

Abstract
Colorimetric protein assays, such as the Coomassie blue G-250 dye-binding (Bradford) and bicinchoninic acid (BCA) assays, are commonly used to quantify protein concentration. The accuracy of these assays depends on the amino acid composition. Because of the extensive use of reductive methylation in the study of proteins and the importance of biological methylation, it is necessary to evaluate the impact of lysyl methylation on the Bradford and BCA assays. Unmodified and reductively methylated proteins were analyzed using the absorbance at 280 nm to standardize the concentrations. Using model compounds, we demonstrate that the dimethylation of lysyl ε-amines does not affect the proteins’ molar extinction coefficients at 280 nm. For the Bradford assay, the response (absorbance per unit concentration) of the unmodified and reductively methylated proteins were similar with a slight decrease in the response upon methylation. For the BCA assay, the responses of the reductively methylated proteins were consistently higher, overestimating the concentrations of the methylated proteins. The enhanced color-formation in the BCA assay may be due to the lower acid dissociation constants of the lysyl ε-dimethylamines, compared to the unmodified ε-amine, favoring Cu(II) binding in biuret-like complexes. The implications for the analysis of biologically methylated samples are discussed.
Keywords: protein, quantitation, Bradford assay, bicinchoninic acid assay, methylation, lysine
1.1 Introduction
The Coomassie blue G-250 dye-binding (Bradford) and bicinchoninic acid (BCA) assays are common colorimetric assays used to determine the concentrations of proteins [1-3]. Both assays produce a colored solution in the visible spectrum in response to protein. The color formation observed in the Bradford assay is a result of interactions between the protein and the Coomassie blue G-250 dye (Fig. 1a). Under acidic conditions, the dye is red in its protonated state. Through electrostatic and hydrophobic interactions with a protein molecule, the anionic (−1 net charge) blue form of the dye is stabilized [4-7]. In the BCA assay, color formation is the result of two reactions. First, cupric ions (Cu+2) are reduced to cuprous ions (Cu+) by the protein under alkaline conditions via the biuret reaction [8]. Then, a complex between Cu+ and two molecules of bicinchoninic acid forms, which gives the characteristic purple color (Fig. 1b) [2]. The intensity of the color formed by these assays is measured by absorbance photometry at 595 nm and 562 nm for the Bradford and BCA assays, respectively. Typically, standard solutions of bovine serum albumin (BSA) are used to produce a calibration curve of absorbance versus mass concentration. Assuming the analyte-proteins react in the same manner as the BSA standard, the unknown concentration can be determined. Unfortunately, the assumption that the assay sensitivity or response (absorbance per unit concentration) is universal is not always valid, and protein-to-protein variability can lead to an over- or under-estimation of the analyte-protein’s concentration [9].
Fig. 1.
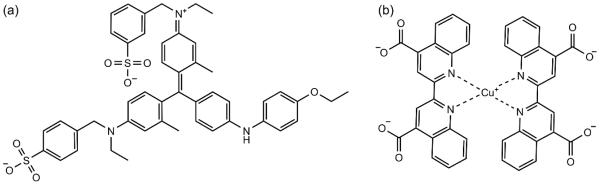
The structures of (a) the Coomassie blue G-250 dye in its blue, anionic (−1 net charge) form and (b) the purple, bicinchoninic acid2-Cu+ complex.
Protein composition can bias the results of Bradford and BCA assays. In the Bradford assay, the dye-protein interaction is influenced by the electrostatic interactions of the sulfonate groups with the basic residues, arginine and lysine [4-5]. An additional factor is the hydrophobic interactions of the dye with tryptophan, phenylalanine, and tyrosine residues [4,10]. Proteins that are largely hydrophobic and/or have a high proportion of arginine and lysine residues give higher absorbance values than the same mass concentration of a protein with less hydrophobic character and/or fewer basic residues. Similarly, the BCA assay can be influenced by the amino acid composition of the analyte-protein. Cysteine-cystine, tyrosine, and tryptophan residues have been shown to participate in the reduction of Cu2+ to Cu+ ions [11]. Proteins that contain a high proportion of these reactive residues will produce higher responses. Differences in the composition of the proteins and the standard, typically BSA, can result in inaccurate concentration determinations.
The influence of amino acid composition on protein concentration assays also includes effects from post-translational modifications (PTMs). PTMs alter the chemical composition of a protein with the addition or cleavage of a functional group and are important for protein regulation and function. Glycosylation is a PTM where a mono- or polysaccharide is added to a protein, typically at asparagine, serine, or threonine residues. Fountoulakis et al. investigated the variability of the Bradford, BCA, and Lowry assays in quantifying several glycosylated proteins in comparison to the unmodified protein [10]. They concluded that glycosylation interfered with the assay responses, resulting in an under-estimation of the glycoprotein concentration with the Bradford assay and an over-estimation of the glycoprotein concentration with the BCA and Lowry assays [10].
Lysine methylation is a common PTM and can be mimicked using the reductive methylation reaction [12]. Lysine methylation is important for protein regulation and signaling [13] and can exist as mono-, di-, or tri-methylation of the ε-amino group. Mono- and di-methylation of lysine residues can be produced chemically using the reductive methylation reaction. Reductive methylation selectively incorporates methyl groups at the lysyl ε-amino groups and the N-terminal α-amino group without disturbing the structure, and often activity, of the protein [12,14]. Formaldehyde reacts with the primary amines to form Schiff bases, which are subsequently reduced to form methyl groups with a reducing reagent, such as dimethylamine-borane complex (DMAB) [15]. In the presence of excess reagents, the reaction adds two methyl groups to each primary amine. Reductive methylation is commonly used in X-ray crystallography because the modification rarely alters the protein’s overall structure and aids crystallization [14]. The modification is also useful for studying proteins by nuclear magnetic resonance (NMR) spectroscopy because 13C-formaldehyde can be used to add 13C-methyl groups as isotopic labels [16-17]. The 13C-methyl groups are sensitive probes for investigating structural and dynamic properties of proteins and their complexes by NMR [16,18-39]. Because of the extensive use of reductive methylation in the study of proteins and the natural occurrence of methylated proteins, it is necessary to evaluate the impact of methylation on the response of commonly used concentration assays. In this study, we investigate the effect of lysine methylation on the determination of protein concentration by the Bradford and BCA assays. Amino acid composition and hydrophobicity can bias the responses of the Bradford and BCA assays, so reductive methylation may likewise alter the accuracy of these assays. Unmodified and reductively methylated hen egg white lysozyme (HEWL), BSA, and ovalbumin were used to test and compare the assays’ responses.
1.2 Experimental
1.2.1 Materials
Acetonitrile, ammonium sulfate, BSA (catalog number A7030), DMAB, formaldehyde, HEWL (catalog number l6876), albumin from chicken egg white (ovalbumin, catalog number A5503, Grade V), potassium phosphate (monobasic and dibasic), sinapinic acid, trifluroacetic acid, L-lysine hydrochloride, and Nε,Nε-dimethyl-L-lysine hydrochloride, anhydrous caffeine, and sodium 3-(trimethylsilyl)-1-propanesulfonic acid (DSS) were purchased from Sigma Aldrich. The BCA assay kit and Coomassie Plus (Bradford) assay reagent were purchased from Fischer Scientific/Pierce. Corning UV-transparent half-area 96-well plates were purchased from VWR. Deuterated water (D2O) was purchased from Cambridge Isotope Laboratories. Bio-Gel P-4 size exclusion chromatography resin was purchased from Bio-Rad. All water used was supplied from a Millipore Direct-Q 3 ultrapure water system.
1.2.2 Sample Pathlength Determination
Because a microplate spectrophotometer was used to measure the absorbance, the pathlength varied with the sample volume. The method of McGown et al. was used to determine the sample pathlength and is an option in the Gen5 software for the BioTek PowerWave XS2 microplate spectrophotometer used in this study [40]. Briefly, the absorbance of water at 977 nm and 900 nm are subtracted and divided by the known absorbance of water at 977 nm and 1 cm (0.180) to give the pathlength in cm. The pathlength of replicate samples of water at 100, 120, and 200 μL were measured in a UV-transparent half-area 96-well plate at 25 or 37 °C. The average pathlength values and 95% confidence intervals are summarized in the supplementary data in Table S1. The average pathlength values were used to normalize the measured absorbances to 1 cm.
1.2.3 Extinction Coefficients at 280 nm
To determine if lysine methylation added significantly to the overall absorbance at 280 nm (A280nm) of a protein, the molar extinction coefficients at 280 nm (ε280’s) of L-lysine (Lys) and Nε,Nε-dimethyl-L-lysine (DM-Lys) were determined in phosphate-buffered saline (PBS) buffer (50 mM potassium phosphate, 100 mM NaCl, pH 7.5). Solutions (50 g/L) of Lys and DM-Lys were prepared by weighing at least 10.0 mg of material and dissolving in the appropriate amount of buffer. The A280nm was measured using 100 μL of sample in a 96-well plate with a BioTek PowerWave XS2 microplate spectrophotometer at 25 °C. The absorbance values were corrected for the background absorbance of buffer and normalized to a 1 cm pathlength using the average pathlength of 100 μL of water (0.568 ± 0.002 cm). To correct for differences in the moisture-content of Lys and DM-Lys, the concentration of each solution was determined using quantitative NMR with an internal standard, anhydrous caffeine [41-42]. A 50.0 mM stock solution of caffeine was prepared in D2O. NMR samples were prepared by diluting the stock solutions to make approximately 2 mM Lys or DM-Lys, 1.00 mM caffeine, and 0.01% DSS (chemical shift reference) in D2O. The samples were analyzed using a 400 MHz Bruker instrument at 25 °C using a relaxation delay of 2 s, an acquisition time of 4 s, a 30° pulse width, and 8 scans. The areas of the caffeine methyl peaks and the Lys or DM-Lys methylene and Hα peaks were determined by integration. The relative peak area was used to calculate the concentration of Lys and DM-Lys in each stock solution. The pathlength-corrected A280nm’s were divided by the concentrations to give the molar ε280’s of Lys and DM-Lys in PBS buffer in units of M−1cm−1.
The mass ε280’s were determined for each unmodified protein in water and in PBS buffer at 25 °C. Stock solutions of each unmodified protein (HEWL, BSA, and ovalbumin) were prepared in triplicate at a concentration of 1.00 g/L by weighing at least 10.0 mg of protein and dissolving in an appropriate volume of water or PBS buffer. Aliquots of the stock solutions were added to a 96-well plate and diluted to a final volume of 100 μL with water or PBS buffer. The final concentrations for BSA and ovalbumin were 0, 200, 400, 600, 800, and 1000 mg/L and for HEWL were 0, 100, 200, 300, 400 and 400 mg/L. These concentration ranges were chosen to ensure that the maximum absorbance values were less than 1.0. The A280nm was measured with a BioTek PowerWave XS2 microplate spectrophotometer after incubating the samples at 25 °C for 5 minutes. The absorbance values were corrected for the background absorbance of water or buffer and normalized to a 1 cm pathlength using the average pathlength of 100 μL of water (0.568 ± 0.002 cm). The slope, intercept, and correlation coefficient (R2) of the absorbance versus the concentration was calculated by fitting the data to a linear equation using the least squares method. Because the absorbance values were corrected for a pathlength of 1 cm, the slope values give the mass ε280’s in units of g−1 L cm−1. The average ε280’s and 95% confidence intervals were calculated for each protein in water and in PBS buffer. The molar ε280’s were calculated using the average molecular weight of the proteins determined by matrix assisted laser desorption ionization mass spectrometry (MALDI MS).
1.2.4 Reductive Methylation
Stock solutions of unmodified HEWL, BSA, and ovalbumin were prepared at a concentration of 5 g/L in 50 mM potassium phosphate buffer, pH 7.5. Reductive methylation of all proteins was performed using 500 μL aliquots of the protein stock solutions. Stock solutions of DMAB (1.0 M) and formaldehyde (1.0 M) were prepared fresh. An aliquot of DMAB was added to each protein in a 20:1 molar ratio (DMAB to protein amino groups) followed by an aliquot of formaldehyde in a 40:1 molar ratio (formaldehyde to protein amino groups). The samples were incubated at 4 °C while shaking for 2 hours. After 2 hours, a second aliquot of DMAB (20:1) and formaldehyde (40:1) were added, followed by a second 2 hour incubation with shaking. A final aliquot of DMAB (10:1) was added to each sample, and the samples were incubated overnight at 4 °C while shaking for a total reaction time of 24 hours. After 24 hours, the reactions were quenched by adding ammonium sulfate to a final concentration of 10 mM. The reductively methylated samples were then exchanged into PBS buffer using a Bio-Gel P4 size exclusion column (7 mL of media) at a flow-rate of 0.2 mL/min. The eluting protein was monitored by absorbance photometry at 280 nm using a Bio-Rad BioLogic DuoFlow F10 Workstation, and the fractions with reductively methylated protein were pooled.
The concentration of the reductively methylated samples was calculated by measuring the A280nm and using the determined ε280’s in PBS buffer for the unmodified proteins. The A280nm of each reductively methylated sample was analyzed in triplicate by diluting 50 μL aliquots of the pooled fractions to 200 μL with PBS buffer in a 96-well plate. Buffer blanks and water were run in triplicate (200 μL). After correcting for the blank absorbance of the buffer, correcting for dilution, and normalizing to a 1 cm pathlength using the average pathlength of 200 μL of water (1.102 ± 0.002 cm), the average absorbance values were divided by the ε280’s to give the concentrations of the reductively methylated samples.
1.2.5 MALDI MS Sample Preparation and Analysis
MALDI MS analysis of the unmodified and reductively methylated proteins was performed to measure the extent of methylation. The proteins (approximately 1 mg) were desalted into water using a Bio-Gel P-4 size exclusion column (7 mL of media) at a flow-rate of 0.2 mL/min. A volume containing approximately 100 pmol of protein (as determined by the A280nm) was dried using a centrifugal evaporator (Savant SPD121P Speedvac Concentrator) and reconstituted in 2 μL of matrix (a saturated solution of sinapinic acid in 30% acetonitrile, 0.1% trifluoroacetic acid). The protein-matrix mixture (1 μL) was spotted on a MALDI target (MTP 384, Bruker) and allowed to dry under ambient conditions. Unmodified and reductively methylated samples were analyzed using a Bruker UltrafleXtreme operating in linear mode with a mass-to-charge ratio (m/z) range of 5,000-70,000 Da. A nitrogen laser was used at 89-92% power, and 2000 laser shots were averaged for each spectrum. Using Origin software, the spectra were smoothed using either the Savitzky-Golay method (5 coefficients, 2nd order polynomial; BSA and HEWL) or a 0.05 Hz low pass, fast Fourier transform filter (ovalbumin). To determine the molecular weights of the proteins, the peaks were either fitted to a Gaussian curve (BSA and ovalbumin) using nonlinear regression analysis or the apex value was used (HEWL).
1.2.6 Bradford Assay
Protein solutions were made at 50.0 mg/L from the stock solutions of unmodified and reductively methylated protein. Aliquots of the 50.0 mg/L solutions (2, 4, 8, 12, 16, and 20 μL) were added to a 96-well plate, and PBS buffer was added to bring the total sample volume to 20 μL. Blank samples of 20 μL of buffer were prepared in triplicate. Coomassie reagent (100 μL) was added to each well, mixed by shaking, and incubated at 25 °C for 5 min. The absorbance at 595 nm (A595nm) was measured with a BioTek PowerWave XS2 microplate spectrophotometer. The absorbance values were corrected by subtracting the average absorbance of the blank samples and normalized by dividing by the average pathlength of 120 μL of water (0.679 ± 0.003 cm). The normalized absorbance values were plotted versus the mass concentration (g/L). The data were fitted to a linear equation using the least squares method. Since the absorbance values were corrected to a 1 cm pathlength, the slopes are equal to the assay responses in units of g−1 L cm−1. The average response and 95% confidence interval were calculated for each sample.
1.2.7 BCA Assay
Protein solutions were made at 250 mg/L from the stock solutions of unmodified and reductively methylated protein solutions. Aliquots of the 250 mg/L solutions (2, 4, 8, 12, 16, and 20 μL) were added to a 96-well plate, and PBS buffer was added to bring the total sample volume to 20 μL. Blank samples of 20 μL of buffer were prepared in triplicate. Fresh BCA reagent was prepared following the manufacturer’s instructions, and 100 μL was added to each well, mixed by shaking, and incubated at 37 °C for 30 min. The absorbance at 562 nm (A562nm) was measured immediately with a BioTek PowerWave XS2 microplate spectrophotometer. The absorbance values were corrected by subtracting the average absorbance of the blank samples and normalized by dividing by the average pathlength of 120 μL of water (0.679 ± 0.003 cm). The normalized absorbance values were plotted versus the mass concentration (g/L). The data were fitted to a linear equation using the least squares method. Since the absorbance values were corrected to a 1 cm pathlength, the slopes are equal to the assay responses in units of g−1 L cm−1. The average response and 95% confidence interval were calculated for each sample.
1.3 Results and Discussion
1.3.1 Reductive Methylation and MALDI-MS Analysis
Unmodified and reductively methylated proteins were analyzed with MALDI MS and compared to assess the extent of methylation. A 100% conversion to the dimethylated species would indicate that all lysines and the N-terminus of the protein had undergone the addition of two methyl groups at the ε-amino groups and the α-amino group. Fig. 2 shows the mass spectra of unmodified and reductively methylated BSA, HEWL, and ovalbumin. The unmodified mass, average mass of the reductively methylated samples, and average fraction methylated is reported in Table 1. BSA and HEWL were reductively methylated reproducibly with 86 ± 3% and 100 ± 1% methylation, respectively, based on mass shifts. Ovalbumin is a mixture of cleaved and full-length proteins, each with an N-linked glycosylation site [43]. The ovalbumin samples consistently ionized poorly in MALDI MS, which is not unusual for glycoproteins. Electrospray ionization MS was attempted, but also gave signals with low signal-to-noise ratios (data not shown). Nonetheless, the data indicate that ovalbumin was highly methylated with percentages of 91 ± 2% and 77 ± 17% for the cleaved and full-length ovalbumin, respectively.
Fig. 2.

MALDI mass spectra of unmodified (a,e,i) and reductively methylated (b,c,d) BSA, (f,g,h) HEWL, and (j,k,l) ovalbumin showing the percentage of dimethylation, the theoretical shift of completely dimethylated protein (dashed gray lines), and the best fit (red lines) to a Gaussian curve for the BSA and ovalbumin samples.
Table 1.
The percentages of complete dimethylation and the molecular weights of unmodified and reductively methylated proteins determined with MALDI MS.
| Protein | Unmodified mass (Da) |
Average reductively methylated mass and standard deviation (Da) |
Change in mass (Da) |
Fraction methylated (%) |
|---|---|---|---|---|
| BSA | 66,165 | 67,611 ± 56 | 1,447 | 86 ± 3 |
| HEWL | 14,297 | 14,494 ± 2 | 197 | 100 ± 1 |
| Ovalbumin (cleaved) |
39,898 | 40,406 ± 12 | 508 | 91 ± 2 |
| Ovalbumin (full-length) |
44,190 | 44,645 ± 98 | 455 | 77 ± 17 |
1.3.2 Extinction Coefficients at 280 nm
The apparent mass ε280’s of the unmodified proteins at 25 °C were determined in water and in PBS buffer. Table 2 summarizes the mass ε280’s, 95% confidence intervals, the literature values reported by the manufacturers [44-47], and the molar ε280’s. The raw data for determining the ε280’s is in the supplementary data in Tables S2-S7. Overall, the apparent mass ε280’s of the protein samples were similar to those from the literature. The apparent ε280’s in buffer and water for BSA and ovalbumin were 10% less than the manufacturer’s values even though the protein content was ≥ 98% (according to the manufacturer). This difference may be due to differences in moisture-content or lot-to-lot variability. The only significant difference (p<0.05) between samples measured in water and in buffer was the apparent ε280’s for HEWL with a 12% difference (asterisk in Table 2). The molar ε280’s and propagated errors are also listed in Table 2 and were calculated by multiplying the mass ε280’s in PBS buffer with the mass of the proteins determined by MALDI MS (Table 1).
Table 2.
Mass and calculated molar extinction coefficients at 280 nm and 25 °C for BSA, HEWL, and ovalbumin [44-47].
| Protein | Literature ε280
(g−1 L cm−1) |
Mass ε280 in PBS (g−1 L cm−1) ± 95% CI |
Mass ε280 in water (g−1 L cm−1) ± 95% CI |
Molar ε280 in PBS (mM−1 cm−1) ± 95% CI |
|---|---|---|---|---|
| BSA | 0.667 [44] | 0.61 ± 0.06 | 0.61 ± 0.01 | 40 ± 4 |
| HEWL | 2.64 [45-46] | 2.26 ± 0.17* | 2.56 ± 0.14* | 32.3 ± 2.4 |
| Ovalbumin | 0.69 – 0.76 [47] | 0.63 ± 0.03 | 0.63 ± 0.02 | 26 ± 1 |
p = 0.004
After the reductive methylation reaction, the protein concentration must be empirically determined because some protein is lost to precipitation and/or adhesion to surfaces. If the dimethyl modifications do not contribute significantly to the A280nm, the ε280’s of the unmodified proteins could be used to determine the concentration of their reductively methylated counterparts. To test this assertion, the molar ε280’s of Lys and DM-Lys were determined and compared. Lys and DM-Lys stock solutions were prepared, and quantitative NMR was used to determine the molar concentrations to correct for moisture in the weighed samples. Integrated 1H NMR spectra of each compound, with caffeine as an internal standard, are shown in the supplementary data in Fig. S1. The blank and pathlength-corrected A280nm’s for the stock solutions were divided by the molar concentrations to give the molar ε280’s of Lys and DM-Lys in PBS buffer of 0.72 M−1cm−1 and 0.70 M−1cm−1, respectively. The magnitude and difference between the measured ε280’s are small compared to the molar ε280’s of the proteins, which range from 26 × 103 to 40 × 103 M−1cm−1 (Table 2). Even for BSA, which has 59 lysine residues, the difference between the A280nm’s of unmodified and reductively methylated BSA is less than 0.01%. These results confirm that the molar ε280’s of the unmodified proteins can be used to determine the concentration of the reductively methylated proteins.
The concentrations of the reductively methylated samples were determined by measuring the A280nm’s and dividing by the apparent ε280’s of the unmodified proteins in PBS buffer. Initially, stock solutions (50 and 250 mg/L) of the reductively methylated proteins were incorrectly made based on the calculated concentration using the mass ε280’s. The mass ε280 is widely reported for proteins because the exact mass of a protein is difficult to obtain (especially if the protein is a mixture), and the common colorimetric protein assays, including the Bradford and BCA assays, assume a composition-independent response. The A280nm of a protein, on the other hand, depends on the composition because the absorbance is primarily due to the number of tyrosine, tryptophan, and cystine residues [48]. Because the A280nm depends on the moles of protein and not the mass, the molar ε280’s of the unmodified and reductively methylated proteins is the same, but the mass ε280’s are different by the ratio of the molecular weights. Therefore, the stock solution concentrations were corrected using the ratio of the masses of the reductively methylated and unmodified proteins determined by MALDI MS (Table 1), effectively using the molar ε280nm’s instead of the mass ε280nm’s to determine the concentrations. The corrected concentrations for the 50 mg/L stock solutions are 50.69, 51.09, and 50.57 mg/L and for the 250 mg/L stock solutions are 253.44, 255.47, and 252.86 mg/L for BSA, HEWL, and ovalbumin, respectively.
1.3.3 Bradford Assay
The Bradford assay responses of the unmodified and reductively methylated proteins are summarized in Fig. 3. Typically, BSA is used as a standard for the Bradford assay, and a calibration curve based on the mass concentration of BSA is used to determine the unknown concentration of a protein. Using the Bradford assay in this manner assumes that the color intensity (A595nm) is composition-independent such that the mass sensitivity or response (g−1 L cm−1) of the assay is the same for every protein. Based on our results in Fig. 3, this assumption is valid for unmodified BSA and HEWL, which have similar responses of 46 ± 5 g−1 L cm−1 and 45 ± 3 g−1 L cm−1, respectively. Unmodified ovalbumin, however, has a significantly lower response of 33 ± 1 g−1 L cm−1. This result is not surprising because ovalbumin is a glycoprotein. A similar decrease in response for glycoproteins in general, and ovalbumin specifically (33% decrease), has already been demonstrated [10,49]. The color production in the Bradford assay occurs when the blue, anionic form of the dye is stabilized, typically through electrostatic and hydrophobic interactions. The ovalbumin glycan comprises approximately 3-4% of the total mass and is mainly composed of neutral, high-mannose- or hybrid-type oligosaccharides [50-51]. While the glycan mass alone does not explain the approximately 27% decrease in sensitivity, it is likely that the glycan competes with the dye by forming hydrogen bonds with basic amino acids, stacking with hydrophobic residues, and/or steric shielding [49]. In addition, ovalbumin is highly phosphorylated at two sites [51], which likely further decreases the affinity for the anionic dye.
Fig. 3.

Results of the Bradford assay plotted as the pathlength-normalized absorbance values (A595nm/cm) versus the protein concentration determined for triplicate samples of unmodified and reductively methylated (a) BSA, (b) HEWL, and (c) ovalbumin at various concentrations, and (d) a bar graph summarizing the Bradford assay responses (g−1 L cm−1, error bars = 95% confidence intervals) with brackets indicating the p-value between pairs of data.
With the Bradford assay, the reductively methylated proteins had similar responses as their unmodified counterparts (Fig. 3). The general trend was a lower response, but only BSA and reductively methylated BSA showed significantly different responses (p > 0.05) among the unmodified/reductively methylated pairs. A greater difference might be expected since lysine is one of the primary amino acids involved in dye binding [4-5,52], but the amine retains its positive charge allowing for electrostatic interactions. The small decrease in response can be explained by a slight change in the affinity of the amino groups for the Coomassie blue G-250 dye upon methylation. The methyl groups may sterically hinder the electrostatic interaction with the dye’s sulfonic groups or decrease the accessibility of the amine by binding hydrophobic groups on the protein. Reductively methylated BSA, in particular, shows a larger difference in response compared to unmodified BSA, likely because it has the highest density of lysine residues with 0.89 lysines per kDa of protein. HEWL and ovalbumin have nearly half this density with 0.42 and 0.46 lysines per kDa of protein, respectively, suggesting that the proportion of methylated lysines in the protein is important to consider when using the Bradford assay.
1.3.4 BCA Assay
The BCA assay responses of the unmodified and reductively methylated proteins are summarized in Fig. 4. The BCA assay responses of the unmodified proteins are 16.6 ± 2.0, 22.7 ± 0.4, and 15.1 ± 0.6 g−1 L cm−1 for BSA, HEWL, and ovalbumin, respectively. Unlike the Bradford assay responses, the BCA assay responses of BSA and ovalbumin are similar, and the response of HEWL is significantly higher. The color formation in the BCA assay is mainly due to Cu2+ reduction by cysteine, cystine, tyrosine, tryptophan, and the backbone amide groups, with the cysteine and cystine residues being the most reactive [2,11,53]. While the color formation cannot be predicted based on the sum of the individual color-producing components [11], in the case of BSA, HEWL, and ovalbumin, there is a correlation between the BCA assay response and the weight percentage of cysteine, cystine, tyrosine, and tryptophan residues (Fig. 5). The similar responses of ovalbumin and BSA are not consistent with results by Fountoulakis et al., where the BCA assay gave an overestimation of 148% for ovalbumin when BSA was used as the calibration standard [10]. The primary difference between our work and Fountoulakis et al. is the temperature of the BCA reaction; we incubated our samples at 37 °C and Fountoulakis et al. at 60 °C. While Fountoulakis et al. observed an overestimation of several glycoproteins using the BCA assay at 60 °C, Noble et al. observed an underestimation of the protein concentration for the glycoprotein RNase B compared to non-glycosylated RNase A using the BCA assay at 37 °C [49]. The temperature of the BCA reaction is clearly important for accurately measuring glycoprotein concentration. It has been shown that increasing the reaction temperature increases the participation of tyrosine, tryptophan, and the backbone amide groups in copper reduction [11], but it may also increase the participation of the glycans. While individual monosaccharides do not interfere strongly in the BCA assay [10], the polydentate effect of a polysaccharide may result in a significant interference at elevated temperatures.
Fig. 4.

Results of the BCA assay plotted as the pathlength-normalized absorbance values (A562nm/cm) versus the protein concentration determined for triplicate samples of unmodified and reductively methylated (a) BSA, (b) HEWL, and (c) ovalbumin at various concentrations, and (d) a bar graph summarizing the BCA assay responses (g−1 L cm−1, error bars = 95% confidence intervals) with brackets indicating the p-value between pairs of data.
Fig. 5.
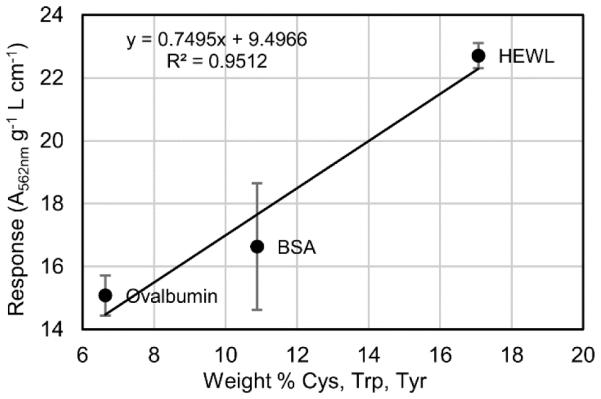
The BCA assay responses of BSA, HEWL, and ovalbumin versus the weight percentage of cysteine, cystine, tryptophan, and tyrosine residues in each protein with the best fit line equation, correlation coefficient (R2), and error bars representing the 95% confidence interval determined for each response from three replicates.
The BCA assay responses of the reductively methylated proteins are consistently higher than the responses of the unmodified proteins for all three test proteins. The responses of the reductively methylated proteins in the BCA assay were 22.4 ± 3.0, 30.1 ± 0.9, and 22.4 ± 2.1 g−1 L cm−1 for BSA, HEWL, and ovalbumin, respectively. As shown in Fig. 4, the responses for the unmodified and reductively methylated proteins increased by 32-49% upon methylation. This change in response may be due to an increase in the affinity of lysyl ε-dimethylamine, compared to lysyl ε-amine, for Cu2+. It has been shown that the lysine side-chain amino group can complex Cu2+ in a biuret-like complex [54-55] and even bridge copper ions [56-57], but the interaction has also been shown to be unfavorable and/or only occurring at alkaline pH [54,58-59]. Alkaline conditions are necessary for binding Cu2+ because the typical acid dissociation constant (pKa) of lysyl ε-amines in proteins is 11.0 ± 0.4 [60-61]. The BCA reaction pH is 11.25, so the lysyl ε-amines exist as protonated and deprotonated species (Fig. 6a). Tertiary amines typically have lower pKa’s than their corresponding primary amines [62], and this is the case for lysyl ε-dimethylamines in proteins. The typical pKa of lysyl ε-dimethylamines in proteins is 10.1 ± 0.3 [16,18,26,30]. This difference in the pKa’s is significant because a larger fraction of the ε-dimethylamines is in the deprotonated state compared to the unmodified ε-amines (Fig. 6a-b) at the BCA assay pH. The deprotonated ε-dimethylamine can form biuret-like complexes with Cu2+ (Fig. 6c) [55], leading to increased copper reduction.
Fig. 6.
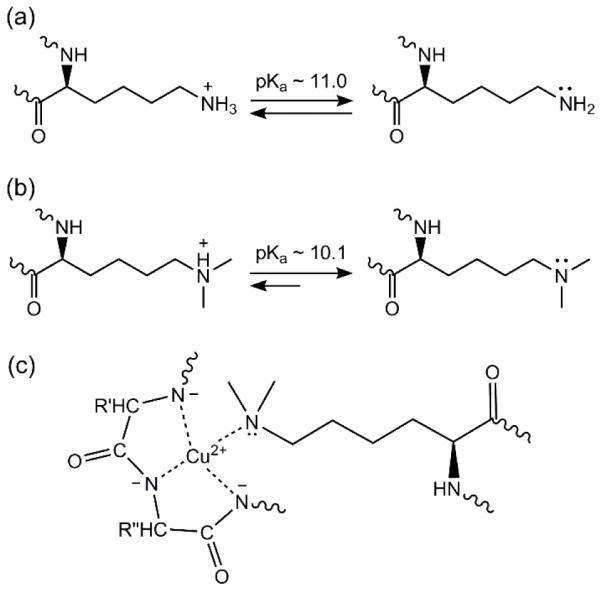
Acid dissociation equilibria of (a) lysyl ε-amines and (b) ε-dimethylamines favoring the deprotonated lysyl ε-dimethylamines under the alkaline conditions (pH 11.25) of the BCA assay, and (c) a model of lysyl ε-dimethylamine in a biuret-like complex with Cu2+ (R, R’, and R’’ represent the side-chains of three amino acids).
Dimethylation of the N-terminal α-amino group may also affect the BCA assay because the N-terminal α-amino group plays an important role in peptide-Cu2+ complexes, anchoring the Cu2+ ion as it chelates neighboring amide groups [55,63]. Unlike the lysyl ε-amines, the pKa of the α-amine is lower with a pKa of 7-8, so nearly all the α-amino- and α-dimethylamino-groups are in the deprotonated, neutral state during the BCA assay. The N-terminal α-amine forms a complex with Cu2+ through the lone pair of electrons on the nitrogen (Fig. 7a). It has been shown that the absence or modification of the N-terminal α-amine decreases a peptide’s affinity for Cu2+ [64], suggesting that reductive methylation of the N-terminal α-amine would decrease the BCA assay response. Alternatively, the small methyl groups may not sterically hinder binding to Cu2+ (Fig. 7b) and could contribute to binding by stabilizing the correct ligand geometry.
Fig. 7.
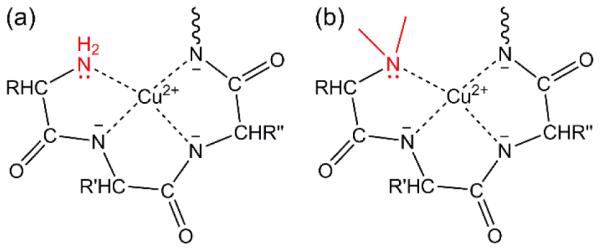
Biuret-like complexes of the N-terminal (a) α-amine and (b) α-dimethylamine with Cu2+ with the α-amino and α-dimethylamino groups colored in red as the anchors of quadridentate ligands (R, R’, and R’’ represent the side-chains of three amino acids).
1.4 Conclusions
This study demonstrates the importance of considering not only the amino acid composition, but also the composition of the post-translational or chemical modifications of a protein when using the Bradford or BCA assay. Reductive methylation can be used to uniformly modify proteins with dimethyl groups at the lysyl ε-amines and N-terminal α-amine. The assay responses to the unmodified proteins showed typical protein-to-protein variation. Results similar to previous studies were observed, including an underestimation of the glycoprotein, ovalbumin, with the Bradford assay and a dependence on the amino acid composition (cysteine, cystine, tyrosine, and tryptophan residues) with the BCA assay. The Bradford assay produced more consistent responses between the unmodified and reductively methylated proteins than the BCA assay. The Bradford assay responses were somewhat smaller for the reductively methylated proteins, suggesting a decrease in the dimethylamine affinity for the Coomassie blue G-250 dye, with the largest decrease for the lysine-rich protein, BSA. The BCA assay response was significantly higher for all three, reductively methylated proteins tested. The lower pKa values of the tertiary ε-dimethylamino groups likely contribute to the increased BCA assay responses, increasing the concentration of the deprotonated ε-dimethylamine and favoring Cu2+ binding.
The results of this study are relevant to biological methylation of lysine residues. Biological methylation can exist as mono-, di-, or tri-methyl-lysine. For the Bradford assay, the extent and proportion of methylation should be considered. If the methyl groups hinder binding to the Coomassie blue G-250 dye, then the assay response will decrease with increasing numbers of methyl groups. The concentration of a heavily tri-methylated protein may be underestimated when analyzed with the Bradford assay. On the other hand, if methylation occurs on a small fraction of lysine residues in a protein, the Bradford assay will likely give the same response as the unmodified protein. The effect of biological methylation on the BCA assay response is more complicated due to the differences in the pKa values of lysyl ε-amines, ε-monomethylamines, and ε-dimethylamines near the assay pH and the lack of an ionizable group on the lysyl ε-trimethylamine. For lysyl ε-amino groups on proteins, the unmodified ε-amine pKa is 11.0 ± 0.4 [60-61], the ε-monomethylamine pKa is 10.9 ± 0.3 [16,30], and the ε-dimethylamine pKa is 10.1 ± 0.3 [16,18,26,30]. These values follow the typical trend, in which the pKa of the secondary amine is the same or higher than the primary amine and the pKa of the tertiary amine is lower than that of the primary amine [62]. Based on the rationale that the lower pKa of the lysyl ε-dimethylamine allows for Cu2+ binding in the BCA assay, the effect of a lysyl ε-monomethylamine would be the same as an unmodified lysyl ε-amine. Similarly, the lack of an ionizable group on a lysyl ε-trimethylamine would result in a decrease in the BCA assay response compared to the unmodified lysyl ε-amine.
In conclusion, the dimethylation from reductive methylation does not greatly alter the response of the Bradford assay, which can be used to determine the concentration of reductively methylated proteins with nearly the same response as the unmodified proteins. The use of a reductively methylated standard may compensate for the slight decrease in the Bradford assay response, but the composition of the standard should match the density of lysine residues in the analyte-protein. The BCA assay response increases upon reductive methylation, providing insight into the role of lysine residues in binding Cu2+. For studying proteins with biological methylation, the Bradford assay is recommended due to the varying effects that mono-, di-, and tri-methylation may have on the BCA assay response.
Supplementary Material
Acknowledgments
Research reported in this publication was partially supported by the NIGMS of the NIH under Award Number R25GM069743 (Louisiana State University Initiative for Maximizing Student Development (IMSD) program); Pamlea Brady thanks the IMSD program for her scholarship. The project described was partially supported by Award Number R00RR024105 from the National Center for Research Resources of the NIH. We thank Dr. Aaron P. Smith for proof reading the manuscript.
Abbreviations
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- PTMs
post-translational modifications
- DMAB
dimethylamine-borane complex
- NMR
nuclear magnetic resonance
- HEWL
hen egg white lysozyme
- DSS
sodium 3-(trimethylsilyl)-1-propanesulfonic acid
- D2O
deuterated water
- A280nm
absorbance at 280 nm
- ε280
extinction coefficient at 280 nm
- Lys
L-lysine
- DM-Lys
Nε,Nε-dimethyl-L-lysine
- PBS
phosphate-buffered saline
- MALDI MS
matrix assisted laser desorption ionization mass spectrometry
- pKa
acid dissociation constant
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- [2].Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- [3].Sedmak JJ, Grossberg SE. A rapid, sensitive, and versatile assay for protein using Coomassie brilliant blue G250. Anal Biochem. 1977;79:544–552. doi: 10.1016/0003-2697(77)90428-6. 10.1016/0003-2697(77)90428-6. [DOI] [PubMed] [Google Scholar]
- [4].Compton SJ, Jones CG. Mechanism of dye response and interference in the Bradford protein assay. Anal Biochem. 1985;151:369–374. doi: 10.1016/0003-2697(85)90190-3. 10.1016/0003-2697(85)90190-3. [DOI] [PubMed] [Google Scholar]
- [5].de Moreno MR, Smith JF, Smith RV. Mechanism studies of Coomassie blue and silver staining of proteins. J Pharm Sci. 1986;75:907–911. doi: 10.1002/jps.2600750919. 10.1002/jps.2600750919. [DOI] [PubMed] [Google Scholar]
- [6].Congdon RW, Muth GW, Splittgerber AG. The binding interaction of Coomassie blue with proteins. Anal Biochem. 1993;213:407–413. doi: 10.1006/abio.1993.1439. 10.1006/abio.1993.1439. [DOI] [PubMed] [Google Scholar]
- [7].Georgiou CD, Grintzalis K, Zervoudakis G, Papapostolou I. Mechanism of Coomassie brilliant blue G-250 binding to proteins: A hydrophobic assay for nanogram quantities of proteins. Anal Bioanal Chem. 2008;391:391–403. doi: 10.1007/s00216-008-1996-x. 10.1007/s00216-008-1996-x. [DOI] [PubMed] [Google Scholar]
- [8].Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- [9].Noble JE, Bailey MJA. Quantitation of protein. In: Richard RB, Murray PD, editors. Methods in enzymology: Guide to protein purification. Academic Press; New York: 2009. pp. 73–95. (8 8) [DOI] [PubMed] [Google Scholar]
- [10].Fountoulakis M, Juranville JF, Manneberg M. Comparison of the Coomassie brilliant blue, bicinchoninic acid and Lowry quantitation assays, using non-glycosylated and glycosylated proteins. J Biochem Biophy Meth. 1992;24:265–274. doi: 10.1016/0165-022x(94)90078-7. 10.1016/0165-022x(94)90078-7. [DOI] [PubMed] [Google Scholar]
- [11].Wiechelman KJ, Braun RD, Fitzpatrick JD. Investigation of the bicinchoninic acid protein assay: Identification of the groups responsible for color formation. Anal Biochem. 1988;175:231–237. doi: 10.1016/0003-2697(88)90383-1. 10.1016/0003-2697(88)90383-1. [DOI] [PubMed] [Google Scholar]
- [12].Means GE, Feeney RE. Reductive alkylation of amino groups in proteins. Biochemistry. 1968;7:2192–2201. doi: 10.1021/bi00846a023. 10.1021/bi00846a023. [DOI] [PubMed] [Google Scholar]
- [13].Lanouette S, Mongeon V, Figeys D, Couture JF. The functional diversity of protein lysine methylation. Mol Syst Biol. 2014;10:1–26. doi: 10.1002/msb.134974. 10.1002/msb.134974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rypniewski WR, Holden HM, Rayment I. Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme: An X-ray analysis at 1.8-Angstrom resolution. Biochemistry. 1993;32:9851–9858. doi: 10.1021/bi00088a041. 10.1021/bi00088a041. [DOI] [PubMed] [Google Scholar]
- [15].Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. In: Carter CW Jr, Sweet R, Abelson JN, Simon MI, editors. Methods in enzymology: Macromolecular crystallography, part A. Academic Press; New York: 1997. pp. 171–179. (12 12) [PubMed] [Google Scholar]
- [16].Jentoft JE, Jentoft N, Gerken TA, Dearborn DG. 13C NMR studies of ribonuclease A methylated with [13C]formaldehyde. J Biol Chem. 1979;254:4366–4370. [PubMed] [Google Scholar]
- [17].Roberson KJ, Macnaughtan MA. Review of methods to assign the nuclear magnetic resonance peaks of reductively methylated proteins. Anal Biochem. 2014;466:76–82. doi: 10.1016/j.ab.2014.08.024. 10.1016/j.ab.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bradbury JH, Brown LR. Determination of dissociation constants of lysine residues of lysozyme by proton-magnetic resonance spectroscopy. Euro J Biochem. 1973;40:565–576. doi: 10.1111/j.1432-1033.1973.tb03228.x. 10.1111/j.1432-1033.1973.tb03228.x. [DOI] [PubMed] [Google Scholar]
- [19].Brown LR, Bradbury JH. Proton-magnetic-resonance studies of lysine residues of ribonuclease A. Euro J Biochem. 1975;54:219–227. doi: 10.1111/j.1432-1033.1975.tb04131.x. 10.1111/j.1432-1033.1975.tb04131.x. [DOI] [PubMed] [Google Scholar]
- [20].Jentoft JE, Gerken TA, Jentoft N, Dearborn DG. [13C]Methylated ribonuclease A. 13C NMR studies of the interaction of lysine 41 with active site ligands. J Biol Chem. 1981;256:231–236. [PubMed] [Google Scholar]
- [21].Gerken TA, Jentoft JE, Jentoft N, Dearborn DG. Intramolecular interactions of amino groups in 13C reductively methylated hen egg-white lysozyme. J Biol Chem. 1982;257:2894–2900. [PubMed] [Google Scholar]
- [22].Hardy RE, Batstonecunningham RL, Dill K. Specific 13C reductive methylation of glycophorin A. Possible relation of the N-terminal amino acid and the lysine residue to MN blood group specificities. Arch Biochem Biophys. 1983;222:222–230. doi: 10.1016/0003-9861(83)90520-9. 10.1016/0003-9861(83)90520-9. [DOI] [PubMed] [Google Scholar]
- [23].Sherry AD, Teherani J. Physical studies of 13C-methylated concanavalin A. pH- and Co2+-induced nuclear magnetic resonance shifts. J Biol Chem. 1983;258:8663–8669. [PubMed] [Google Scholar]
- [24].Goux WJ, Teherani J, Sherry AD. Amine inversion in proteins: A 13C-NMR study of proton-exchange and nitrogen inversion rates in Ne,Ne,Na,Na-[13C]tetramethyllysine, Ne,Ne,Na,Na-[13C]tetramethyllysine methyl-ester, and reductively methylated concanavalin A. Biophys Chem. 1984;19:363–373. doi: 10.1016/0301-4622(84)87019-2. 10.1016/0301-4622(84)87019-2. [DOI] [PubMed] [Google Scholar]
- [25].Sherry AD, Keepers J, James TL, Teherani J. Methyl motions in 13C-methylated concanavalin as studied by 13C magnetic resonance relaxation techniques. Biochemistry. 1984;23:3181–3185. doi: 10.1021/bi00309a011. 10.1021/bi00309a011. [DOI] [PubMed] [Google Scholar]
- [26].Dick LR, Sherry AD, Newkirk MM, Gray DM. Reductive methylation and 13C NMR-studies of the lysyl residues of fd gene 5 protein: Lysines 24, 46, and 69 may be involved in nucleic acid binding. J Biol Chem. 1988;263:18864–18872. [PubMed] [Google Scholar]
- [27].Dick LR, Geraldes CFGC, Sherry AD, Gray CW, Gray DM. 13C NMR of methylated lysines of fd gene 5 protein: Evidence for a conformational change involving lysine 24 upon binding of a negatively charged lanthanide chelate. Biochemistry. 1989;28:7896–7904. doi: 10.1021/bi00445a052. 10.1021/bi00445a052. [DOI] [PubMed] [Google Scholar]
- [28].Gluck M, Sweeney WV. 13C-NMR of Clostridium pasteurianum ferredoxin after reductive methylation of the amines using [13C]formaldehyde. Biochim Biophys Acta. 1990;1038:146–151. doi: 10.1016/0167-4838(90)90197-n. 10.1016/0167-4838(90)90197-n. [DOI] [PubMed] [Google Scholar]
- [29].Huque ME, Vogel HJ. Carbon-13 NMR studies of the lysine side chains of calmodulin and its proteolytic fragments. J Protein Chem. 1993;12:695–707. doi: 10.1007/BF01024928. [DOI] [PubMed] [Google Scholar]
- [30].Zhang MJ, Vogel HJ. Determination of side chain pKa values of lysine residues in calmodulin. J Biol Chem. 1993;268:22420–22428. [PubMed] [Google Scholar]
- [31].Moore GR, Cox MC, Crowe D, Osborne MJ, Rosell FI, Bujons J, Barker PD, Mauk MR, Mauk AG. Ne,Ne-dimethyl-lysine cytochrome c as an NMR probe for lysine involvement in protein-protein complex formation. Biochem J. 1998;332:439–449. doi: 10.1042/bj3320439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ashfield JT, Meyers T, Lowne D, Varley PG, Arnold JRP, Tan P, Yang JC, Czaplewski LG, Dudgeon T, Fisher J. Chemical modification of a variant of human MIP-1 alpha; implications for dimer structure. Protein Sci. 2000;9:2047–2053. doi: 10.1110/ps.9.10.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Macnaughtan MA, Kane AM, Prestegard JH. Mass spectrometry assisted assignment of NMR resonances in reductively 13C-methylated proteins. J Am Chem Soc. 2005;127:17626–17627. doi: 10.1021/ja056977r. 10.1021/ja056977r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Abraham SJ, Hoheisel S, Gaponenko V. Detection of protein-ligand interactions by NMR using reductive methylation of lysine residues. J Biomol NMR. 2008;42:143–148. doi: 10.1007/s10858-008-9274-y. 10.1007/s10858-008-9274-y. [DOI] [PubMed] [Google Scholar]
- [35].Larda ST, Bokoch MP, Evanics F, Prosser RS. Lysine methylation strategies for characterizing protein conformations by NMR. J Biomol NMR. 2012;54:199–209. doi: 10.1007/s10858-012-9664-z. 10.1007/s10858-012-9664-z. [DOI] [PubMed] [Google Scholar]
- [36].Hattori Y, Furuita K, Ohki I, Ikegami T, Fukada H, Shirakawa M, Fujiwara T, Kojima C. Utilization of lysine 13C-methylation NMR for protein-protein interaction studies. J Biomol NMR. 2013;55:19–31. doi: 10.1007/s10858-012-9675-9. 10.1007/s10858-012-9675-9. [DOI] [PubMed] [Google Scholar]
- [37].Roberson KJ, Brady PN, Sweeney M, Macnaughtan MA. Methods to identify the NMR resonances of the 13C-dimethyl N-terminal amine on reductively methylated proteins. JoVE. 2013;82:e50875. doi: 10.3791/50875. 10.3791/50875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Moebius K, Nordsieck K, Pichert A, Samsonov SA, Thomas L, Schiller J, Kalkhof S, Pisabarro MT, Beck-Sickinger AG, Huster D. Investigation of lysine side chain interactions of interleukin-8 with heparin and other glycosaminoglycans studied by a methylation-NMR approach. Glycobiology. 2013;23:1260–1269. doi: 10.1093/glycob/cwt062. 10.1093/glycob/cwt062. [DOI] [PubMed] [Google Scholar]
- [39].Xie Q, Fulton DB, Andreotti AH. A selective NMR probe to monitor the conformational transition from inactive to active kinase. ACS Chem Biol. 2015;10:262–268. doi: 10.1021/cb5004702. 10.1021/cb5004702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McGown EL, Nelson JW, Williams J, Fraser C. UV-VIS spectrophotometry: Automated determination of pathlength in microplate samples. Am Lab. 1997;29:21–24. [Google Scholar]
- [41].Craigie JS, MacKinnon SL, Walter JA. Liquid seaweed extracts identified using 1H NMR profiles. J App Phyc. 2008;20:665–671. 10.1007/s10811-007-9232-1. [Google Scholar]
- [42].Godejohann M, Preiss A, Mugge C. Quantitative measurements in continuous flow HPLC/NMR. Anal Chem. 1998;70:590–595. doi: 10.1021/ac970630s. 10.1021/ac970630s. [DOI] [PubMed] [Google Scholar]
- [43].Wright HT. Ovalbumin is an elastase substrate. J Biol Chem. 1984;259:14335–14336. [PubMed] [Google Scholar]
- [44].Peters T., Jr . Serum albumin. In: Putnam FW, editor. The plasma proteins: Structure, function, and genetic control. Academic Press; New York: 1975. pp. 133–181. (3 3) [Google Scholar]
- [45].Aune KC, Tanford C. Thermodynamics of denaturation of lysozyme by guandine hydrochloride. I. Dependence on pH at 25 degrees. Biochemistry. 1969;8:4579–4585. doi: 10.1021/bi00839a052. 10.1021/bi00839a052. [DOI] [PubMed] [Google Scholar]
- [46].Davies RC, Neuberge A, Wilson BM. Dependence of lysozyme activity on pH and ionic strength. Biochim Biophys Acta. 1969;178:294–299. doi: 10.1016/0005-2744(69)90397-0. 10.1016/0005-2744(69)90397-0. [DOI] [PubMed] [Google Scholar]
- [47].Fasman GD. Practical handbook of biochemistry and molecular biology. CRC Press; Boca Raton: 1989. [Google Scholar]
- [48].Gill SC, Vonhippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- [49].Noble JE, Knight AE, Reason AJ, Di Matola A, Bailey MJA. A comparison of protein quantitation assays for biopharmaceutical applications. Mol Biotech. 2007;37:99–111. doi: 10.1007/s12033-007-0038-9. 10.1007/s12033-007-0038-9. [DOI] [PubMed] [Google Scholar]
- [50].Harvey DJ, Wing DR, Kuster B, Wilson IBH. Composition of N-linked carbohydrates from ovalbumin and co-purified glycoproteins. J Am Soc Mass Spectrom. 2000;11:564–571. doi: 10.1016/S1044-0305(00)00122-7. 10.1016/S1044-0305(00)00122-7. [DOI] [PubMed] [Google Scholar]
- [51].Yang Y, Barendregt A, Kamerling JP, Heck AJR. Analyzing protein micro-heterogeneity in chicken ovalbumin by high-resolution native mass spectrometry exposes qualitatively and semi-quantitatively 59 proteoforms. Anal Chem. 2013;85:12037–12045. doi: 10.1021/ac403057y. 10.1021/ac403057y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Qian X, Dong H, Hu X, Tian H, Guo L, Shen Q, Gao X, Yao W. Analysis of the interferences in quantitation of a site-specifically PEGylated exendin-4 analog by the Bradford method. Anal Biochem. 2014;465:50–52. doi: 10.1016/j.ab.2014.06.009. 10.1016/j.ab.2014.06.009. [DOI] [PubMed] [Google Scholar]
- [53].Chen JG, Logman M, Weber SG. Effect of peptide primary sequence on biuret complex formation and properties. Electroanalysis. 1999;11:331–336. 10.1002/(sici)1521-4109(199905)11:5<331::aid-elan331>3.0.co;2-#. [Google Scholar]
- [54].Remelli M, Conato C, Agarossi A, Pulidori F, Mlynarz P, Kozlowski H. Copper complexes of dipeptides with L-Lys as C-terminal residue: A thermodynamic and spectroscopic study. Polyhedron. 2000;19:2409–2419. 10.1016/s0277-5387(00)00539-8. [Google Scholar]
- [55].Sigel H, Martin RB. Coordinating properties of the amide bond. Stability and structure of metal ion complexes of peptides and related ligands. Chem Rev. 1982;82:385–426. 10.1021/cr00050a003. [Google Scholar]
- [56].Fusch G, Hillgeris EC, Lippert B. Interaction of the lysine side-chain amino group with Cu(II) in (glycyl-L-lysine2-)Cu. Inorg Chim Acta. 1994;217:33–38. 10.1016/0020-1693(93)03740-2. [Google Scholar]
- [57].Kushwaha SSS, Verma N. A solution study of copper(II)-dipeptide complexation with tyrosinate and lysinate as bridging residues. J Indian Chem Soc. 2005;82:503–506. [Google Scholar]
- [58].Conato C, Contino A, Maccarrone G, Magri A, Remelli M, Tabbi G. Copper(II) complexes with L-lysine and L-ornithine: Is the side-chain involved in the coordination? A thermodynamic and spectroscopic study. Thermochim Acta. 2000;362:13–23. 10.1016/s0040-6031(00)00633-x. [Google Scholar]
- [59].Gyurcsik B, Vosekalna I, Larsen E. Copper(II) complexes of oligopeptides. An equilibrium and spectroscopic study on the copper(II) Lys-Leu-Ala-His-Phe-Gly system. Acta Chem Scand. 1997;51:49–58. 10.3891/acta.chem.scand.51-0049. [Google Scholar]
- [60].Kesvatera T, Jonsson B, Thulin E, Linse S. Measurement and modelling of sequence-specific pKa values of lysine residues in calbindin D9k. J Mol Biol. 1996;259:828–839. doi: 10.1006/jmbi.1996.0361. 10.1006/jmbi.1996.0361. [DOI] [PubMed] [Google Scholar]
- [61].Andre I, Linse S, Mulder FA. Residue-specific pKa determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: Application to apo calmodulin. J Am Chem Soc. 2007;129:15805–15813. doi: 10.1021/ja0721824. 10.1021/ja0721824. [DOI] [PubMed] [Google Scholar]
- [62].Hall NF, Sprinkle MR. Relations between the structure and strength of certain organic bases in aqueous solution. J Am Chem Soc. 1932;54:3469–3485. 10.1021/ja01348a001. [Google Scholar]
- [63].Sanna D, Agoston CG, Micera G. Potentiometric and spectroscopic studies on the copper(II) complexes formed by oligopeptides containing histidine with a protection at the terminal amino group. Polyhedron. 2001;20:937–947. S. I. 10.1016/S0277-5387(01)00747-1. [Google Scholar]
- [64].Sovago I, Kallay C, Varnagy K. Peptides as complexing agents: Factors influencing the structure and thermodynamic stability of peptide complexes. Coord Chem Rev. 2012;256:2225–2233. 10.1016/j.ccr.2012.02.026. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.