

Abstract
Nuclear factor-kappa B (NF-κB) is a ubiquitously expressed protein complex regulating the transcription of genes involved in inflammation and pain. Increased NF-κB activity in immune and nervous system cells is linked to several chronic pain conditions in humans as well as inflammation- and nerve injury-evoked pain in animals. A recent in vitro study further demonstrates that increased NF-κB activity in astrocytes decreases transcription of catechol-o-methyltransferase (COMT), an enzyme that inactivates catecholamines that cause pain. The purpose of the present study was to examine the relationship between systemic and astrocytic NF-κB activity, pain, and COMT expression in an animal model of inflammation. Results demonstrated that administration of the inflammatory stimulant complete Freund’s adjuvant (CFA) led to increased pain and decreased COMT protein expression in an NF-κB-dependent manner. Specifically, we found that rats and mice receiving intraplantar CFA exhibited increased behavioral responses to mechanical and thermal heat stimuli. CFA-evoked pain was blocked in rats receiving a pre-emptive systemic dose of the NF-κB inhibitor MG132 and exacerbated in IKKca mice with constitutive NF-κB activity in astrocytes. Furthermore, we observed NF-κB-linked reductions in COMT expression in midbrain at 6h and 1d following CFA in rats and at 1h and 1d in forebrain and midbrain following CFA in IKKca mice. Collectively, these results demonstrate that systemic and astrocytic NF-κB activity drive inflammatory pain and regulate the expression of COMT in forebrain and midbrain structures.
Keywords: complete Freund’s adjuvant (CFA), MG132, astrocytes, catecholamines, tumor necrosis factor-α (TNFα)
1. Introduction
The signaling complex nuclear factor-κB (NF-κB) is a key regulator of molecules and pathways important for inflammation and pain. NF-κB can exist as homo- or heterodimers composed of the Rel family proteins including p65, p50, p52, RelB, and c-Rel, with the p65/p50 heterodimer being the canonical form.1 In its inactive state, NF-κB is bound in the cytoplasm bound to inhibitors of κB (IκBα). Upon activation by pro-inflammatory cytokines (e.g., interleukin-1β; IL-1β and tumor necrosis factor α; TNFα), growth factors (e.g., epidermal growth factor; EGF)2, or adjuvants that stimulate toll-like receptors 2 and 4 (e.g., complete Freund’s adjuvant; CFA)3, the IκB kinase (IKK) phosphorylates IκBα, tagging it for degradation. As a result, IκBα releases the p65/p50 complex, which subsequently translocates to the nucleus and initiates transcription of κB-associated inflammatory genes such as TNFα, IL-1β, and cyclooxogenase-2 (COX-2) that directly or indirectly influence pain.1,4
Activation of NF-κB in humans is implicated in a variety of pain conditions such as rheumatoid arthritis, migraine, and nerve injury.5 In patients with rheumatoid arthritis, NF-κB-linked proteins are upregulated in synovial tissues.6 In addition, patients that experience chronic migraines express increased IκBα in blood monocytes concurrent with increased migraine intensity.7 Accordingly, NF-κB is upregulated in the dorsal root ganglia and spinal cord of animals experiencing inflammatory and neuropathic pain.8-11 Inflammation-evoked pain is attenuated by knockdown of p50,12 gene silencing of p65,13 or by administration of a proteasome inhibitor that prevents IκBα dissociation from p50 and p65.14 Similarly, pain following nerve injury is reversed by administration of an NF-κB decoy that prevents activated NF-κB from binding to a consensus DNA fragment and initiating transcription.15,16
While NF-κB is ubiquitously expressed in a variety of cell types,17 its contribution to pain is driven in large part by its signaling in astrocytes. Astrocytes are glial cells located in the central nervous system that participate in the transmission of neuronal signaling and provide neuroprotection during synaptic remodeling or neurodegeneration.2 In animal studies, astrocytic NF-κB is upregulated following viral coat protein-induced inflammation (e.g. gp120),11 CFA-induced inflammation,11,13 and nerve injury.8 Likewise, loss of astrocytic NF-κB signaling attenuates pain following formalin administration18 or spinal nerve injury.9 While these studies point to the contribution of astrocytic NF-κB activity in pain, more research is required to identify downstream pain-relevant molecules.
Recent work by our group suggests that activation of NF-κB in astrocytes may produce pain by regulating the expression of catechol-o-methyltransferase (COMT), an enzyme that inactivates catecholamines and modulates pain.19 Specifically, we found in vitro evidence demonstrating that TNFα-induced activation of NF-κB in a human astrocyte (H4) cell line led to binding of p65 to the P2 promoter region of the Comt gene, thereby reducing COMT mRNA and protein expression. Reduced COMT expression and activity is associated with heightened pain sensitivity in patients with chronic pain conditions.20,21 Furthermore, pharmacological inhibition of COMT increases pain in animals.22,23 Therefore, COMT may represent a novel molecular target for NF-κB that dictates pain behaviors.
Building on in vitro work demonstrating that NF-κB downregulates COMT expression together with in vivo work demonstrating the role of COMT in pain, the purpose of the present study was to examine the relationship between systemic and astrocyte-specific NF-κB activity, pain, and COMT expression in an animal model of inflammation. Specifically, we evaluated the effects of NF-κB inhibition in rats or overexpression in mice on pain behavior and COMT expression following local administration of CFA. We hypothesized that induction of NF-κB by the pro-inflammatory stimulant CFA would result in enhanced pain and decreased COMT protein expression.
2. Materials and Methods
2.1 Animals
Seventy-eight adult male Sprague-Dawley rats (250-320g; Charles River Laboratories, Raleigh, NC) were used in behavior and molecular experiments. Eighty-eight male C57Bl6 expressing constitutively active form of IκB kinase (IKK) in GFAP+ cells (IKK constitutive activity, hence “IKKca”) and littermate control (Co) mice (25-35 g; a gift from the lab of Dr. Ken McCarthy) were used in behavior and molecular experiments. All procedures were approved by the University of North Carolina Animal Care and Use Committee and adhered to the guidelines of the Committee for Research and Ethical Issues of the IASP.24
2.2 General Experimental Methods
To evaluate the role of NF-κB in regulating inflammatory pain and COMT expression in rats, separate groups received an intraperitoneal (i.p.) injection of the NF-κB inhibitor MG132 (10 mg/kg) or vehicle (10% DMSO in 0.9% saline) one hour prior to a unilateral intraplantar (i.pl.) injection of CFA (200 μl) or saline (200 μL). In mice, separate groups of IKKca or Co mice received a unilateral i.pl. injection of CFA (20 μL) or incomplete Freund’s adjuvant (IFA; 20 μL). IFA is identical in its chemical composistion to CFA, but does not contain Mycobacterium tuberculosis, which are responsible for the induction of inflammation. Behavioral responses to mechanical and thermal stimuli were reassessed at acute (1h, 6h, 1d), subchronic (3d, 5d, 7d), and chronic (9d, 11d, and 13d) phases of inflammation, as outlined by Raghavendra et al., 2004.25 In all studies, the experimenter was blinded to the experimental conditions.
2.3 Assessment of Mechanical and Thermal Pain Sensitivity
Rats and mice were handled and habituated to the testing environment for 4d prior to establishing baseline responsiveness to mechanical and thermal stimuli. First, animals were placed in plexiglass cages positioned over an elevated mesh stainless steel platform and habituated to the environment for 20 min. Mechanical allodynia and hyperalgesia were assessed using a repetitive presentation of either an innocuous or noxious stimulus, as used in Ringkamp et al., 199926 and Fecho et al., 2005.27 Mechanical allodynia was assessed by placing a normally innocuous von Frey filament (3.632g for rat; 0.166g for mouse) onto the plantar surface of the hindpaw ten times for 1s with an inter-stimulus interval of 1s. Mechanical hyperalgesia was assessed by placing a normally noxious von Frey filament (15g for rat; 1.494g for mouse) onto the plantar surface of the hindpaw ten times for 1s with an inter-stimulus interval of 1s. Mechanical allodynia or hyperalgesia was defined as an increase in the frequency ([number of paw withdrawals/10] × 100%) of paw withdrawals evoked by stimulation with the von Frey monofilaments compared to baseline.
Thermal hyperalgesia was assessed using the Hargreaves method as described previously.22 Briefly, animals were placed on an elevated heated glass platform maintained at 30°C and habituated to the environment for 20 min. A radiant beam of light was applied to the plantar surface of the hindpaw and the latency to withdrawal for a maximum of 20s was recorded. Withdrawal latency was measured in duplicate for each paw, with an inter-stimulus interval of 5 min. If the difference between the duplicate measures was >4s, a third measurement was taken. The two most consistent measurements were included in the analysis. Thermal hyperalgesia was defined as a decrease in paw withdrawal latency compared to baseline.
2.4 Assessment of COMT Protein Expression in Nervous System Tissues
COMT expression was measured in tissues collected from separate groups of rats sacrificed at 6h, 1d, or 5d following experimental treatment and from mice sacrificed at 1h, 6h, 1d, 3d, 5d, or 11d following experimental treatment. The specific tissues collected include the dorsal root ganglia (DRG), spinal cord, and brain regions (hindbrain [locus coeruleus and periaqueductal gray], midbrain [thalamus and hypothalamus] or forebrain [primary somatosensory cortex and nucleus accumbens]).
Total protein from each discrete tissue region was diluted in tissue protein extraction reagent (TPER; Thermo Scientific; Rockford, IL) with protease inhibitor (Pierce; Rockford, IL) and homogenized using the Precellys Homogenizer system (MD, USA). Soluble protein concentrations were assessed and normalized using a BCA Protein Assay Kit (Pierce; Rockford, IL), run at 10-50 μg on precast 12% Tris-Glycine gels (Bio-Rad; Hercules, CA), and transferred onto PVDF membranes using the iBlot Dry Blotting system (Invitrogen; Waltham, MA). Following transfer, blots were blocked using 5% w/v bovine serum albumin diluted in TBST for 1h and incubated in either anti-COMT primary antibody (BD Biosciences; San Diego, CA) diluted at 1:7,500 in blocking buffer solution or anti-β-actin primary antibody (Cell Signaling; Danvers, MA) diluted at 1:3,000 at 4°C overnight. Chemiluminescence was detected on a ImageQuant TL system and analyzed using ImageQuant TL 3000 software (GE Healthcare; Piscataway, NJ). Densitometry volume was analyzed such that COMT densitometry values were normalized to β-actin values.
2.5 Statistical Analysis
Behavioral data were analyzed by one-way analysis of variance (ANOVA) for repeated measures. Post hoc comparisons were performed using the Bonferroni’s test. Normalized protein data were analyzed by unpaired t-test with post-hoc tests. P < 0.05 was considered to be statistically significant.
3. Results
3.1 NF-κB inhibition attenuates inflammation-evoked increases in pain sensitivity in rats
To evaluate the role of NF-κB in regulating pain sensitivity in the CFA model of inflammation, separate groups of rats received i.p. administration of the NF-κB inhibitor MG132 or vehicle 1h prior to i.pl. administration of CFA or saline. Behavioral responses to mechanical and thermal stimuli were then assessed during acute, subchronic, and chronic phases of inflammation.
Prior to the induction of inflammation, responsiveness to mechanical and thermal stimuli did not differ between groups. Following administration of i.pl. CFA, rats exhibited mechanical allodynia, mechanical hyperalgesia, and thermal hyperalgesia in the ipsilateral paws. CFA-induced increases in pain sensitivity were observed 1h following administration and lasted throughout the 13-day testing period. Administration of MG132 prior to CFA attenuated inflammation-evoked mechanical allodynia and thermal hyperalgesia during acute, subchronic, and chronic phases (F9,112 = 6.06, P < 0.0001; Figure 1A and F9,112 = 6.027, P < 0.0001; Figure 1C, respectively). Pre-emptive administration of MG132 also attenuated inflammation-evoked mechanical hyperalgesia during subchronic and chronic phases (F9,112 = 13.19, P < 0.0001; Figure 1B). No differences in behavioral responsiveness to mechanical and thermal stimuli were observed following MG132 administration in the absence of CFA or in the contralateral noninjected paws.
Figure 1.
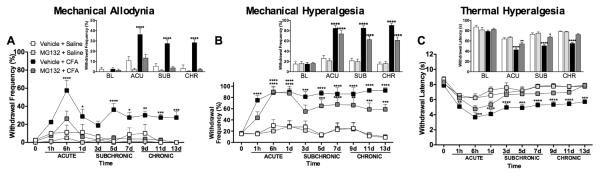
Blockade of NF-κB reduces CFA-induced mechanical and thermal pain in rats. Intraplantar administration of CFA (200 μL) increases (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia. Pretreatment with MG132 (20 mg/kg dosage) attenuates mechanical allodynia and thermal hyperalgesia compared to Vehicle + Saline group. N=8 per group. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 different from Vehicle + Saline for larger graphs and different from Vehicle for inset graphs.
3.2 Constitutive activity of astrocytic NF-κB prolongs CFA-induced pain in mice
We next sought to determine if constitutive activity of the astrocytic NF-κB pathway would alter inflammatory pain behaviors. Separate groups of IKKca and Co mice received i.pl. CFA or IFA. Behavioral responses to mechanical and thermal stimuli were then assessed during acute, subchronic and chronic phases of inflammation.
Compared to their Co littermates, IKKca mice displayed prolonged increases in mechanical allodynia (F27,457 = 2.130, P<0.001; Figure 2A) and mechanical hyperalgesia (F27,456 = 3.247, P<0.0001; Figure 2B) in the affected ipsilateral paw beginning on day 3, which lasted throughout the 13-day testing period (Figure 2A-C). Furthermore, while we observed no changes in mechanical or thermal pain in the unaffected contralateral paw of Co mice, we did observed increased mechanical allodynia and hyperalgesia in the contralateral paw of IKKca mice beginning on day 3, during the subchronic phase of inflammatory pain, and lasting until the end of the behavioral paradigm. We did not observe significant differences in thermal hyperalgesia in IKKca and Co mice. As expected, no differences in pain behaviors were observed between IKKca and Co mice following IFA administration for mechanical allodynia (F9,162=0.270, P=0.982), mechanical hyperalgesia (F9,162=0.926, P=0.504), or thermal hyperalgesia (F9,178=0.496, P=0.876) (Supplementary Figure 1A-C).
Figure 2.
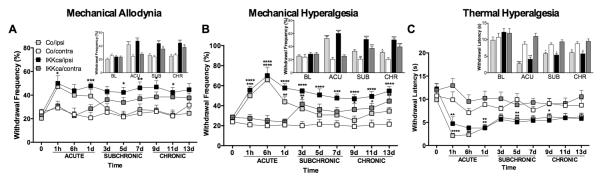
Mice expressing constitutively active IKK exhibit enduring pain following CFA treatment compared to Co. Intraplantar administration of CFA (20 μL) increases (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia. While CFA-mediated (A) mechanical allodynia and (B) mechanical hyperalgesia subside in Co mice, IKKca mice exhibit prolonged mechanical allodynia and hyperalgesia compared to Co contra. No differences were observed in thermal hypersensitivity following CFA administration between IKKca and Co mice. N=10-13 per group. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 different from Co/ contra.
3.3 NF-κB inhibition blocks inflammation-evoked reductions in COMT protein expression in rats
After establishing that NF-κB activity mediated pain behavior in the CFA model of inflammation, we sought to assess the role of NF-κB in regulating the expression of COMT. Separate groups of rats received an i.p. dose of the NF-κB inhibitor MG132 or vehicle 1h prior to i.pl. administration of CFA or saline and were sacrificed at 6h, 1d, and 5d after CFA injection. Tissues from forebrain (nucleus accumbens, primary somatosensory cortex) and midbrain (thalamus and hypothalamus), hindbrain, and lumbar spinal cord were collected for analysis of COMT protein levels. CFA administration produced noticeable reductions in forebrain COMT protein expression levels at 6h and 1d (Figure 3). Administration of MG132 prior to CFA normalized reductions in COMT expression in forebrain. No differences in COMT expression were observed in hindbrain or spinal cord tissue samples (data not shown).
Figure 3.

CFA inflammation reduces COMT expression in rat midbrain via an NF-κB mechanism. Western blot analysis of COMT expression in rat forebrain lysates after intraplantar administration of CFA and MG132 prior to CFA. N=3/group. Data are mean ± SEM.
3.4 Constitutive activity of astrocytic NF-κB reduces COMT protein expression in mice
Finally, we sought to determine if animals expressing astrocyte-specific IKKca exhibited altered COMT expression following CFA. Separate groups of IKKca and Co mice were administered CFA and tissue was collected at acute, subchronic and chronic time points from discrete pain-associated brain regions, spinal cord and dorsal root ganglia for COMT protein expression analysis. As the trends in COMT expression levels were similar according to brain region (e.g. hindbrain, midbrain and forebrain), the data were averaged in this manner. Analyses showed significant group differences in COMT expression between IKKca and Co mice in forebrain (t12=4.014, P<0.002) and midbrain (t12=3.676, P<0.01). Specifically, similar to the results obtained in rats receiving the NF-κB inhibitor, IKKca mice exhibited significant decreases in COMT expression in midbrain (thalamus and hypothalamus) as well as in forebrain (nucleus accumbens and somatosensory cortex). We then assessed if there were significant differences at specific timepoints. Compared to Co mice, IKKca mice expressed significantly less COMT in forebrain at 1h (t12=3.10, P<0.003) (Figure 4A,C) and in midbrain at 1h (t11=3.04, P<0.01) and 1d (t12=3.14, P<0.008) post-CFA (Figure 4B,D).
Figure 4.

CFA inflammation reduces COMT expression in forebrain and midbrain of IKKca mice. (A,C) Western blot analysis of COMT expression in mouse forebrain and midbrain of IKKca and Co mice receiving CFA (B,D) Representative Western blot depicting IKKca and Co mice following CFA injection in forebrain and midbrain tissues. N=3-4/group. Data are mean ± SEM. *P<0.05.
4. Discussion
The present study employed pharmacologic and genetic approaches in an animal model of inflammation to examine the relationship between NF-κB, pain, and COMT expression. We found that animals with inhibited NF-κB activity exhibited decreased CFA-induced pain, while those with enhanced astrocytic NF-κB activity exhibited increased CFA-induced pain. Furthermore, we provide the first systems-level demonstration showing that CFA-induced inflammation leads to reductions in COMT protein expression via an NF-κB mechanism.
NF-κB is widely recognized as a master switch that is essential for immune responses. An emerging literature shows that NF-κB plays an important role in the central nervous system, where it can drive pain associated with injury and inflammation. For example, animals with p50 knockout12 or p65 knockdown13 show reduced pain following administration of intraplantar formalin or intradermal CFA, respectively. CFA contains Mycobacterium tuberculosis, which can directly stimulate toll-like receptors-2 and -4, which in turn signal through MyD88 to increase the stimulation various inflammatory cascades, including the NF-κB pathway. Administration of MG132, which induces upstream IκBα nuclear translocation to more fully block NF-κB-mediated transcription,28 reduces pain following systemically-administered CFA. The present study extended these findings by testing the ability of MG132 to prevent the development of mechanical and thermal hypersensitivity following intraplantar CFA. Accordingly, we found that rats receiving a pre-emptive dose of MG132 exhibited reduced hypersensitivity to mechanical and thermal stimuli for the duration of the two-week testing paradigm. Because CFA acts through
Based on evidence demonstrating that nociceptive stimuli induce NF-κB activity in astrocytes, we were particularly interested in understanding if astrocytic NF-κB signaling regulated CFA-induced pain. Astrocytic NF-κB regulates the expression of genes linked to immune responses, triggering the transcription of TNF-α, IL-1β and COX-2.12 In turn, TNFα or IL-1β can increase the transmission of pain signals by directly lowering the activation threshold for nociceptors.29,30 Astrocytic NF-κB can also strengthen plasticity-related events that increase the transmission of pain. For example, in a study assessing memory performance, mice expressing a dominant negative isoform of IκBα in astrocytes, thereby inhibiting astrocytic NF-κB activity, showed deficits in memory tasks that correlated with decreased expression of postsynaptic density protein 95 (PSD95).31 In the present study, we found that mice expressing a constitutively active form of IKK in astrocytes displayed increased pain in the CFA-injected ipsilateral paw that lasted throughout the duration of the two-week testing paradigm. Interestingly, the IKKca mice also displayed increased mechanical allodynia and hyperalgesia in the unaffected contralateral paw beginning during the subchronic phase of injury. This may be a result of a phenomenon known as spreading.32 It should also be noted, though, that repetitive mechanical stimuli, as used in this study, may result in increased activation of spinally-located second-order sensory neurons, possibly a result of temporal summation.26,33 Given that IKKca and Co mice did not show significant differences in mechanical or thermal sensitivity at baseline or after IFA injection, a pro-inflammatory event, such as CFA administration, may be required for the emergence of a behavioral phenotype, in this case prolonged mechanical hypersensitivity. To our knowledge, this is the first gain-of-function study to demonstrate that enhanced astrocytic NF-κB activity prolongs the duration of inflammatory pain as well as its ability to spread contralaterally.
While NF-κB can influence the transcription of genes involved in inflammation and plasticity, another gene that contains a known κB binding site is COMT. Following NF-κB induction by TNFα, binding of p65 to the known P2-COMT promoter in a human astrocyte cell line (H4 cells) in vitro dampens COMT mRNA and protein expression.19 In line with these findings, we observed NF-κB-linked reductions in COMT expression at 6h and 1d following CFA in midbrain in rats and 1h and 1d following CFA in forebrain and midbrain in mice (Figure 5). COMT expression likely was not downregulated during the entirety of the inflammation period because NF-κB activity is known to fluctuate during an inflammatory event.34 COMT expression in brain normalized at later stages of inflammation. Furthermore, inflammation-linked COMT expression in mice and rats is tissue- and, in this case, brain region-specific.35 At present, few studies have assessed how COMT expression is regulated, particularly in the brain. Genetic studies have predicted that promoters for the COMT gene contain binding motifs for transcription factors including Sp1, AP-2 and NF-D. Furthermore, altered COMT expression in the forebrain has been associated with administration of mood stabilizing drugs, nutritional deficiencies, as well as a mechanism mediated by the estrogen receptor. 35
Figure 5.

Schematic depicting how NF-κB mediated transcription of TNFα may regulate COMT expression. Our in vivo data, together with in vitro findings from Tchivileva et al., 2009, suggest an NF-κB-dependent mechanism in which a pro-inflammatory stimulus such as TNFα or CFA can initiate NF-κB, thereby permitting nuclear translocation of p65, which subsequently binds to the P2 promoter of COMT and prevents its transcription.
Reductions in COMT expression can increase pain sensitivity in both humans and animals. In humans, decreased COMT expression and/or activity as a result of genetic variance have been linked to increased susceptibility to temporomandibular joint disorders20 and fibromyalgia.36,37 In animals, decreased COMT activity, as a result of pharmacological inhibition, increases pain sensitivity.22,38 Heightened pain sensitivity persists after the cessation of COMT inhibitor administration, suggesting that even transient reductions in COMT expression, such as those observed in brain in this study, may result in heightened pain states.39 In addition to increasing pain sensitivity, decreased COMT activity also results in the release of pro-inflammatory molecules including TNFα, which is required for the development of COMT-dependent pain.23 As discussed earlier, TNFα may also contribute to NF-κB activation and downstream alterations in COMT expression, thereby driving COMT-dependent pain. Indeed, these studies highlight the complexity of overlapping regulatory pathways that may be involved in the potentiation of pain and/or COMT expression.
Here, we demonstrate that COMT protein expression is altered as a result of NF-κB- mediated inflammation. At present, drugs that block NF-κB activity, such as IκB inhibitors,5 or indirectly block the initiation of NF-κB, such as the TNFα antagonist etanercept, are currently in use for individuals experiencing chronic pain conditions such as fibromyalgia and rheumatoid arthritis. Thus, future studies will assess how NF-κB and/or COMT play a role in mediating pain behaviors in other models of inflammatory and neuropathic pain. Furthermore, given that sex differences appear to affect the etiology of inflammatory pain, it will be important to test this relationship in females.
5. Conclusion
This study highlights the role of NF-κB in regulating inflammatory pain and COMT expression in the CNS. Pharmacological inhibition (which quiets NF-κB activity) attenuates inflammatory pain, while constitutive activation of IKK in astrocytes (which enhances NF-κB activity) leads to lasting inflammatory pain. Negatively correlated with pain phenotypes, COMT expression was decreased in animals with greater NF-κB activity. This is the first demonstration that inflammation can influence COMT expression in the brain in an NF-κB-dependent method.
Supplementary Material
Supplementary Figure 1. Co and IKKca mice exhibit heightened (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia behaviors at 1h and 6h following IFA. There are no group differences in IFA-induced pain behavior, which resolves by day 1 or 3. Data are mean ± SEM.
Rats receiving a systemic NF-κB inhibitor exhibit reduced CFA inflammatory pain.
Mice with constitutively active astrocytic NF-κB exhibit lasting inflammatory pain.
COMT expression decreased in animals with greater NF-κB activity.
The first evidence that inflammation influences COMT expression in brain via NF-κB.
Acknowledgments
We thank Dr. Ken McCarthy and his laboratory for the IKKca mice used in this study. This work was funded by the NIH/NINDS R01NS072205 to A.G.N., NIH/NINDS R01NS072205S1 to F.A.O., and NIH/NIDCR T90DE021986 to J.E.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Bibliography
- 1.Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol. 2009;1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–99. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Lee KM, et al. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur J Neurosci. 2004;19:3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- 4.Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–7. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- 5.Niederberger E, Geisslinger G. The IKK-NF-kappaB pathway: a source for novel molecular drug targets in pain therapy? FASEB J. 2008;22:3432–42. doi: 10.1096/fj.08-109355. [DOI] [PubMed] [Google Scholar]
- 6.Marok R, et al. Activation of the transcription factor nuclear factor-KB in human inflamed synovial tissue. Arthritis Rheumatol. 1996;39:583–591. doi: 10.1002/art.1780390407. [DOI] [PubMed] [Google Scholar]
- 7.Sarchielli P, et al. NF-kappaB activity and iNOS expression in monocytes from internal jugular blood of migraine without aura patients during attacks. Cephalalgia. 2006;26:1071–9. doi: 10.1111/j.1468-2982.2006.01164.x. [DOI] [PubMed] [Google Scholar]
- 8.Lee MK, et al. Behavioral evidence for the differential regulation of p-p38 MAPK and p-NF-kappaB in rats with trigeminal neuropathic pain. Mol Pain. 2011;7:57. doi: 10.1186/1744-8069-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma W, Bisby MA. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 1998;797:243–254. doi: 10.1016/s0006-8993(98)00380-1. [DOI] [PubMed] [Google Scholar]
- 10.Wu LC, Goettl VM, Madiai F, Hackshaw KV, Hussain SR. Reciprocal regulation of nuclear factor kappa B and its inhibitor ZAS3 after peripheral nerve injury. BMC Neurosci. 2006;7:4. doi: 10.1186/1471-2202-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ledeboer A, et al. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur J Neurosci. 2005;22:1977–86. doi: 10.1111/j.1460-9568.2005.04379.x. [DOI] [PubMed] [Google Scholar]
- 12.Niederberger E, et al. Impaired acute and inflammatory nociception in mice lacking the p50 subunit of NF-kappaB. Eur J Pharmacol. 2007;559:55–60. doi: 10.1016/j.ejphar.2006.11.074. [DOI] [PubMed] [Google Scholar]
- 13.Luo JG, et al. Activation of spinal NF-kappaB/p65 contributes to peripheral inflammation and hyperalgesia in rat adjuvant-induced arthritis. Arthritis Rheumatol. 2014;66:896–906. doi: 10.1002/art.38328. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed AS, et al. Proteasome inhibitor MG132 modulates inflammatory pain by central mechanisms in adjuvant arthritis. International Journal of Rheumatic Diseases. 2014 doi: 10.1111/1756-185X.12353. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 15.Inoue G, et al. Injection of nuclear factor-kappa B decoy into the sciatic nerve suppresses mechanical allodynia and thermal hyperalgesia in a rat inflammatory pain model. Spine. 2006;31:2904–2908. doi: 10.1097/01.brs.0000248424.46652.67. [DOI] [PubMed] [Google Scholar]
- 16.Sakaue G, et al. NF-kappa B decoy suppresses cytokine expression and thermal hyperalgesia in a rat neuropathic pain model. Neuroreport. 2001;12:2079–2084. doi: 10.1097/00001756-200107200-00008. [DOI] [PubMed] [Google Scholar]
- 17.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu ES, Zhang YP, Sagen J, Yang ZQ, Bethea JR. Transgenic glial nuclear factor-kappa B inhibition decreases formalin pain in mice. Neuroreport. 2008;18:713–717. doi: 10.1097/WNR.0b013e3280d9e869. [DOI] [PubMed] [Google Scholar]
- 19.Tchivileva IE, et al. Characterization of NF-kB-mediated inhibition of catechol-O-methyltransferase. Mol Pain. 2009;5:13. doi: 10.1186/1744-8069-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diatchenko L, et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14:135–43. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 21.Smith SB, et al. Epistasis between polymorphisms in COMT, ESR1, and GCH1 influences COMT enzyme activity and pain. Pain. 2014;155:2390–9. doi: 10.1016/j.pain.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nackley AG, et al. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both β2- and β3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartung JE, Ciszek BP, Nackley AG. beta2- and beta3-adrenergic receptors drive COMT-dependent pain by increasing production of nitric oxide and cytokines. Pain. 2014;155:1346–55. doi: 10.1016/j.pain.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain (Amsterdam) 16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 25.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–73. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 26.Ringkamp M, et al. Lumbar sympathectomy failed to reverse mechanical allodynia-and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain. 1999;79:143–153. doi: 10.1016/s0304-3959(98)00186-9. [DOI] [PubMed] [Google Scholar]
- 27.Fecho K, Nackley AG, Wu Y, Maixner W. Basal and carrageenan-induced pain behavior in Sprague-Dawley, Lewis and Fischer rats. Physiol Behav. 2005;85:177–86. doi: 10.1016/j.physbeh.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 28.Vu HY, et al. Proteasome inhibitors induce apoptosis of prostate cancer cells by inducing nuclear translocation of IκBα. Arch Biochem Biophys. 2008;475:156–63. doi: 10.1016/j.abb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gudes S, et al. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-alpha-mediated increase in nociceptors excitability. J Neurophysiol. 2015;113:601–19. doi: 10.1152/jn.00652.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Binshtok AM, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–73. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bracchi-Ricard V, et al. Astroglial nuclear factor-kappaB regulates learning and memory and synaptic plasticity in female mice. J Neurochem. 2008;104:611–23. doi: 10.1111/j.1471-4159.2007.04993.x. [DOI] [PubMed] [Google Scholar]
- 32.Radhakrishnan R, Moore SA, Sluka KA. Unilateral carrageenan injection into muscle or joint induces chronic bilateral hyperalgesia in rats. Pain. 2003;104:567–77. doi: 10.1016/s0304-3959(03)00114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrero JF, Cervero F. Changes in nociceptive reflex facilitation during carrageenan-induced arthritis. Brain Research. 1996;717:62–68. doi: 10.1016/0006-8993(95)01585-x. [DOI] [PubMed] [Google Scholar]
- 34.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 35.Nissinen E. Basic aspects of Catechol-O-methyltransferase and the clinical applications of its inhibitors. Elsevier Science; 2010. [Google Scholar]
- 36.Gursoy S, et al. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23:104–7. doi: 10.1007/s00296-002-0260-5. [DOI] [PubMed] [Google Scholar]
- 37.Barbosa FR, et al. Influence of catechol-O-methyltransferase (COMT) gene polymorphisms in pain sensibility of Brazilian fibromialgia patients. Rheumatol Int. 2012;32:427–30. doi: 10.1007/s00296-010-1659-z. [DOI] [PubMed] [Google Scholar]
- 38.Kambur O, et al. Inhibitors of catechol-O-methyltransferase sensitize mice to pain. Br J Pharmacol. 2010;161:1553–65. doi: 10.1111/j.1476-5381.2010.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartung JE, Nackley AG. 127. B2-and B3-adrenergic receptors drive the development of COMT-dependent pain by increasing release of NO and innate immune cytokines. Brain, Behavior, and Immunity. 2014;40(Supplement):e37. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Co and IKKca mice exhibit heightened (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia behaviors at 1h and 6h following IFA. There are no group differences in IFA-induced pain behavior, which resolves by day 1 or 3. Data are mean ± SEM.