

Abstract
Amphetamine (AMPH) and its more potent enantiomer S(+)AMPH are psychostimulants used therapeutically to treat attention deficit hyperactivity disorder and have significant abuse liability. AMPH is a dopamine transporter (DAT) substrate that inhibits dopamine (DA) uptake and is implicated in DA release. Furthermore, AMPH activates ionic currents through DAT that modify cell excitability presumably by modulating voltage-gated channel activity. Indeed, several studies suggest that monoamine transporter-induced depolarization opens voltage-gated Ca2+ channels (CaV), which would constitute an additional AMPH mechanism of action. In this study we co-express human DAT (hDAT) with Ca2+ channels that have decreasing sensitivity to membrane depolarization (CaV1.3, CaV1.2 or CaV2.2). Although S(+)AMPH is more potent than DA in transport-competition assays and inward-current generation, at saturating concentrations both substrates indirectly activate voltage-gated L-type Ca2+ channels (CaV1.3 and CaV1.2) but not the N-type Ca2+ channel (CaV2.2). Furthermore, the potency to achieve hDAT-CaV electrical coupling is dominated by the substrate affinity on hDAT, with negligible influence of L-type channel voltage sensitivity. In contrast, the maximal coupling-strength (defined as Ca2+ signal change per unit hDAT current) is influenced by CaV voltage sensitivity, which is greater in CaV1.3- than in CaV1.2-expressing cells. Moreover, relative to DA, S(+)AMPH showed greater coupling-strength at concentrations that induced relatively small hDAT-mediated currents. Therefore S(+)AMPH is not only more potent than DA at inducing hDAT-mediated L-type Ca2+ channel currents but is a better depolarizing agent since it produces tighter electrical coupling between hDAT-mediated depolarization and L-type Ca2+ channel activation.
Keywords: Monoamine transporters, neurotransmitter transport, stimulants, serotonin, MDMA, excitability, L-type Ca2+ channels
Graphical Abstract
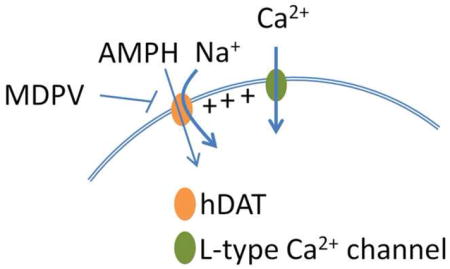
1. Introduction
The dopamine transporter (DAT) is a Na+/Cl−-dependent symporter expressed in dopaminergic neurons; its principal function is to limit dopamine receptor signaling by restricting the extracellular concentration of dopamine (DA) [1, 2]. Amphetamine (AMPH) is a DAT substrate and its more potent enantiomer, S(+)AMPH, is used therapeutically to treat attention deficit hyperactivity disorder and narcolepsy [2, 3]. AMPH competes with and diminishes DA uptake. In addition, intracellular AMPH disrupts DA’s internal stores and induces the reverse transport of DA through DAT, increasing extracellular DA concentration [4, 5]. Accordingly, the activation of dopaminergic pathways in the brain accounts for both the therapeutic properties and addictive liability of AMPH and its active derivatives [2, 6, 7].
An additional level of complexity for AMPH’s action in cells is the generation of DAT-mediated, AMPH-induced inward currents [8–11]. Although substrate-induced currents through monoamine transporters are widely accepted [12–15] and they have been implicated in neurotransmitter depletion in the brain [16], the physiological significance of such currents are still under debate [17, 18]. Recently, we showed that depolarization induced by serotonin (5HT) or S(+)3,4-methylenedioxymethamphetamine (MDMA, ecstasy) in skeletal muscle cells engineered to express the human serotonin transporter (hSERT) activates the L-type Ca2+ channel CaV1.1 [19]. Similarly, hSERT-mediated depolarization activates the L-type Ca2+ channel CaV1.3 in HEK cells, whereas hSERT activation is unable to open the N-type Ca2+ channel CaV2.2 under identical experimental conditions [19]. The L-type Ca2+ channels are important modulators of signal transduction and excitability in excitable cells. In particular, CaV1.3 and CaV1.2 have been extensively studied upstream of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cAMP response element-binding protein (CREB) signaling pathways in neurons [20–22]. Furthermore, the lower-threshold L-type CaV1.3 channel is implicated in pace-making in dopaminergic neurons, and in neuroendocrine cells, such as adrenal chromaffin cells [23, 24]. Since L-type channels CaV1.2 and CaV1.3 are expressed with monoamine transporters in several excitable cells [23–29], determining a functional interaction between these two classes of proteins could constitute an additional molecular mechanism of AMPH action.
In the present study we co-expressed the human DAT (hDAT) with CaV1.2, CaV1.3 or CaV2.2 in Flp-InTM T-RExTM 293 cells, and measured the effect of S(+)AMPH- or DA-induced DAT currents on CaV activation. These experiments were designed to study the interplay between two variables: 1) the affinity of S(+)AMPH and DA on hDAT, and 2) the voltage sensitivity of the Ca2+ channels studied, in achieving effective hDAT-CaV coupling. The results show that, regardless of the compound affinity on hDAT, DA and S(+)AMPH can couple indirectly to both L-type channels (CaV1.2 and CaV1.3) but not to the N-type channel (CaV2.2) under identical conditions. In addition, whereas the potency to achieve hDAT-CaV electrical coupling is dominated by substrate-hDAT affinity, the coupling-strength, defined as the Ca2+ signal change per unit hDAT current, is influenced by the sensitivity of Ca2+ channels to voltage. Moreover, S(+)AMPH showed larger coupling-strength compared to DA at concentrations that induced relatively small hDAT-mediated currents. These results suggest that S(+)AMPH- and DA-induced currents through hDAT are qualitatively different, because the S(+)AMPH-induced current is pharmacologically and electrically stronger at activating L-type channels.
2. Materials and methods
Generation of Flp-InTm T-RExTM cells expressing the human dopamine transporter (Flp-hDAT cells) and CaV channel transfection
The generation of the hDAT stable inducible cell line (Flp-hDAT) was done using the Flp-InTm T-RExTM 293 system (Life Technologies). The hDAT cDNA (accession number: NM_001044) was subcloned into the pcDNA5/FRT/TO plasmid and the targeted single site recombination and cell selection were performed as described previously [19]. The Ca2+ channels used in this study were CaV2.2 (α1B, Addgene #26570), CaV1.3 (α1D, Addgene #26576), CaV1.2 (α1C accession number: NM_001136522), β3 (Addgene #26574) and α2δ1 (Addgene #26575). All these plasmids were kindly provided by Dr. Diane Lipscombe (Department of Neuroscience, Brown University, Providence, Rhode Island, USA) except CaV1.2, which was kindly provided by Dr. Manfred Grabner (Department of Medical Genetics, Molecular, and Clinical Pharmacology, Innsbruck Medical University, Innsbruck, Austria). The CaV1.2 cDNA was subcloned into the pcDNA6 expression plasmid thus all α1 subunitsare expressed under the same background vector. EGFP expression plasmid was used as a transfection marker. The cells were co-transfected with the DNA ratio α1:β3:α2δ1:EGFP = 1:1:1:0.2 using Fugene 6 (Promega) as the transfection reagent.
Immunofluorescence
Sample fixation and labeling was performed as described earlier [30]. The primary antibody used was a rat monoclonal-anti DAT (Santa Cruz Biotechnology, Cat# sc-32258) and the secondary antibody used was Alexa Fluor 555 goat anti-rat IgG (Invitrogen, Cat# A21434). The nuclei were stained with DAPI. The specimens were visualized in a Zeiss 710 confocal microscope.
[3H]DA Uptake
Flp-DAT cells were counted and 1 × 106 cells were exposed to different concentrations of DA where 1% of the total concentration consisted of [3H]DA. The uptake reaction was performed for 10 min at 37°C in an external solution containing (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4. Non-specific uptake was determined adding 10 μM methylenedioxypyrovalerone (MDPV, a potent hDAT blocker) [31, 32]. After the incubation period, cells were centrifuged, washed once with PBS, centrifuged again and the cell pellets were resuspended in Ecoscint H (National Diagnostics, Atlanta, GA, USA); radioactivity was measured in a liquid scintillation counter.
Dose response-experiments were fit to the following expression:
| Eq. 1 |
Where x is the concentration of the tested compound, Y(x) is the response measured, Ymax is the maximal response, EC50 is the concentration that yields half-maximal response, and n is the Hill slope parameter. Competition assays were carried out adding a variable concentration of cold DA or cold S(+)AMPH to a constant 10 μM DA solution containing 1% [3H]DA. The inhibition constant (Ki) was estimated using the Cheng-Prusoff equation.
Electrophysiology
Determination of hDAT substrate-induced currents
Patch pipettes made from borosilicate glass capillary tubing and coated with Sylgard were filled with the following internal solution (in mM): 133 K Gluconate, 5.9 NaCl, 1 CaCl2, 0.7 MgCl2, 10 EGTA, 10 HEPES, pH adjusted to 7.2 with KOH. In this condition the pipettes showed a tip resistance of ~4 MΩ. The external solution used was (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4. Patch-clamp recordings were performed under constant perfusion at 35°C (AutoMate Scientific) and currents were acquired using an Axopatch 200A amplifier, Digidata 1322A acquisition system and Clampex 8.2 software (Molecular Devices); current traces were acquired at 1 kHz at −60 mV holding potential. Drugs were applied at various concentrations following a 30 μM dopamine pre-pulse. Holding currents for all traces were subtracted and divided by the DA pre-pulse peak current for cell to cell comparison.
Determination of Ca2+ currents
The Ca2+ currents were determined in HEK293T cells transfected with CaV1.2, CaV1.3 or CaV2.2 plus β3, α2δ1, and EGFP as described previously [19]. The external solution used was (in mM): 155 tetraethylammonium (TEA)-Cl, 5 CaCl2, 10 Hepes, pH 7.4 with TEA-OH. The internal solution composition was (in mM): 130 CsCl, 10 Cs-EGTA, 1 CaCl2, 4 MgATP and 10 HEPES, pH adjusted to 7.3 with CsOH. The effective serial resistance was corrected to 80% using the built-in circuit of the Axopatch 200B amplifier (remaining voltage error < 1.2 mV). The leak current was subtracted using a – P/6 protocol. The microelectrodes were made from 8520 glass capillary (Warner Instruments, #64-0817), fire polished, and Sylgard coated. The electrodes tip resistance was ~2.5 MΩ when filled with the internal solution. The whole-cell patch-clamp parameters of the recordings were: cell capacitance = 22.7 ± 2.3 pF, access resistance = 5.3 ± 0.4 MΩ, and time constant (τ) = 121 ± 16.4 μs (n = 23). The current was set to zero using the “pipette offset” command of the amplifier when the pipette was immersed in the external solution and no additional correction to the liquid-junction potential was performed. The recorded signals were acquired at 10 kHz and filtered at 5 kHz.
The voltage dependence of the ICa was fit to the following expression:
| Eq. 2 |
Where Gmax is the maximal conductance, V is the test potential, V1/2 is the potential at which G = 1/2 Gmax, k represent a slope parameter, and Vr is the reversal potential.
Determination of intracellular Ca2+
Ca2+ determinations were done using the Ca2+ sensitive dye Fura-2AM and visualized in an epifluorescence microscope following the procedure and using the equipment described previously [19]. The measurements were done with constant perfusion at 35°C using an external solution with composition (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4. The Fura-2 signal was acquired switching the excitation wavelength between 340/10 to 380/10 nm using a monochromator as described previously [19], dichroic mirror 490LP and an emission filter 510/40 nm. The acquisition frequency was 3 Hz. All images were background subtracted and the Ca2+ signals are shown as F340/380/F0. For dose-response experiments the test values were normalized by the mean of the maximal value of control DA pulses.
Statistics
The data are expressed as mean ± s.e.m. Comparison between two groups of data were made by unpaired two-tailed t-test, and when multiple groups were analyzed, one-way ANOVA followed by Tukey’s post test was used; p < 0.05 was considered significant.
3. Results
Immunostaining in conjunction with confocal microscopy showed membrane localization of hDAT in Flp-hDAT cells three days after doxycycline induction, whereas the parental Flp-InTM T-RExTM 293 (Flp-In) cells showed no hDAT expression (insert, Fig. 1A). In addition, Flp-hDAT cells have specific [3H]DA uptake (EC50 = 2.73 ± 0.49 μM, Fig. 1A). Uptake competition assay using cold S(+)AMPH or cold DA yielded inhibition constants (Ki) equal to 0.24 ± 0.03 and 2.06*** ± 0.67 μM respectively (*** = p < 0.001 t-test n ≥ 9, Fig. 1B).
Figure 1. S(+)AMPH is more potent than DA as an hDAT substrate.
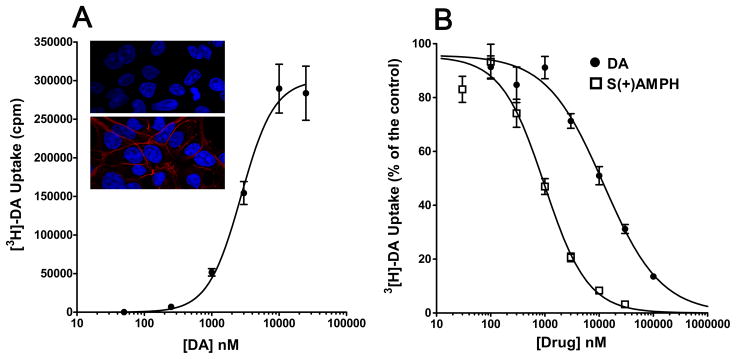
A. Confocal images show hDAT expression in Flp-hDAT cells (red stain in bottom panel) but not in parental Flp-InTm T-RExTM cells (top panel). DAPI nuclear staining is depicted in blue. Flp-hDAT cells show specific [3H] DA uptake with the following fitting parameters (see Eq. 1): EC50 = 2.73 ± 0.49 μM, Hill slope = 1.8 ± 0.5 (n = 18). B. Competition of [3H]DA uptake using cold dopamine or S(+)AMPH yielded the following fitting parameters (Eq. 1): IC50 = 12.05 ± 1.79 μM, Hill Slope = 0.8 ± 0.1 (n = 9) and IC50 = 0.98*** ± 0.12 μM, Hill slope = 1.1 ± 0.1 (n = 11, ***p < 0.001 vs. IC50 DA competition, t-test) for DA and S(+)AMPH, respectively.
To determine the extent to which S(+)AMPH- or DA-induced currents in hDAT could activate voltage-gated Ca2+ channels, we measured Ca2+ signals using the Ca2+ sensitive dye Fura-2 in Flp-hDAT cells transfected with CaV1.2, CaV1.3, or CaV2.2. The perfusion of saturating concentrations of DA (10 μM) or S(+)AMPH (5 μM) for 5 s at 35°C induced equivalent Ca2+ signals in Flp-hDAT cells expressing CaV1.3 (CaV1.3 in Fig. 2). Similar results were obtained when Flp-hDAT cells expressing CaV1.2 were exposed to these agents (CaV1.2 in Fig. 2). The CaV1.3- and CaV1.2-mediated signals were blocked by isradipine (2 μM), a potent L-type Ca2+ channel blocker (CaV1.3 and CaV1.2 in Fig. 2). In contrast, the intracellular Ca2+ concentration did not change in Flp-hDAT cells expressing CaV2.2 when exposed to DA or S(+)AMPH under the same conditions. The sequential exposure to high-K+ external solution yielded a convincing Ca2+ transient demonstrating adequate expression of CaV2.2 on these cells (CaV2.2 in Fig. 2). In control assays, in which the α1 subunit of CaV channels is absent in the transfection mix, neither DA, S(+)AMPH nor high-K+ mobilized Ca2+ in Flp-hDAT cells (control in Fig. 2).
Figure 2. S(+)AMPH or DA activates CaV1.2 and CaV1.3, but not CaV2.2.

(Upper and middle panel) Intracellular Ca2+ determinations in Fura-2AM loaded Flp-hDAT cells evaluated by fluorescence microcopy, under constant perfusion and at 35°C. Flp-hDAT cells were co-transfected with CaV1.3, CaV1.2 or CaV2.2 plus β3, α2δ and EGFP plasmids. The α1 subunit was omitted from the plasmid transfection mix for the control condition. Transfected cells were identified by their EGFP signal and then briefly exposed to dopamine 10 μM (DA), S(+)AMPH 5 μM, high potassium external solution 130 mM (K+, equimolar substitution of Na+) or 4Br-A23187 (calcium ionophore, 5 μM) as indicated in the timeline of each panel. Isradipine (2 μM) averts Ca2+ signals induced by both hDAT substrates. Each trace constitutes the mean ± s.e.m. of n ≥ 81 cells per condition. (Lower panel) Voltage dependence of CaV1.2, CaV1.3 and CaV2.2- mediated Ca2+ currents: HEK293T cells were co-transfected with β3, α2δ, and EGFP expression plasmids plus alternatively CaV1.3, CaV1.2 or CaV2.2 plasmids. The Ca2+ current (ICa) recordings were carried out at room temperature under constant perfusion. Test pulses in 5 mV steps for CaV2.2 or 10 mV steps for CaV1.2 and CaV1.3 were applied from a holding potential of −80 mV. Representative responses are shown for CaV1.3 (light grey circle), CaV1.2 (dark grey triangle) and CaV2.2 (black square) and the magnitude of the test potentials are indicated in mV. The peak current density for the voltage steps were fit to Eq. 2 and yielded the following parameters: Gmax = 497 ± 86, 560 ± 128 and 631 ± 77 (pS/pF); V1/2 = −25.6 ± 1.0, −3.2 ± 0.8 and 5.5 ± 1.0 mV (***p < 0.001, one-way ANOVA, indicated in the figure); k = 6.7 ± 0.2, 7.7 ± 0.2 and 4.7 ± 0.1 (mV) for CaV1.3 (n = 8), CaV1.2 (n = 7) and CaV2.2 (n = 8), respectively.
To study the voltage sensitivity of Ca2+ channels in more detail, CaV1.2, CaV1.3, or CaV2.2 were transiently transfected in HEK293T cells and Ca2+ currents were measured under whole-cell voltage-clamp (Fig. 2). In our experimental conditions the test potential that yielded half of the maximal conductance (V1/2 in Eq. 2, see experimental procedures section) was −25.6 ± 1.0 mV (n = 8), −3.2 ± 0.8 mV (n = 7) and +5.5 ± 1.0 mV (n = 8) for CaV1.3, CaV1.2 and CaV2.2, respectively (Fig. 2). These results show that although these channels have been categorized as “high-voltage activated” [33], they have notable differences in their response to changes in membrane potential.
To rule out a direct activation of CaV channels by DA or S(+)AMPH, intracellular Ca2+ was determined in parental Flp-In (no hDAT expression) expressing the L-type Ca2+ channels. Neither DA nor S(+)AMPH induce changes in resting Ca2+ level in these cells, whereas high K+-induced Ca2+ transients reveal the normal expression of Ca2+ channels (Fig. 3A and 3B). MDPV is an abused drug that was found to exert long-lasting inhibitory action of the substrate- induced-DAT conductance and DAT-transport. Indeed, MDPV has higher potency than cocaine blocking both hDAT-mediated currents and DA transport [31, 32, 34]. The perfusion of MDPV (1 μM) abolished S(+)AMPH- or DA-induced Ca2+ transients in Flp-hDAT cells expressing either CaV1.2 or CaV1.3 (Fig. 3C and 3D); whereas high K+-induced Ca2+ signals were refractory to MDPV treatment. Li+ ions leak through monoamine transporters in the absence of substrates, and the inward current carried by Li+ can be as large as the current induced by substrates in Na+-based external solution [8, 12, 14, 35]. To test whether depolarization mediated by hDAT currents is responsible for CaV activation, cells expressing the CaV channels were briefly exposed to an external solution in which Na+ was replaced with equimolar Li+. As expected, Li+ induced Ca2+ transients in Flp-hDAT cells expressing CaV1.2 or CaV1.3 but not in parental Flp-In cells expressing Ca2+ channels. In contrast, high-K+ solution produced Ca2+ signals in parental Flp-In and in Flp-hDAT cells expressing these Ca2+ channels (Fig. 3E and 3F). Together these results strongly suggest that depolarization is the cause of activation of L-type Ca2+ channels in Flp-hDAT cells exposed to hDAT substrates.
Figure 3. hDAT-mediated depolarization activates L-type Ca2+ channels in Flp-hDAT cells.

Intracellular Ca2+ concentration was determined by fluorescence microscopy in Flp-hDAT or the parental Flp-InTM T-RExTM 293 (Flp-In) cells (no hDAT expression) cells co-transfected with CaV1.2 (A, C, E) or CaV1.3 (B, D,F) plus β3, α2δ and EGFP plasmids, using the Ca2+ sensitive dye Fura-2AM. The experiments were carried out under constant perfusion at 35°C. The transfected cells were identified by their EGFP signal. A, B. Cells were briefly exposed to DA 10 μM, S(+)AMPH 5 μM or high potassium external solution 130 mM (K+) as indicated in each panel. C, D. The blockade of hDAT with methylenedioxypyrovalerone (MDPV, 1 μM) prevented Ca2+ signals induced by hDAT substrates. E, F. Cells were exposed to external solution containing Li+ (equimolar substitution of Na+) or external solution with high potassium as indicated in the timeline of each panel. Li+-induced Ca2+ signals only take place in cells expressing hDAT. Traces represent the mean ± s.e.m. of n ≥ 30 cells per condition.
Dose-response experiments measuring Ca2+ signals in Flp-hDAT cells expressing CaV1.2 or CaV1.3 showed that S(+)AMPH has greater potency than DA at activating both L-type Ca2+ channels (Fig. 4). In addition, although the difference between DA-elicited EC50 values in CaV1.2- and CaV1.3-expressing cells showed statistical significance (916 ± 54.3 nM vs 693 ± 25.0 nM, p < 0.001 one way-ANOVA, Fig. 4), this small difference is not expected to be biologically relevant. For S(+)AMPH, the EC50 values were not significantly different in cells expressing CaV1.2 or CaV1.3 (Fig. 4). These data show that S(+)AMPH is more potent than DA at achieving electrical coupling between hDAT and L-type Ca2+ channels. Moreover, channel voltage sensitivity, measured in Fig. 2, only modestly influences the compound potency (EC50) to activate the hDAT- L-type Ca2+ channel electrical coupling.
Figure 4. S(+)AMPH is more potent than DA producing Ca2+ signals in L-type Ca2+ channel- expressing Flp-hDAT cells.

Intracellular Ca2+ signals were monitored using the calcium sensitive dye Fura-2AM and epifluorescence microscopy. Three days prior to each experiment Flp-hDAT cells were co-transfected with CaV1.3 or CaV1.2 plus β3, α2δ and EGFP plasmids. The EGFP was used as transfection marker. (A, B, C, D) The potency of each compound was calculated using a two-pulse protocol, where one fixed saturating concentration pulse of dopamine works as an internal calibration and a variable concentration pulse was applied to get a dose-response curve. The traces represent the mean ± s.e.m. of n ≥ 48 cells per concentration. E. The dose-response curve was obtained fitting the responses to the Eq.1; cells expressing CaV1.3 the EC50 values and Hill slope were: 693 ± 25 and 102*** ±16 nM (***p < 0.001 one way ANOVA) and 2.5 ± 0.2 and 0.9*** ± 0.1 (***p < 0.001 one way ANOVA) for dopamine and S(+)amphetamine, respectively. Cells expressing CaV1.2 the EC50 values and Hill slope were: 916 ± 54 and 144*** ± 11 nM (***p < 0.001 one way ANOVA) and 1.7 ± 0.2 and 1.2 ± 0.1 for DA and S(+)AMPH, respectively, and (F) for better comparison the EC50 values are plotted.
The activation of the L-type Ca2+ channels described above requires membrane depolarization. In this study the membrane depolarization is mediated by substrate-induced hDAT currents (IhDAT). To study how IhDAT activates CaV1.2 and CaV1.3, the concentration-dependence of IhDAT was measured. S(+)AMPH had greater potency than DA inducing inward currents in Flp-hDAT cells clamped to −60 mV, (Fig. 5); however, S(+)AMPH and DA produced comparable current amplitudes at saturating concentrations (Fig. 5) similar to a previous report [36] (non-normalized currents are shown as supplementary material). To study the relationship between Ca2+ signals and hDAT inward currents, the Ca2+ signal amplitudes fitted in Fig. 4E were plotted against the fitted hDAT-inward currents of Fig. 5C, which were both evoked by the same compound concentrations (Fig. 5D). Whereas DA generated sigmoidal curves for both CaV1.2 and CaV1.3, S(+)AMPH generated a quasi hyperbolic shape for CaV1.3 and a slightly sigmoidal curve for CaV1.2 (Fig. 5D). These curves show that S(+)AMPH, compared to DA, generates a relatively larger Ca2+ transient in response to smaller hDAT inward currents. To better visualize this difference, the first derivative of the curves shown in Fig. 5D is plotted in Fig. 5E. The value of the slope at every IhDAT defines the “coupling-strength” since it quantifies the sensitivity of the system to generate a Ca2+ response per unit hDAT inward current. Regardless of the compound tested, CaV1.3-expressing cells showed higher maximal coupling-strength than CaV1.2-expressing cells (Fig. 5E) suggesting that CaV1.3, which is more sensitive to voltage than CaV1.2 (Fig. 2), requires less depolarization mediated by IhDAT to open. Lastly, S(+)AMPH consistently showed higher coupling-strength in response to smaller IhDAT magnitudes than DA for both L-type Ca2+ channels (Fig. 5E) suggesting that S(+)AMPH- induced currents are more effective than DA-induced currents at depolarizing and activating L-type Ca2+ channels.
Figure 5. S(+)AMPH-induced currents are electrically favored to activate L-type Ca2+ channels.

Ionic currents were determined by voltage-clamp in whole cell configuration under constant perfusion at 35°C. (A and B) Flp-hDAT cells clamped at −60 mV were exposed to a constant DA calibration pulse and a variable pulse of DA or S(+)AMPH; representative traces for the indicated concentrations tested are depicted in each panel. C. The full dose-response curves were fit to Eq.1 and yield the following parameters: EC50 = 1.44 ± 0.24 μM and 0.28** ± 0.04 μM (**p < 0.01, t-test) and Hill Slope = 1.1 ± 0.1 and 1.0 ± 0.1 for DA and S(+)AMPH, respectively. Each point indicates mean ± s.e.m. of n ≥ 5 current determinations for each concentration. D. The fitted S(+)amphetamine and dopamine Ca2+ signals on Fig. 4E were plotted as a function of the fitted hDAT currents and (E) the coupling-strength index was computed as the first derivative of the Ca2+ signal – hDAT current curves.
4. Discussion
Voltage-gated Ca2+ channels are composed of the main α1 subunit and the auxiliary α2δ, β and γ subunits [37]. The α1 subunit contributes to the ionic pore and voltage sensor structures, while the others modulate expression, targeting, and function [33, 38, 39]. Neurons and neuroendocrine cells express several α1 isoforms. The biophysical properties, location and biochemical partners of the α1 subunits regulate Ca2+ influx for specific purposes. For example, the opening of L-type Ca2+ channels mobilize Ca2+, which in turn activates signaling pathways and gene expression in neurons [21], and Ca2+ entry through CaV2.2 is coupled to neurotransmitter release [40, 41]. The α1 isoforms differ in their voltage sensitivity, for instance, CaV3 (T-type) channels are activated by slight depolarization (low-voltage activated), whereas CaV1 (L-type) and CaV2 (N-, P/Q- and R- type) channels activate upon strong depolarization (high-voltage activated) [33]. Despite this categorization, a broad range of voltage sensitivity exists between CaV1 and CaV2 channels; as shown in Fig. 2, CaV1.3 is more sensitive to depolarization than CaV1.2 [42] and CaV2.2 is the least sensitive channel [43]. For cells expressing monoamine transporters most, if not all, co-express CaV1.3 and CaV1.2, a functional interplay between transporters and channels is plausible. Indeed, DAT-mediated Ca2+ channel activation has been described in neurons [44] and AMPH activates Ca2+ channels by a NET-dependent mechanism in PC12 cells [45]. Moreover, DAT-mediated currents depolarize neurons [9] and at low concentrations DAT substrates increase electrical excitability of dopaminergic neurons through a mechanism dependent on DAT’s substrate-induced current [46, 47]. Interestingly, CaV1.3 is involved in excitability, accordingly CaV1.3-knockout mice showed altered pace-making activity in ventral tegmental area dopaminergic neurons [23], leading to the hypothesis that monoamine transporter currents may modulate excitability in neurons through a mechanism that involves L-type Ca2+ channels. In addition affecting excitability, as mentioned above, L-type Ca2+ channels are also involved in signaling pathway activation. One example of interest is CaMKII, which has been well characterized as an effector of Ca2+ currents downstream of L-type Ca2+ channels [21, 22]. Interestingly, DAT is a CaMKII substrate and phosphorylated DAT favors the reverse transport of dopamine [48, 49], constituting a possible mechanism by which electrical activity and L-type Ca2+ channels may modulate DAT states and dopamine release.
Membrane depolarization induced by monoamine transporters could potentially modulate any voltage-gated channel including not only Ca2+, but also Na+ and K+ channels. The functional interaction between transporters and voltage-gated channels described here can depend on several factors such as relative levels of protein expression and clustering of the implicated proteins in membrane microdomains. In addition, biophysical characteristics of the voltage-gated channels, such as voltage sensitivity, and activation/inactivation kinetics, may be decisive factors in the effective electrical coupling with transporters. For instance, tetrodotoxin-sensitive voltage-gated Na+ channels were not involved in the membrane depolarization induced by serotonin or MDMA in myotubes expressing hSERT since the small but sustained hSERT-mediated depolarization may inactivate the fast Na+ channels [19]. In another example, the high-voltage-activated Ca2+ channel CaV2.2 is not activated by substrate-induced depolarization through hSERT or hDAT in our heterologous expression system ([19] and Fig. 2), probably because greater depolarization is required for its activation. Case-specific studies would be required to characterize the modulation of specific voltage-gated channels by transporters in native systems.
In the present manuscript we explore how hDAT substrates could modulate Ca2+ channel activity in cells; in particular, DAT-mediated substrate-induced membrane depolarization could reach the level of CaV channel activation. We tested the functional interaction between hDAT and a single CaV channels using heterologous expression, which would be difficult to achieve in native systems. Experiments in Fig. 2 and Fig. 3 show that DA- and AMPH- can induce Ca2+ signals in cells expressing L-type CaV channels, but these signals required the simultaneous expression of functional hDAT protein. Thus, neither DA nor AMPH can activate L-type Ca2+ channels directly (Fig. 3A). Accordingly, both DA and AMPH induced reproducible inward (depolarizing) currents in Flp-hDAT cells (Fig. 5A and 5B), suggesting that depolarization is the cause of L-type CaV channel opening upon hDAT activation.
It is well known that hDAT has constitutive conductance to Li+ in the absence of substrate [8, 12, 14, 35]. Since the intracellular concentration of this ion is zero, the Nernst equilibrium potential for Li+ during high-Li+ perfusion is extremely positive. Thus, the cell membrane would depolarize in cells expressing hDAT when exposed to Li+. As shown in Fig 3C, Flp-hDAT cells must undergo strong depolarization when exposed to Li+, as is evidenced by activation of CaV channels. These data clearly show that hDAT inward current can activate CaV channels even in the absence of DA or AMPH, reinforcing the idea that CaV channel activation is a consequence of the membrane depolarization and not a direct action of DA or AMPH at CaV channels.
Similarly to hSERT [19], hDAT substrate-induced currents are coupled to CaV1.3 channel activation. Unexpectedly, CaV1.2 despite its ~20 mV right shift in voltage dependence compared to Cav1.3 (Fig. 2) showed robust electrical coupling with hDAT (Fig. 2). On the other hand, CaV2.2, which has a ~30 mV right shift in the voltage dependence compared to CaV1.3, showed no Ca2+ channel activation (Fig. 2). These results strongly suggest that L-type but not N-type Ca2+ channels are within the activation range of hDAT-mediated depolarization under our experimental conditions. Interestingly, the significant difference in voltage sensitivity between CaV1.2 and CaV1.3 plays a little role in the potency (EC50) by which S(+)AMPH or DA activates the channels via hDAT mediated depolarization (Fig. 4). These results suggest that the hDAT mediated depolarization only activates Ca2+ channels when the activation threshold falls within the range of depolarization induced by IhDAT. In addition the potency at which a hDAT substrate activate the overall coupling process is dominated by the affinity of the substrate at hDAT.
To quantify the strength required for DA- or S(+)AMPH-induced currents to depolarize the cell membrane and activate Ca2+ channels, we introduced the coupling-strength index, which measures the Ca2+ signal induced per unit IhDAT. Cells expressing CaV1.3 consistently showed higher coupling-strength than CaV1.2-expressing cells for both hDAT substrates tested. These observations are consistent with the higher voltage sensitivity of CaV1.3 over CaV1.2. At lower IhDAT the activated Ca2+ conductance may favor further depolarization, thus potentiating the coupling-strength, whereas at higher IhDAT amplitudes internal Ca2+ may induce channel inactivation decreasing the coupling-strength. In any case, the coupling-strength would decrease at higher substrate concentration because the system would reach saturation. The coupling-strength vs. IhDAT curve (Fig. 5E) is left-shifted for S(+)AMPH compared to DA indicating that S(+)AMPH-induced currents are better at activating Ca2+ channels than DA at lower hDAT current amplitudes, further supporting the idea that the S(+)AMPH-induced current is qualitatively different than the DA-induced current. These hDAT currents may differ in kinetics and ionic composition, which in turn could affect the coupling-strength; e.g., faster currents would recruit Ca2+ channels more quickly leading to greater coupling-strength. Time-resolved kinetics that use piezoelectric rapid solution exchange showed that AMPH produces an instantaneous peak current up to several times larger than the steady-state current. The DA-induced current has similar kinetics profile, however, its peak current amplitude is significantly smaller than the one seen for AMPH [50]. The amplitude of the instantaneous current may play a role in the coupling-strength of hDAT substrates. The ionic composition of the currents induced by hDAT substrates is not fully resolved: substrate-induced currents appear to involve both Na+ and Cl− conductance. In the context of the HEK cell environment, the presumably low intracellular Cl− concentration and the resting membrane potential of ~−50 mV [51] renders depolarization due to increased Cl− conductance unlikely. We and others have recorded substrate-induced inward currents at −60 mV in low-chloride internal solution ([52] and Fig. 5). Under these ionic conditions, the Cl− current would be outward, and thus, Na+ must be the charge carrier. In some neurons, higher internal Cl− concentration, high input resistance and lower resting membrane potential would account for depolarization induced by elevation in Cl− permeability [53]. Indeed, Cl− conductance through DAT is implicated in depolarization of midbrain rat dopaminergic neurons and in Caenorhabditis elegans dopaminergic neurons [9, 46]. It is not clear whether different substrates can induce currents with different ionic composition through hDAT but, in any case, conditions in which an inward current is increased would lead to higher coupling-strength due to stronger depolarization.
In summary, our results suggest that pharmacologically, S(+)AMPH is more potent than DA at activating hDAT-mediated depolarizing currents, leading to L-type Ca2+ channel activation, and the S(+)AMPH-induced current is more tightly coupled than DA to open L-type Ca2+ channels.
Supplementary Material
Highlights.
Amphetamine (AMPH) and dopamine (DA) induce depolarizing currents through the DA transporter (DAT).
DAT substrate-induced depolarization activates L-type but not N-type Ca2+ channels (CaV).
AMPH is more potent than DA when activating L-type CaV.
The activation of L-type CaV displays higher coupling-strength with AMPH- than DA-induced currents.
AMPH action likely involves L-type CaV activation.
Acknowledgments
The authors would like to thank Dr. Richard A. Glennon (Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA) for insightful discussions and for providing methylenedioxypyrovalerone used in this work. This study was supported by the National Institute of Health R01 DA033930 (L.J.D.).
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–12. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 2.Seeman P, Madras BK. Anti-hyperactivity medication: methylphenidate and amphetamine. Mol Psychiatry. 1998;3:386–96. doi: 10.1038/sj.mp.4000421. [DOI] [PubMed] [Google Scholar]
- 3.Mignot E. Perspectives in narcolepsy research and therapy. Curr Opin Pulm Med. 1996;2:482–7. [PubMed] [Google Scholar]
- 4.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–33. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–98. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 6.Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9:557–69. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- 7.Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol. 2007;64:1575–9. doi: 10.1001/archneur.64.11.1575. [DOI] [PubMed] [Google Scholar]
- 8.Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997;17:960–74. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvelli L, McDonald PW, Blakely RD, Defelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci U S A. 2004;101:16046–51. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeFelice LJ, Goswami T. Transporters as channels. Annu Rev Physiol. 2007;69:87–112. doi: 10.1146/annurev.physiol.69.031905.164816. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Menchaca AA, Solis E, Jr, Cameron K, De Felice LJ. S(+)amphetamine induces a persistent leak in the human dopamine transporter: molecular stent hypothesis. British journal of pharmacology. 2012;165:2749–57. doi: 10.1111/j.1476-5381.2011.01728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mager S, Min C, Henry DJ, Chavkin C, Hoffman BJ, Davidson N, Lester HA. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12:845–59. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 13.Galli A, Blakely RD, DeFelice LJ. Norepinephrine transporters have channel modes of conduction. Proc Natl Acad Sci U S A. 1996;93:8671–6. doi: 10.1073/pnas.93.16.8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin F, Lester HA, Mager S. Single-channel currents produced by the serotonin transporter and analysis of a mutation affecting ion permeation. Biophys J. 1996;71:3126–35. doi: 10.1016/S0006-3495(96)79506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonders MS, Amara SG. Channels in transporters. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 16.Baumann MH, Bulling S, Benaderet TS, Saha K, Ayestas MA, Partilla JS, Ali SF, Stockner T, Rothman RB, Sandtner W, Sitte HH. Evidence for a role of transporter-mediated currents in the depletion of brain serotonin induced by serotonin transporter substrates. Neuropsychopharmacology. 2014;39:1355–65. doi: 10.1038/npp.2013.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quick MW. Regulating the conducting states of a mammalian serotonin transporter. Neuron. 2003;40:537–49. doi: 10.1016/s0896-6273(03)00605-6. [DOI] [PubMed] [Google Scholar]
- 18.Sulzer D, Galli A. Dopamine transport currents are promoted from curiosity to physiology. Trends Neurosci. 2003;26:173–6. doi: 10.1016/S0166-2236(03)00063-8. [DOI] [PubMed] [Google Scholar]
- 19.Ruchala I, Cabra V, Solis E, Jr, Glennon RA, De Felice LJ, Eltit JM. Electrical coupling between the human serotonin transporter and voltage-gated Ca(2+) channels. Cell calcium. 2014;56:25–33. doi: 10.1016/j.ceca.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Ca1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–63. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–24. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Harding M, Pittman A, Dore J, Striessnig J, Rajadhyaksha A, Chen X. Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J Neurophysiol. 2014;112:1119–30. doi: 10.1152/jn.00757.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2010;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durante P, Cardenas CG, Whittaker JA, Kitai ST, Scroggs RS. Low-threshold L-type calcium channels in rat dopamine neurons. J Neurophysiol. 2004;91:1450–4. doi: 10.1152/jn.01015.2003. [DOI] [PubMed] [Google Scholar]
- 26.Phillips JK, Dubey R, Sesiashvilvi E, Takeda M, Christie DL, Lipski J. Differential expression of the noradrenaline transporter in adrenergic chromaffin cells, ganglion cells and nerve fibres of the rat adrenal medulla. J Chem Neuroanat. 2001;21:95–104. doi: 10.1016/s0891-0618(00)00113-7. [DOI] [PubMed] [Google Scholar]
- 27.Sukiasyan N, Hultborn H, Zhang M. Distribution of calcium channel Ca(V)1.3 immunoreactivity in the rat spinal cord and brain stem. Neuroscience. 2009;159:217–35. doi: 10.1016/j.neuroscience.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Takada M, Kang Y, Imanishi M. Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Eur J Neurosci. 2001;13:757–62. doi: 10.1046/j.1460-9568.2001.01435.x. [DOI] [PubMed] [Google Scholar]
- 29.Schroeter S, Levey AI, Blakely RD. Polarized expression of the antidepressant-sensitive serotonin transporter in epinephrine-synthesizing chromaffin cells of the rat adrenal gland. Mol Cell Neurosci. 1997;9:170–84. doi: 10.1006/mcne.1997.0619. [DOI] [PubMed] [Google Scholar]
- 30.Eltit JM, Li H, Ward CW, Molinski T, Pessah IN, Allen PD, Lopez JR. Orthograde dihydropyridine receptor signal regulates ryanodine receptor passive leak. Proc Natl Acad Sci U S A. 2011;108:7046–51. doi: 10.1073/pnas.1018380108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cameron KN, Kolanos R, Solis E, Jr, Glennon RA, De Felice LJ. Bath salts components mephedrone and methylenedioxypyrovalerone (MDPV) act synergistically at the human dopamine transporter. British journal of pharmacology. 2013;168:1750–7. doi: 10.1111/bph.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolanos R, Solis E, Jr, Sakloth F, De Felice LJ, Glennon RA. “Deconstruction” of the abused synthetic cathinone methylenedioxypyrovalerone (MDPV) and an examination of effects at the human dopamine transporter. ACS chemical neuroscience. 2013;4:1524–9. doi: 10.1021/cn4001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–5. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 34.Baumann MH, Partilla JS, Lehner KR, Thorndike EB, Hoffman AF, Holy M, Rothman RB, Goldberg SR, Lupica CR, Sitte HH, Brandt SD, Tella SR, Cozzi NV, Schindler CW. Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology. 2013;38:552–62. doi: 10.1038/npp.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borre L, Andreassen TF, Shi L, Weinstein H, Gether U. The second sodium site in the dopamine transporter controls cation permeation and is regulated by chloride. J Biol Chem. 2014;289:25764–73. doi: 10.1074/jbc.M114.574269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C. Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem. 1998;71:1289–97. doi: 10.1046/j.1471-4159.1998.71031289.x. [DOI] [PubMed] [Google Scholar]
- 37.Szpyt J, Lorenzon N, Perez CF, Norris E, Allen PD, Beam KG, Samso M. Three-dimensional localization of the alpha and beta subunits and of the II-III loop in the skeletal muscle L-type Ca2+ channel. J Biol Chem. 2012;287:43853–61. doi: 10.1074/jbc.M112.419283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–25. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 39.Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82:24–45. doi: 10.1016/j.neuron.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 40.Currie KP. Inhibition of Ca2+ channels and adrenal catecholamine release by G protein coupled receptors. Cell Mol Neurobiol. 2010;30:1201–8. doi: 10.1007/s10571-010-9596-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mochida S, Yokoyama CT, Kim DK, Itoh K, Catterall WA. Evidence for a voltage-dependent enhancement of neurotransmitter release mediated via the synaptic protein interaction site of N-type Ca2+ channels. Proc Natl Acad Sci U S A. 1998;95:14523–8. doi: 10.1073/pnas.95.24.14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–41. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- 43.Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–51. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaarmann A, Gandhi S, Gourine AV, Abramov AY. Novel pathway for an old neurotransmitter: dopamine-induced neuronal calcium signalling via receptor-independent mechanisms. Cell Calcium. 2010;48:176–82. doi: 10.1016/j.ceca.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Kantor L, Zhang M, Guptaroy B, Park YH, Gnegy ME. Repeated amphetamine couples norepinephrine transporter and calcium channel activities in PC12 cells. J Pharmacol Exp Ther. 2004;311:1044–51. doi: 10.1124/jpet.104.071068. [DOI] [PubMed] [Google Scholar]
- 46.Ingram SL, Prasad BM, Amara SG. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat Neurosci. 2002;5:971–8. doi: 10.1038/nn920. [DOI] [PubMed] [Google Scholar]
- 47.Branch SY, Beckstead MJ. Methamphetamine produces bidirectional, concentration-dependent effects on dopamine neuron excitability and dopamine-mediated synaptic currents. J Neurophysiol. 2012;108:802–9. doi: 10.1152/jn.00094.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–29. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 49.Steinkellner T, Yang JW, Montgomery TR, Chen WQ, Winkler MT, Sucic S, Lubec G, Freissmuth M, Elgersma Y, Sitte HH, Kudlacek O. Ca(2+)/calmodulin-dependent protein kinase IIalpha (alphaCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem. 2012;287:29627–35. doi: 10.1074/jbc.M112.367219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erreger K, Grewer C, Javitch JA, Galli A. Currents in response to rapid concentration jumps of amphetamine uncover novel aspects of human dopamine transporter function. J Neurosci. 2008;28:976–89. doi: 10.1523/JNEUROSCI.2796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodwin JS, Larson GA, Swant J, Sen N, Javitch JA, Zahniser NR, De Felice LJ, Khoshbouei H. Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J Biol Chem. 2009;284:2978–89. doi: 10.1074/jbc.M805298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schicker K, Uzelac Z, Gesmonde J, Bulling S, Stockner T, Freissmuth M, Boehm S, Rudnick G, Sitte HH, Sandtner W. Unifying concept of serotonin transporter-associated currents. J Biol Chem. 2012;287:438–45. doi: 10.1074/jbc.M111.304261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.