIH661 cells. Cells were cooled for 30 minutes on ice prior to compound treatment at time 0, and cells were either fixed directly or warmed to 37°C for 5 minutes and then fixed, followed by immunostaining for nucleus/DNA Hoechst stain (blue) and α-tubulin (green). All compounds were used at 500 nmol/L. D, BRD4 bromodomains 1 and 2 time-resolved FRET assay. E, Western blot of lysates from MM1S cells treated for 6 hours with compound at the indicated concentrations.
In some cases, we find that clusters enriched for a particular protein target also contain probes with unrelated target annotations, suggesting previously unreported targets or activities for these compounds. This type of analysis led to our initial hypothesis that STF-31, a putative GLUT1 inhibitor, was instead an NAMPT inhibitor (25). Here, we identified a cluster of compounds known to interfere with mitosis, comprising (i) inhibitors of microtubule dynamics, including both drugs (e.g., paclitaxel) and probes (e.g., nakiterpiosin; ref. 26), and (ii) inhibitors of the mitotic kinases PLK1 (e.g., GSK461364) and KIF11 (SB-743921; Fig. 3A). Interestingly, small molecules nominally targeting the MET proto-oncogene (tivantinib), the SRC proto-oncogene (KX2-391), PDE4A, B, and C (ML030), and ERG (YK-4-279) were also present in this cluster. Recently, the cytotoxicity of tivantinib was tied to the disruption of tubulin polymerization both in biochemical and cellular assays (27). We evaluated the effects of several of these small molecules on tubulin polymerization in vitro and found that KX2-391, ML030, NVP-231, LY2183240, and YK-4-279 modulated tubulin polymerization (Fig. 3B; Supplementary Fig. S5C and S5D). Furthermore, cells treated with KX2-391 and YK-4-279 showed decreased α-tubulin levels, indicating disruption of microtubule dynamics (Supplementary Fig. S5E). To decouple the observed tubulin polymerization effects from cytotoxic effects, we further validated our findings in a short-term cellular assay assessing microtubule network regrowth after cold treatment. Similar to nocodazole treatment, KX2-391, ML030, NVP-231, ceranib-2, and YK-4-279 reduced microtubule filament regrowth, indicating modulation of microtubule assembly (Fig. 3C; Supplementary Fig. S6). Overall, these data suggest that the small molecules found in this particular cluster likely induce cytotoxicity or growth inhibition in the profiling experiment by targeting microtubule dynamics or mitotic arrest.
Using a similar approach, we identified a cluster containing three compounds selective for the bromodomain and extraterminal (BET) family proteins (BRDT, BRD2, BRD3, BRD4), as well as LRRK2-in-1, an LRRK2 inhibitor discovered by ATP-competitive kinase screening (Fig. 3A; ref. 28). We evaluated whether LRRK2-in-1 also inhibited BRD4 bromodomains 1 and 2 using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay and found that LRRK2-in-1 inhibited the BRD4-acetylated peptide interaction in vitro, with comparable affinity to GSK525762A (I-BET; Fig. 3D). Notably, two other reported LRRK2 inhibitors (CZC 54252 and GSK2578215A) showed no activity in the assay. To evaluate the cellular effects of LRRK2-in-1, MYC levels were examined in the multiple myeloma CCL MM1S after compound treatment. MYC levels were diminished in samples treated with known bromodomain inhibitors at comparable levels to those treated with LRRK2-in-1 (Fig. 3E). Overall, these data suggest that LRRK2-in-1 is also a bromodomain inhibitor and that small-molecule clustering can be used as a complementary method to identify dual kinase–bromodomain inhibitors (29).
Using ACME Analysis to Identify Optimal Contexts for Dual IGF1R–ALK Inhibition
By uncovering unreported mechanisms of action for biologic probes, ACME analysis can identify the small-molecule target profile likely to be most effective in a particular context. Illustratively, the analysis revealed sensitivity of a group of neuroblastoma CCLs to IGF1R inhibitors (Fig. 4A; Supplementary Fig. S7A and S7B). Recently, the anti-IGF1R antibody cixutumumab has shown some clinical efficacy in a phase II clinical trial, including patients with neuroblastoma (30). We note that the four IGF1R inhibitors included in our study clustered with NVP-TAE684, a small molecule nominally targeting ALK (31). NVP-TAE684 was discovered in a Ba/F3 NPM–ALK fusion protein model, and initial experiments suggested that, despite comparable biochemical inhibition of ALK, IGF1R, and INSR, NVP-TAE684 was selective for the NPM–ALK fusion in cellular assays (Supplementary Fig. S7C; ref. 32). Our results suggest IGF1R inhibition may also contribute to the effects of NVP-TAE684 in neuroblastoma cells. To evaluate this hypothesis, we first showed that treatment with NVP-TAE684 leads to loss of phospho-IGF1R in NB1 cells (Fig. 4B). We then evaluated the sensitivity of a number of neuroblastoma CCLs to NVP-TAE684, BMS-754807 (IGF1R inhibitor), and crizotinib (ALK inhibitor). NVP-TAE684 has greater effects on viability of neuroblastoma CCLs compared with crizotinib, further strengthening the notion that IGF1R inhibition contributes to its effects on cell viability (Fig. 4C; refs. 32, 33). In contrast, NVP-TAE684 and BMS-754807 have similar killing potential in the neuroblastoma CCLs, except for the CCL NB1, which is not in the identified neuroblastoma cluster.
Figure 4.
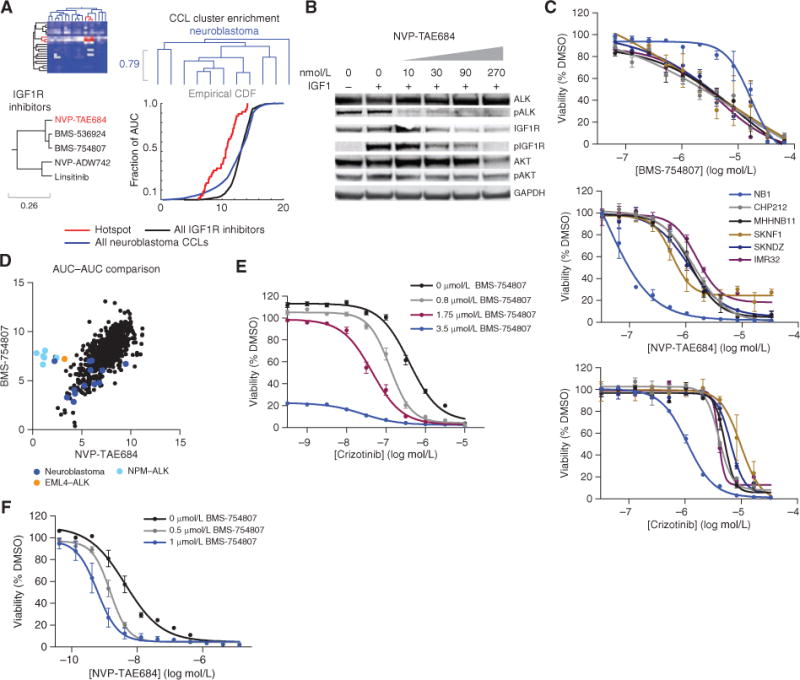
ACME analysis associates the sensitivity of neuroblastoma CCLs to IGF1R inhibitors to reveal the exquisite sensitivity of ALK-overexpressed neuroblastoma CCLs to dual IGF1R and ALK inhibition. A, the three enrichments for IGF1R inhibitors and neuroblastoma CCLs: row and column dendrograms and empirical cumulative distribution function (CDF) of the AUC distributions. The purity of the compound cluster is 0.8 and the confdence is 1. The purity of the CCL cluster is 0.55 and the confdence is 0.43. On the compound dendrogram segment, IGF1R inhibitors (black) and NVP-TAE684 (inhibiting ALK; red) are depicted. B, Western blot of lysates from NB1 cells that were serum-starved overnight, followed by 3-hour treatment with NVP-TAE684 at the indicated concentrations, and then 10-minute stimulation with IGF1. Experiments were repeated twice. These data confrm loss of phophorylated (p) IGF1R upon treatment with NVP-TAE684. C, confrmation of profling results for BMS-754807, crizotinib, and NVP-TAE684. The average of two replicates from two independent experiments is shown. D, AUC-AUC comparison for NVP-TAE684 and BMS-754807 with neuroblastoma CCLs (dark blue), CCLs with NPM-ALK rearrangement (light blue), and EML4-ALK rearrangements (orange) highlighted. E, sensitization of crizotinib by cotreatment with BMS-754807. Two independent experiments were performed. The average of one experiment with 5 to 7 replicates is shown. F, sensitization of NVP-TAE684 by cotreatment with BMS-754807. Two independent experiments were performed. The average of one experiment with 5 to 7 replicates is shown.
To better understand why NB1 behaves differently than other neuroblastoma CCLs, we compared the sensitivity profiles of BMS-754807 and NVP-TAE684 and found that NB1 responds similarly to CCLs harboring ALK fusions (Fig. 4D; Supplementary Fig. S7D). Notably, NB1 has the highest expression of ALK among CCLs tested, leading us to hypothesize that neuroblastoma CCLs with ALK overexpression may be more vulnerable to dual inhibition of ALK and IGF1R than to inhibition of either protein alone (Supplementary Fig. S7E). Consistent with this hypothesis, cotreatment with crizotinib plus BMS-754807 significantly shifted the IC50 compared with crizotinib or BMS-754807 alone in the NB1 CCL, as did cotreatment with NVP-TAE684 plus BMS-754807 (Fig. 4E and F). Furthermore, the neuroblastoma CCL KELLY, which harbors an activating ALK mutation (F1174L), clusters with NB1 (33, 34). KELLY also responds better to cotreatment with BMS-754807 plus crizotinib, as do CCLs carrying ALK fusion proteins (Supplementary Fig. S7F and S7G). Two additional small molecules with similar reported polypharmacology, LDK-378 and AZD3463, gave comparable results for NB1, although the remainder of the neuroblastoma CCLs were less responsive to LDK-378 (Supplementary Fig. S7H). Finally, we found that treatment with dual ALK–IGF1R inhibitors reduces phospho-AKT levels in the ALK-overexpressed neuroblastoma CCL NB1 compared with treatment with IGF1R or ALK inhibitors alone, suggesting that ALK overexpression could be a potential mechanism of resistance to IGF1R therapies (Supplementary Fig. S7I; ref. 35). ALK amplification is a common event in neuroblastoma and has recently been tied to poorer prognosis (36). This analysis reveals a clinically actionable combination that may provide a necessary therapeutic effect (37).
Using ACME Analysis to Identify Synergistic Combinations
ACME analysis may associate a particular genetic or lineage feature to more than one small-molecule target (Fig. 5A). We hypothesized that treatments with enhanced efficacy for a particular cluster of CCLs might be obtained by combining compounds arising from the different clusters identified by ACME analysis. This is especially important for compounds such as MEK inhibitors, which predominantly exert a cytostatic effect in vitro and have not produced significant clinical responses as single agents (38). Furthermore, it has been hypothesized that combinations may lead to more durable patient response (3).
Figure 5.
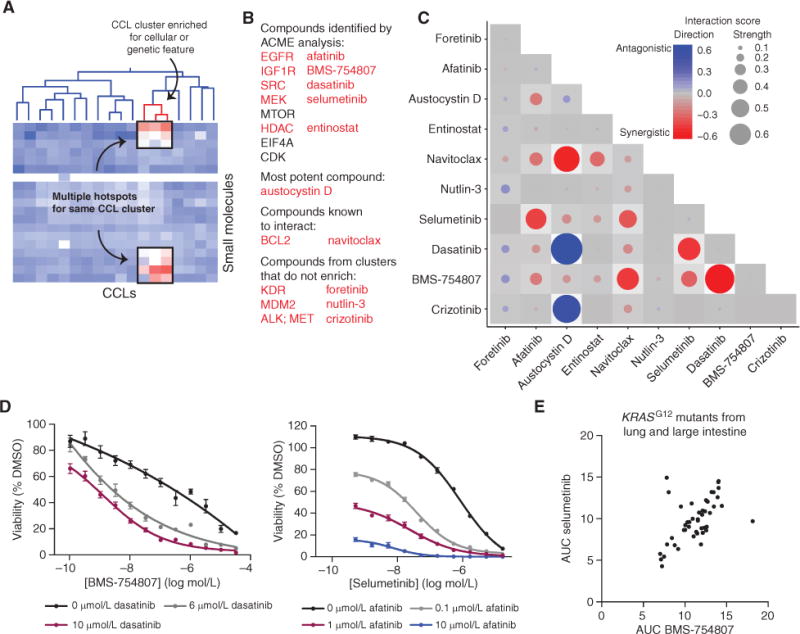
ACME analysis suggests vulnerabilities of a specific genetic context of KRAS-mutant CCLs to combination treatments. A, cartoon example of multiple hotspots for a single CCL cluster. B, the 10 compounds probed in the combination screen in LS513 cells and the rationale for their choice. Five were identified by ACME analysis. C, results from combination screen. Two biological and two technical replicates of the high-throughput screen were performed. The averages from the two biological replicates are shown. The synergy threshold is −0.17, and the antagonism threshold is +0.23. D, validation of the synergistic combinations identified. Two independent experiments were performed. The average of one experiment with 5 to 7 replicates is shown for each combination. E, AUC comparison of BMS-754807 and selumetinib in KRASG12-mutant CCLs in lung and large-intestine lineages.
To explore this potential application of ACME analysis to our sensitivity dataset, we examined small-molecule interactions in a group of CCLs enriched for KRAS mutations. This CCL cluster was associated with multiple compound clusters, linking sensitivity of these CCLs to MEK, IGF1R, SRC, EGFR, MTOR, PIK3CA, and HDAC inhibitors (e.g., Supplementary Fig. S8A). We selected exemplary compounds from five of these enriched target classes to test in a combination panel, namely BMS-754807; dasatinib, a multikinase inhibitor from the SRC cluster; afatinib, an irreversible EGFR inhibitor; selumetinib; and entinostat, an HDAC inhibitor (Fig. 5B). We also included navitoclax, which is known to synergize with MEK inhibitors in KRAS-mutant CCLs but was relatively nontoxic alone (AUC = 13.9; ref. 38), and austocystin D, one of the most effective compounds in these CCLs (Supplementary Fig. S8B; ref. 39). Three additional compounds were chosen from target classes that were cytotoxic but were not identified by ACME analysis for this specific CCL cluster: foretinib, crizotinib, and nutlin-3.
We devised an efficient high-throughput method to probe all pairwise combinations (45 total) of this set of 10 compounds in the KRASG12D-mutant CCL LS513 (Fig. 5B). Using this approach, we uncovered 10 synergistic combinations and two antagonistic combinations, where each combination had significantly more or less efficacy than additive (Fig. 5C; Supplementary Fig. S8C). Overall, inhibitors suggested by the resource accounted for 13 of the 20 (65%) synergistic partners identified. Whereas all of the compounds suggested by ACME participated in at least one synergistic interaction, the three compounds that were not identified by ACME did not produce synergistic combinations. As expected, the interaction between selumetinib and navitoclax is synergistic (38); intriguingly, navitoclax also synergized with BMS-754807 (Supplementary Fig. S8D). Other synergistic relationships between dasatinib and BMS-754807, dasatinib and selumetinib, afatinib and navitoclax, selumetinib and BMS-754807, and afatinib and selumetinib were suggested by this approach (Fig. 5D; Supplementary Fig. S8D). Given the similarity in response of LS513 to MEK and IGF1R inhibitors, we delved deeper into the sensitivity of these CCLs to IGF1R inhibitors and MEK inhibitors. We found a strikingly high correlation between sensitivity to BMS-754807 and selumetinib in lung and large intestine CCLs harboring the KRASG12 mutation. These inhibitor classes appear to target the same genetic vulnerability in these mutants, implying a mutation-specific response to these small molecules (Fig. 5E; Supplementary Fig. S8E; ref. 40). These preliminary data suggest that ACME analysis of profiling data may be useful in nominating compounds for combination testing.
DISCUSSION
Precision cancer therapies can achieve strong responses in specific patient populations by targeting vulnerabilities particular to the underlying genetic alterations and lineage features in the cancer. Developing new effective therapies and matching them to the right patients requires both an understanding of the vulnerabilities that cancers acquire as a result of their genetic and cellular features, and identification of the small-molecule targets which, when modulated, can elicit a patient response. CCL sensitivity profiling has emerged as an unbiased method to associate responses to small-molecule treatment with potential predictive markers. Here, we report a sensitivity dataset that includes responses to a broader range of small-molecule mechanisms of action than previously examined. Furthermore, rather than focusing on identifying predictors of sensitivity to compounds individually, we developed and automated an analytical methodology, ACME, to examine shared patterns of response for CCLs and small molecules. By synthesizing information from multiple CCLs and multiple compounds, we were able to strengthen associations of CCL sensitivity to a given genetic or cellular feature and a protein target, and make our interpretation more robust to identify, for example, previously uncharacterized polypharmacology of individual probes. ACME analysis may help de-noise potential confounding factors, such as discrepancies in mutation calling (41) or differences in growth media preference for each CCL. Importantly, our approach also identifies new associations that may merit deeper investigation for their clinical relevance, such as the efficacy of dual ALK– IGF1R inhibitors in neuroblastoma CCLs.
In addition to identifying potential cancer vulnerabilities, CCL profiling can uncover previously unreported protein targets for small molecules and drugs that may mediate their effects on cell viability. Developing small-molecule modulators with selectivity for targets of interest remains a major challenge for drug discovery. Here, we show that ACME analysis can identify unexpected mechanisms of action for small molecules, such as the inhibitory activity of LRRK2-in-1 on bromodomains. When a compound has multiple mechanisms of action, ACME analysis can also provide insight into which protein target may be primarily responsible for mediating the compound’s killing potential. As information about relevant small-molecule targets can be critical in interpreting CCL sensitivity experiments, we expect this dataset will be valuable to the cancer community that is using small molecules to investigate cancer-relevant pathways.
Combination treatments may mitigate the development of the drug resistance often observed with single-treatment therapies (42), yet principled methods to identify effective combinations have yet to be defined. Unbiased combination screening has been used to successfully identify acquired mechanisms of resistance to targeted therapies (43), though the scale of experiments required to assess combinations prohibits the routine use of this approach. Here, we developed a high-throughput method to rapidly assess interactions between small-molecule treatments. Using this method, we proposed 10 synergistic compound combinations. Corroborating our results, several of the synergistic pairs were previously identified with alternative screening methods, such as the combination of IGF1R and MEK inhibitors (44, 45). Furthermore, two inhibitor classes suggested by ACME analysis that we did not test, those targeting MTOR and PIK3CA, have also been explored in combination treatments in KRAS-mutant CCLs (46, 47). Although we assessed a small set of pairwise combinations, these results demonstrate the potential ability of profiling data to inform selection of effective combinations. Interestingly, the interactions identified in our studies appear to be specific to the genomic context of the CCLs, as the synergy between crizotinib and BMS-754807 in the neuroblastoma CCL NB1 did not recapitulate in the KRAS-mutant CCL LS513. This context specificity of combination therapies, mirroring the context specificity of targeted single agents, further emphasizes the need for systematic approaches such as CCL profiling to link sensitivity to the genetic and cellular features of cancer.
This study has focused on only a small number of relationships between compound features and cancer features. To enable wider analysis by the cancer research community, we created a second generation of our publicly available Cancer Therapeutics Response Portal (9). CTRP v2 is based on a foundation of quantitative sensitivity data on an unprecedented scale. As new “omic” characterizations of the CCLs become publicly available, such as metabolite levels, protein levels, or epigenetic markers, we intend to incorporate these features and their analysis in subsequent versions of the CTRP. Furthermore, combinations of “omic” features and/or pathway annotations of the CCLs could lead to greater insights into the sensitivity of the CCLs to small-molecule treatments; for example, Haibe-Kains and colleagues found the most consilience between datasets with pathway-based correlations (10). Efforts to address these types of connectivity analyses are currently under way, and further analyses will be incorporated into the public portal, including enrichment (9) and correlation analyses.
METHODS
CCL Profiling
An Informer Set of 481 small molecules (Supplementary Table S1) was tested for sensitivity in 860 publicly available human CCLs. Using an automated platform, CCLs were plated at a density of 500 cells per well in white, opaque tissue-culture–treated Aurora 1536-well MaKO plates (Brooks Automation) in the provider-recommended growth media (Supplementary Tables S2 and S10). Compounds were added 24 hours after plating by acoustic transfer using an Echo 555 (Labcyte Inc.). The effects of small molecules were measured over a 16-point concentration range (2-fold dilution) in duplicate. As a surrogate for viability, cellular ATP levels were assessed 72 hours after compound transfer using CellTiterGlo (Promega) with detection on a ViewLux Microplate Imager (PerkinElmer).
Sensitivity Data Processing
Log-transformed duplicate data were averaged during normalization of luminescence values to vehicle (DMSO) treatment and background (media-only) wells, and at each compound concentration, a D-score was calculated (9), which after re-exponentiation we used as the percent-viability (PV) score. A total of 6,239,426 average PV response-point measurements were computed across all cell lines, compounds, and concentrations. Prior to curve fitting, we applied two QC heuristics to concentration–response data: (i) we checked whether the highest 1 or 2 concentrations returned an otherwise low-viability signal back toward the DMSO signal by 20% or more (indicating a likely problem with compound solubility at these concentrations); (ii) we computed a sum, δ, of the absolute change in response across all pairs of adjacent concentrations. If this heuristic value remained below an acceptable threshold (δ < n/8, where n is the number of concentrations), we fit all points without censoring. Otherwise, we used Cook’s distance (48) during the curve-fit procedure to censor outlier points, but did not allow the highest two concentration points to be censored this way.
Curves were fit with nonlinear sigmoid functions, forcing the low-concentration asymptote to 1 (appropriate for DMSO treatment) using a three 3-parameter sigmoidal curve fit. If the predicted IC50 was higher than the highest concentration used, we refit with the bottom asymptote forced to 0, using a 2-parameter sigmoidal curve fit to avoid problematic curves with unrealistic IC50s and effect sizes. In total, we attempted 397,710 concentration–response curve fits, of which 397,681 fits succeeded (99.993% success; average 15.6 concentration points each; 6,198,098 PV points total).
The AUC for each compound–CCL pair was calculated by numerically integrating under the 16-point concentration–response curve (Supplementary Table S3). Subsequent to curve fitting and AUC calculation, we performed visual inspection of sets of curves to determine additional QC heuristics (Supplementary Methods). In all, 395,263 concentration–response curves were kept for further analysis (99.39% of those fit, 99.38% of total attempted). Primary sensitivity data are available from the NCI Cancer Target Discovery and Development (CTD2) Data Portal (ctd2.nci.nih.gov/dataPortal/).
ACME Analysis
We developed ACME analysis to automatically identify sets of related compounds to which sets of related CCLs are specifically sensitive. After clustering compounds across all CCLs, and CCLs across all compounds, regions of the resulting clustered heatmap (“hotspots”) are tested statistically for fulfillment of three criteria: a hotspot must (i) be enriched in low AUCs (i.e., high sensitivities); (ii) correspond to a compound cluster enriched for a target protein; and (iii) correspond to a CCL cluster enriched for a CCL mutation or lineage annotation.
Correlation and Clustering
We developed a method to perform correlation-based clustering on a matrix with missing data. First, we calculated pairwise correlation coefficients using Pearson correlation. We used Fisher z-transformation to normalize the correlations based on the number of non-empty elements shared by each pair, resulting in a z-score for each correlation whenever two sets of responses shared at least 4 elements (16). The Fisher-transformed z-scores were converted back to correlation distances using a monotonic double-sigmoid transformation:
which uses the distribution of linkage distances to emphasize variation in the most relevant part of the resulting dendrogram without changing dendrogram connectivity. For this transformation, we used k = 3 and derived α by tuning a single constraint, forcing the double sigmoid to pass through the point (zlim, −0.95). Because the z-scores from Fisher transformation are approximately normally distributed, we can map them to P values using the cumulative distribution function for the normal distribution, ϕ(μ,σ)(x). To get zlim, we find pllim such that , where plim = ϕ(0,1)(zlim), pmin = ϕ(0,1)(zmin), and zmin is the minimum z-score in the dataset (16). Finally, we generated clusters by complete linkage of the resulting distances and stored all cluster node memberships for further analysis.
Sensitivity Enrichment
For a clustered M × N matrix with corresponding M-2 “row” nodes and N-2 “column” nodes (we did not consider singleton or root nodes), we identified areas of sensitivity enrichment as the intersection of row cluster m and column cluster n for which the Wilcoxon rank-sum test comparing AUCs in the intersection with the remaining AUCs from m and n is significant [i.e., comparing AUCs in (m ^ n) with AUCs in (m ⊕ n); Supplementary Fig. S3C]. This definition accounts for broadly active small molecules and frequently sensitive CCLs in creating the appropriate null distribution for each putative sensitivity hotspot. The resulting output was corrected using the Benjamini-Hochberg procedure (49) to account for multiple hypothesis testing.
Cluster Node Enrichment
To compute enrichment of dendrogram nodes in annotation terms (protein target for compound row nodes; lineage or mutation for cell line column nodes), we consider only those annotations with ≥2 occurrences in the entire dataset. For enrichment of dendrogram nodes in annotation terms, we use a method similar to CLEAN (14). Specifically, for each of P annotation terms, we consider (M-2) × P or (N-2) × P possible chances for a dendrogram node to be enriched in a term, applying a Fisher exact test for each such comparison. P values resulting from Fisher exact tests were corrected using the Benjamini-Hochberg procedure (49) to account for multiple hypothesis testing, treating each distinct Fisher exact test as a separate hypothesis. Annotations are listed in Supplementary Tables S4, S5, and S6. Final ACME analysis outputs appear in Supplementary Table S7. See Supplementary Methods for filtering heuristics of ACME analysis output.
Cellular Validation and Follow-up Assays
The small molecules and CCLs used in validation assays are listed in Supplementary Tables S8, S9, and S10. CCLs for large-scale profiling were obtained from the Broad Biological Samples Platform, thawed, and tested for sensitivity in tranches between January 2012 and February 2013. When a reference SNP genotype was available for a CCL through the CCLE project (8), we set aside a sample for SNP genotyping by Fluidigm as described (50). At the time of publication, we have thus far positively matched 82.5% of the 664 CCLs analyzed in this study to their reference genotype (Supplementary Table S2). As additional samples are matched, we will provide updated information and analyses reflecting any changes at the CTD2 Data Portal (ctd2.nci.nih.gov/dataPortal/) and in CTRP v2 (9). Small molecules were transferred using a CyBi-Well Vario pin-transfer machine 24 hours after plating. Sensitivity was measured using CellTiterGlo 72 hours after the addition of small molecules as previously described (9). DMSO-normalized concentration-response curves were generated using four-parameter sigmoidal nonlinear regression in GraphPad Prism. For the combination treatment follow-up experiments, cells were plated at 1,000 cells per well in 20 μL. After 24 hours, 10 μL of additional media containing a small molecule or DMSO was added. The second small molecule was added using pin transfer.
Combination Screening
Viability measurements were performed as described above (2-fold serial dilution). A 1:1 mixture of the top concentration of the individual compounds, followed by serial dilution (2-fold), was used for combinations. Experiments were run in duplicate (r = 0.84; P < 6.4 × 10−16; Spearman correlation). We determined the highest observed inhibition in response to compound A, B, or A+B. Next, we observed the distance from no inhibition to this inhibition level on the A+B concentration-response curve. Using a Loewe Additivity Model (51), which assumes a compound does not interact with itself, and individual concentration-response curves, we calculated an expected distance from no inhibition to this inhibition level on the A+B concentration response. We computed a compound-interaction score [log2(observed/expected)] for each compound pair. In this setting, a compound-interaction score of 0 is interpreted as an additive compound pair, whereas significantly negative and positive scores mean synergistic or antagonistic compound pairs, respectively. In order to establish our experimental error, we conducted self–self compound-interaction controls where each compound is mixed with itself as if it were a combination pair, which yielded a mean of 0.01 and an SD of 0.07. We used a 99% confidence interval, giving −0.17 as our synergy cutoff and +0.23 as our antagonism cutoff. We also required the combination score in both replicates. This analysis results in 10 synergistic compound pairs and two antagonistic pairs (Supplementary Table S11).
Western Blotting
Western blotting was conducted following a previously reported protocol (8). Primary antibodies used were: phospho-ALK p-Y1604 (Abcam; ab62185), phospho-AKT p-S473 (Invitrogen; 700392), phospho-IGF1R p-Y1331 (Cell Signaling Technology; 3021), GAPDH (Cell Signaling Technology; 2118S), ALK (Invitrogen; 51-3900), IGF1R (Cell Signaling Technology; 3027), AKT (Cell Signaling Technology; 9272), and MYC (Cell Signaling Technology; 9402S).
IGF1 Stimulation
Cells were plated into 6-well dishes or 10-cm plates. Upon reaching 75% confluence, cells were serum-starved overnight. The next morning, cells were treated with compound at the indicated concentrations for 3 hours, followed by IGF1 stimulation for 10 minutes (Gibco; PHG0078; 10 ng/mL). After 10 minutes, the cells were lysed for Western blotting.
Tubulin Polymerization Assay
Tubulin polymerization assays were run using a fluorescent porcine tubulin kit (Cytoskeleton; BK011P), according to the manufacturer’s conditions with either the standard conditions (Supplementary Fig. S5A) or inhibitor conditions (Supplementary Fig. S5B). The following final concentrations were used for each compound: nocodazole, 3 μmol/L; paclitaxel, 3 μmol/L; all other inhibitors, 10 μmol/L.
Microtubule Regrowth Assay
NCIH661 cells were plated in NUNC Lab-Tek II 4-chamber slides. After 24 hours, slides were removed from the incubator and put on ice for 30 minutes. Following ice treatment, cells were treated with compound or control and either fixed directly (aspirate media, 4% formaldehyde) or incubated for 5 minutes at 37°C. After 5 minutes, the media were aspirated and the cells were fixed (Supplementary Methods). Imaging was performed on a Zeiss Cell Observer. Antibodies used were as follows: α-tubulin (Cell Signaling Technologies; 3873P), phalloidin CF568-conjugated (VWR, 89138-134), and FITC 488.
BRD4 TR-FRET Assay
The TR-FRET bromodomain peptide displacement assay was run according to the manufacturer’s conditions (Cayman Chemical; 600520). Each compound was run in duplicate.
Supplementary Material
SIGNIFICANCE.
We present the largest CCL sensitivity dataset yet available, and an analysis method integrating information from multiple CCLs and multiple small molecules to identify CCL response predictors robustly. We updated the CTRP to enable the cancer research community to leverage these data and analyses.
Acknowledgments
The authors thank the following colleagues and centers for contributing compounds: D. Adams, Boston University, P. Brown, C. Chen, J. Clardy, Centro Nacional de Investigaciones Oncológicas (CNIO), E.J. Corey, the Drug Synthesis and Chemistry Branch (Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, NCI), Eutropics, J. Gutterman, E. Holson, Karyopharm, M. Meyerson, A. Myers, J. Porco, J. Qi, Sanford-Burnham, M. Serrano-Wu, M. Shair, B. Stockwell, L. Walensky, X. Wang, and D. Zaharevitz. They also thank S. Chattopadhay, J. Law, G. Schaefer, M. Stewart, S. Wang, and V. Viswanathan for helpful discussions; A. Vrcic and the Broad Compound Management team for handling the Informer Set; Karen Emmith, Jacob Aseidu, and the CSofT informatics group for development and support of cell-line and data-tracking software; J. Boehm, A. Tsherniak, and the Broad Cancer Program for training; the Broad Biological Samples Platform for providing CCLs; and Levi Garraway and the Broad–Novartis CCLE team.
Grant Support: This work was supported by the NCI’s Cancer Target Discovery and Development Network (grant number U01CA176152, awarded to S.L. Schreiber), the Turkish Academy of Sciences GEBIP Programme (M. Cokol), as well as Stiftelsen Bengt Lundqvist Minne for 2013 postdoctoral support (to B. Seashore-Ludlow). S.L. Schreiber is an Investigator at the Howard Hughes Medical Institute.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: B. Seashore-Ludlow, M.G. Rees, J.H. Cheah, C.S.-Y. Hon, A.F. Shamji, P.A. Clemons, S.L. Schreiber
Development of methodology: B. Seashore-Ludlow, M.G. Rees, J.H. Cheah, M. Cokol, N.E. Bodycombe, M. Palmer, A.F. Shamji, P.A. Clemons
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): B. Seashore-Ludlow, M.G. Rees, J.H. Cheah, E.V. Price, M.E. Coletti, C.K. Soule, B. Munoz, M. Palmer, A.F. Shamji
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): B. Seashore-Ludlow, M.G. Rees, J.H. Cheah, M. Cokol, N.E. Bodycombe, B. Alexander, N. Kuru, T. Liefeld, V. Dančík, J.E. Bradner, A.F. Shamji, P.A. Clemons
Writing, review, and/or revision of the manuscript: B. Seashore-Ludlow, M.G. Rees, M. Cokol, M.E. Coletti, J.D. Kotz, A.F. Shamji, P.A. Clemons, S.L. Schreiber
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): B. Seashore-Ludlow, J.H. Cheah, E.V. Price, C.K. Soule, J. Gould, B. Alexander, A. Li, P. Montgomery, M.J. Wawer, T. Liefeld, J.A. Bittker, A.F. Shamji, P.A. Clemons
Study supervision: B. Seashore-Ludlow, J.H. Cheah, M. Palmer, A.F. Shamji, P.A. Clemons, S.L. Schreiber
Other (performance of high-throughput screening): V. Jones, C.K. Soule
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
References
- 1.Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31:1803–5. doi: 10.1200/JCO.2013.49.4799. [DOI] [PubMed] [Google Scholar]
- 2.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 3.Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–26. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 4.Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241–53. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 5.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 6.McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104:19936–41. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. www.broadinstitute.org/ccle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. doi: 10.1016/j.cell.2013.08.003. www.broadinstitute.org/ctrp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJ, et al. Inconsistency in large pharmacogenomic studies. Nature. 2013;504:389–93. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–6. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wawer MJ, Li K, Gustafsdottir SM, Ljosa V, Bodycombe NE, Marton MA, et al. Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc Natl Acad Sci U S A. 2014;111:10911–6. doi: 10.1073/pnas.1410933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freudenberg JM, Joshi VK, Hu Z, Medvedovic M. CLEAN: CLustering Enrichment ANalysis. BMC Bioinformatics. 2009;10:234. doi: 10.1186/1471-2105-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iskar M, Zeller G, Blattmann P, Campillos M, Kuhn M, Kaminska KH, et al. Characterization of drug-induced transcriptional modules: towards drug repositioning and functional understanding. Mol Syst Biol. 2013;9:662. doi: 10.1038/msb.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dancik V, Carrel H, Bodycombe NE, Seiler KP, Fomina-Yadlin D, Kubicek ST, et al. Connecting small molecules with similar assay performance profles leads to new biological hypotheses. J Biomol Screen. 2014;19:771–81. doi: 10.1177/1087057113520226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wawer MJ, Jaramillo DE, Dancik V, Fass DM, Haggarty SJ, Shamji AF, et al. Automated structure-activity relationship mining: connecting chemical structure to biological profiles. J Biomol Screen. 2014;19:738–48. doi: 10.1177/1087057114530783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19:2257–64. doi: 10.1158/1078-0432.CCR-12-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 20.Subramaniam D, He AR, Hwang J, Deeken J, Pishvaian M, Hartley ML, et al. Irreversible multitargeted ErbB family inhibitors for therapy of lung and breast cancer. Curr Cancer Drug Targets. 2015;14:775–93. doi: 10.2174/1568009614666141111104643. [DOI] [PubMed] [Google Scholar]
- 21.Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, et al. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol Cancer Ther. 2011;10:2115–23. doi: 10.1158/1535-7163.MCT-11-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bozec A, Peyrade F, Milano G. Molecular targeted therapies in the management of head and neck squamous cell carcinoma: recent developments and perspectives. Anticancer Agents Med Chem. 2013;13:389–402. [PubMed] [Google Scholar]
- 23.Wang Q, Zhu H, Xiao Z, Zhang W, Liu X, Zhang X, et al. Expression of epidermal growth factor receptor is an independent prognostic factor for esophageal squamous cell carcinoma. World J Surg Oncol. 2013;11:278. doi: 10.1186/1477-7819-11-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood. 2009;114:1607–17. doi: 10.1182/blood-2009-01-199307. [DOI] [PubMed] [Google Scholar]
- 25.Adams DJ, Ito D, Rees MG, Seashore-Ludlow B, Puyang X, Ramos AH, et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem Biol. 2014;9:2247–54. doi: 10.1021/cb500347p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei JH, Seemann J. Nakiterpiosin targets tubulin and triggers mitotic catastrophe in human cancer cells. Mol Cancer Ther. 2010;9:3375–85. doi: 10.1158/1535-7163.MCT-10-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aoyama A, Katayama R, Oh-Hara T, Sato S, Okuno Y, Fujita N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance. Mol Cancer Ther. 2014;13:2978–90. doi: 10.1158/1535-7163.MCT-14-0462. [DOI] [PubMed] [Google Scholar]
- 28.Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7:203–5. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10:305–12. doi: 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2014;61:452–6. doi: 10.1002/pbc.24605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104:270–5. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 33.Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, et al. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J. 2011;440:405–13. doi: 10.1042/BJ20101796. [DOI] [PubMed] [Google Scholar]
- 34.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014;32:2727–34. doi: 10.1200/JCO.2013.54.0674. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Zhou C, Sun Q, Cai R, Li Y, Wang D, et al. ALK amplification and protein expression predict inferior prognosis in neuroblastomas. Exp Mol Pathol. 2013;95:124–30. doi: 10.1016/j.yexmp.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20:1027–34. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marks KM, Park ES, Arefolov A, Russo K, Ishihara K, Ring JE, et al. The selectivity of austocystin D arises from cell-line-specific drug activation by cytochrome P450 enzymes. J Nat Prod. 2011;74:567–73. doi: 10.1021/np100429s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodaz H, Hacibekiroglu I, Erdogan B, Turkmen E, Tozkir H, Albayrak D, et al. Association between specific KRAS mutations and the clinicopathological characteristics of colorectal tumors. Mol Clin Oncol. 2015;3:179–84. doi: 10.3892/mco.2014.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudson AM, Yates T, Li Y, Trotter EW, Fawdar S, Chapman P, et al. Discrepancies in cancer genomic sequencing highlight opportunities for driver mutation discovery. Cancer Res. 2014;74:6390–6. doi: 10.1158/0008-5472.CAN-14-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–92. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 43.Axelrod M, Gordon VL, Conaway M, Tarcsafalvi A, Neitzke DJ, Gioeli D, et al. Combinatorial drug screening identifes compensatory pathway interactions and adaptive resistance mechanisms. Oncotarget. 2013;4:622–35. doi: 10.18632/oncotarget.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3:548–63. doi: 10.1158/2159-8290.CD-12-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4:42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook RD. Influential observations in linear regression. J Am Stat Assoc. 1979;74:169–74. [Google Scholar]
- 49.Benjamini Y, Hochberg B. Controlling true discovery rate: a practical and powerful approach to multiple hypothesis testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 50.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Scientific Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–85. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.