

Abstract
Friedreich ataxia (FRDA) is a member of the Repeat Expansion Diseases, a group of genetic conditions resulting from an increase/expansion in the size of a specific tandem array. FRDA results from expansion of a GAA/TTC-tract in the first intron of the frataxin gene (FXN). The disease-associated tandem repeats all form secondary structures that are thought to contribute to the propensity of the repeat to expand. The subset of these diseases that result from a CGG/CCG-repeat expansion, such as Fragile X syndrome, also express a folate-sensitive fragile site coincident with the repeat on the affected chromosome. This chromosome fragility involves the generation of chromosome/chromatid gaps or breaks, or the high frequency loss of one or both copies of the affected gene when cells are grown underfolate stress or as we showed previously, in the presence of an inhibitor of the ATM checkpoint kinase. Whether Repeat Expansion Disease loci containing different repeats form similar fragile sites was not known. We show here that the region of chromosome 9 that contains the FXN locus is intrinsically prone to breakage in vivo even in control cells. However, like FXS alleles, FRDA alleles show significantly elevated levels of chromosome abnormalities in the presence of an ATM inhibitor, consistent with the formation of a fragile site.
Keywords: Friedreich ataxia, FXN, frataxin, repeat expansion, GAA/TTC-triplet repeat, chromosome fragility
Graphical abstract
1. Introduction
Friedreich ataxia (FRDA) belongs to a group of genetic conditions known as the Repeat Expansion Diseases. These diseases all arise from a poorly understood process that results in an expansion or increase in the size of a tandem repeat tract. In the case of FRDA the repeat unit is GAA/TTC and the repeat tract is located in the first intron of the frataxin (FXN) gene, a gene situated at 9q21.1. Expansion results in epigenetic modifications that down-regulate the transcription of affected alleles [1–5]. The result of this downregulation is a progressive neurodegeneration that leads to gait and balance difficulties that often results in patients becoming wheelchair-bound at an early age. FRDA has a high early mortality resulting from an associated hypertrophic cardiomyopathy [6].
All of the repeats associated with the Repeat Expansion Diseases form secondary structures that are thought to play a role in expansion and in some cases these secondary structures are also thought to be responsible for the underlying pathology [7] Those Repeat Expansion Diseases resulting from expansion of CGG/CCG-repeat tracts, such as Fragile X syndrome (FXS), are also associated with the expression of a fragile site coincident with the repeat [8–12]. Fragile sites are often characterized as chromosomal regions with constrictions, gaps or breaks that are visible during metaphase in cells grown in the presence of specific chemical inducers (reviewed in [13]). One hallmark of these regions is that they are prone to chromosome rearrangement and translocation. Many fragile sites have been described in human genomes some of which are common, being found in large segments of the population, whilst rare fragile sites are only seen in a restricted group of individuals. The chemicals that induce fragility at a particular site are thought to reflect the properties of the DNA sequence of that site. For example, chemicals that affect the intranuclear levels of dCTP induce fragility at CGG/CCG-repeats, whilst A/T-specific DNA ligands such as distamycin A induce fragility at long A/T-rich repeat tracts [14]. While the molecular basis of chromosome fragility is still not completely understood, the prevailing idea is that individual loci are fragile because they are slow to complete replication, either because they impede replication fork progression in some way or are located in regions that only replicate slowly and late, and that this effect is exacerbated in the presence of inducers that further delay the completion of replication such that cells enter mitosis before replication is complete. While many categories of fragile sites have been described that are induced by folate-stress, distamycin, bromodeoxyuridine, 5-azacytidine or aphidicolin, it is possible that there are fragile sites for which a specific inducer has not yet been identified.
Work in yeast has demonstrated that, like the FXS repeats [15, 16], the FRDA repeats impede DNA replication [17] and are fragile [18], as measured in an assay for chromosome breaks. Furthermore, studies in yeast also demonstrate that chromosomal breakage and rearrangements at GAA-repeats are induced in replication-deficient cells [19]. However, breakage of both repeats occurs at frequencies that are many orders of magnitude lower than that of typical fragile sites in humans. It is thus unclear to what extent the events in yeast reflect chromosome fragility in humans. To see whether FRDA repeats are also prone to breakage in humans we took advantage of our observation that an inhibitor of the ATM checkpoint kinase is able to increase fragility at the FX fragile site (FRAXA) [20] as well as at a number of common fragile sites [21]. It is likely to do so because inhibition of the G2/M checkpoint allows cells to enter mitosis before the problem with replication of the FXS allele is resolved. Here we show that the region of chromosome 9 that contains the FXN gene shows evidence of past chromosome rearrangement and translocation even in normal cells that may be related to the proximity of the FXN gene to the pericentric heterochromatin. However, in addition to these events, FRDA alleles but not normal alleles show a significant increase in chromosome abnormalities when the ATM checkpoint kinase is inhibited.
2. Materials and Methods
2.1 Cell lines and Reagents
Lymphoblastoid cell lines from normal (GM06865, GM06895, GM15851) individuals and those with FRDA (GM15850 and GM16209) were obtained from Coriell Cell Repository (Camden, NJ). GM06865 and GM06895 have 9 and 15 repeats on each allele respectively [5]. GM15851 is unaffected brother of GM15850 and has <10 GAA-repeats [22]. GM15850 has 650 repeats on one allele and 1030 repeats on the other, while GM16209 has 800 repeats on both alleles. Lymphoblastoid cells were grown in RPMI 1640 supplemented with 10% FBS and 1x antibiotic-antimycotic liquid consisting of penicillin, streptomycin and fungisone (Life Technologies, Carlsbad, CA).
A BAC clone containing the FXN locus (RP11-876N18) was obtained from Empire Genomics (Buffalo, NY). This clone contains a 205 kb fragment corresponding to bases 71,641,920-71,846,939 of chromosome 9 of the GRC37/hg19 assembly (70,831,739 to 71,036,759 of GRC36/hg18 assembly) including the entire FXN gene and ~71% of the TJP2 gene. The ATM kinase inhibitor KU55933 was obtained from Calbiochem (now EMD-Millipore, Temecula, CA) and dissolved in DMSO. It was used at a final concentration of 10 μM for 20–22 hours.
2.2 Fluorescence in situ hybridization (FISH) to metaphase spreads
2.2.1. Probe preparation
RP11-876N18BAC DNA was labeled with Biotin-Nick Translation mix (Roche Applied science) as per the manufacturer’s instruction. The probe (100–200 ng) was mixed with 10 μg of human Cot1 DNA (Life Technologies) and 1 μg of Salmon sperm DNA (Sigma-Aldrich) and ethanol precipitated. The pellet was resuspended in 7 μl of deionized formamide (Sigma-Aldrich) and incubated at 37°C for 1 hour with shaking. Seven microliters of master mix containing 20% Dextran sulfate (Millipore) and 2X SSC (Quality Biological, Inc., Gaithersburg, MD) was added to the probe and incubated at 37°C for 1 hour with shaking. The probe was denatured at 80°C for 8 minutes and pre-annealed at 37°C for 1 hour without shaking before using it in the hybridization reaction.
2.2.2 Cell treatment and hybridization
Lymphoblastoid cells from control and FRDA patients cells were treated with either DMSO or 10 μM KU55933, an ATM inhibitor, in duplicate for 20–22 hours. The cultures were then treated with 100 ng/ml KaryoMAX® Colcemid™ solution (Life Technologies) for two hours at 37°C and metaphase spreads prepared by dropping the solution onto microscope slides according to standard procedures. The slide was treated with 1 μl of pretreatment solution (Dako North America, Inc., Carpinteria, CA) diluted in ml of 1X TBS pH 7.5 (Quality Biological, Inc.) and incubated at room temperature for 10 minutes. The slide was washed with 1X TBS and then dehydrated in a chilled ethanol series, 70%, 85% and 100% and air-dried. The slide was then denatured at 70°C for 5 minutes in denaturing solution (70% formamide/2X SSC pH 7.0), passed through the ethanol series again and air-dried. Fourteen microliters of the pre-annealed FISH probe was added and the spread covered with a 22×22 mm coverslip that was then sealed with Elmer’s rubber cement (Electron Microscopy Sciences, Hatfield, PA). Hybridization was carried out at 37°C overnight in a moist chamber. The slide was then washed three times in a prewarmed solution of 50% formamide/2X SSC (pH 7.0) for 5 minute each at 45°C, followed by three washes with prewarmed 0.1X SSC at 60°C for 5 minutes each. The slides were then dipped in 4XSSC/0.1% Tween 20 solution, and incubated with 200 μl of blocking solution (3% BSA/4X SSC/0.1% Tween 20) in a moist chamber at 37°C for 30 minutes. Excess blocking solution was removed by dipping the slide in 4XSSC/0.1% Tween 20. The slide was then incubated with 200 μl of alexa-555-streptavidin (Life Technologies) diluted 1:200 in 1%BSA/4X SSC/0.1% Tween 20 in a moist chamber for 45 minutes at 37°C in the dark. After washing three times with prewarmed 4X SSC/0.1% Tween 20 and twice with 2X SSC, the slide was mounted with fluorogel (Electron Microscopy Sciences)solution containing 10 μg/ml DAPI, sealed and stored in dark prior to microscopy. Images were acquired on a Leica DM5500B microscope (Leica Biosystems Inc., Buffalo Grove, IL) using iVision-Mac™ (4.5.2) software (BioVision Technologies, Exton, PA). For each experiment a total of 100 metaphase spreads were analyzed. FISH signals that were slightly different in intensity, either stronger or fainter than the normal signal, were not scored as abnormal.
2.3 Sequence analysis
The sequence contained in the RP11-876N18 BAC clone was aligned with the human genome sequence (UCSC; hg19). To determine the extent of DNA flexibility of the probe region, we generated a Python script (TwistFlex) that summed the average fluctuations in the twist angle for each of the possible dinucleotides as previously described (http://margalit.huji.ac.il/TwistFlex/Home.html;[23]). The analysis was performed in overlapping windows of 100 bp. Dinucleotide values were summed along the window and averaged by the window length. The data was plotted as a custom track in the UCSC Genome Browser. Windows with values of >13.7° were considered as flexibility peaks in accordance with previous definitions [23]. Custom tracks were also generated for the ORIs from the GSM927238 dataset [24]. The bed file associated with the Dataset was converted to hg19 using the liftOver tool (UCSC Genome Bioinformatics) and the ORI peaks called using the MACS program [25]. These peaks were then processed into a Custom Track for viewing on the UCSC Genome Browser. A script was written to generate 20 randomly selected intervals the same size as RP11-876N18 from the autosomes, excluding regions from the centromeres. Ten intervals from this list that contained at least one gene and lacked any major segmental duplications were used for further analysis. Using the Repeat Masker data table from the UCSC Genome Browser (http://genome.ucsc.edu/) and the fast a sequence from Ensembl (http://www.ensembl.org/index.html), the total number of SINEs, the maximum number of SINEs in any 2 kb region were calculated. The TwistFlex characteristics for each region were also calculated as described above. The CpG islands for each interval were visualized in the UCSC genomic browser and the number determined manually. The number of ORIs in each region present in HeLa cells was determined manually using IGViewer and the custom track generated from the GSM927238 dataset described above [24].
3. Results
3.1 The region of chromosome 9 carrying the FXN gene is prone to chromosomal rearrangements in both control and patient cells
We examined the FXN locus in control and patient cells using a BAC probe that contained a 205 Kb region of chromosome 9. This region includes the entire FXN gene and ~71% of the adjacent tight junction protein 1 (TJP2) gene located at 9q21.11. At the FMR1 locus chromosomal rearrangements are only detected in cells from individuals with FXS in response to induction of the fragile site [20], whereas both normal and FRDA patient cells showed a high frequency of chromosomal abnormalities involving the 9q21 region even under normal growth conditions (Fig. 1). To quantify the FISH abnormalities, each sister chromatid was scored separately since the FISH signals were not always concordant. Of the abnormal FISH signals observed 76–90% represented duplications and the remainder deletions. Chromosomal breakage or translocations were only very rarely seen. Examples of discordant FISH signals are displayed in Fig 1B with 1B(i) showing a chromosome with one normal chromatid and one chromatid showing a duplication of the FXN region, 1B(ii) showing a chromosome with one normal chromatid and a chromatid with a large deletion of the FXN region and 1B(iii) showing a chromosome with one chromatid that has a duplication of the probe region and a sister chromatid with a deletion of that region. The high instance of these chromosomal abnormalities is consistent with the presence of a constitutively expressed fragile site or some other recombination prone sequence in the FXN-TJP2region even on normal chromosomes.
Fig. 1. Both normal and FRDA cells show a high frequency of spontaneous chromosomal abnormalities in the region of the FXN gene.

A) The percentage of abnormal chromosomes seen in normal and patient lymphoblastoid cells. B) Examples of the chromosomal abnormalities observed in these cells. (i) Chromosome with a duplication on one sister chromatid. (ii) Chromosome with a deletion on one sister chromatid. (iii) Chromosome with a duplication on one chromatid and a deletion on the other. Chromosomes were stained with DAPI (blue) and FXN signal is shown in red. Chromosomal duplications made up 76–90% of the chromosomal abnormalities with the remainder being deletions of some or all of the probe region.
3.2 The FXN locus is not enriched for factors typically associated with fragile sites
Various factors have been proposed to characterize fragile sites including a certain CpG-island density [26], highly AT-rich sequences, the presence of sequences with non-B-DNA forming potential or the enrichment for regions with high flexibility [23]. Other factors such as a paucity of origins of replication (ORIs) or impaired replication dynamics [27–29], whether the locus is late replicating and enrichment for Alu repeats [30] or simple sequence repeats [26] have also been suggested to be important determinants of chromosome fragility.
We therefore examined the sequence of the probe region in some detail. This region has a G/C-content of 44.3% with 5 very small CpG islands distributed more or lessevenly across the region. This compares to the average G/C content of chromosome 9 of 41.4%. It has a number of relatively strong ORIs within 100 Mb that are seen in many different cell types including HeLa (Fig. 2). Relatively few flexibility peaks are seen (Fig. 2) as assessed by our implementation of the Twistflex algorithm[23] as described in the Materials and Methods (Section 2.3).
Fig. 2. Sequence characteristics of the FXN locus.

A ~150 kb region that includes the FXN gene showing the locations and levels of simple repeats, origins of replication, regions with twist flex peaks and %GC that were extracted from the USCS database (http://genome.ucsc.edu/cgi-bin/hgBlat) using the Feb .2009 (GRCh37/hg19) assembly of the human genome or determined as described in the Materials and Methods (Section 2.3).
This region does contain a variety of repeated sequences of various types (Fig. 2 and 3). Since simple repeats are prone to deletion during cloning in bacteria and very long repeats are difficult to map to the genome accurately, the possibility of the presence of longer repeats cannot be definitively excluded. However, none of the simple repeats in this region in the hg19 build of the human genome are unusual either in terms of length or sequence. The region covered by the probe and particularly the intergenic region is enriched for a variety of different SINEs and LINEs. In particular, RepeatMasker analysis (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker) showed that Alu elements make up 48% of the intergenic region compared to 21% and 10% of the next two intergenic regions centromeric to FXN. Furthermore, 71% of the FXN-TJP2 intergenic region was made up of different repeated sequence family members compared to 56% and 50% of the two more centromeric intergenic regions.
Fig. 3. Sequence characteristics of the pericentromeric region of Chromosome 9 immediately centromeric to the FXN locus.
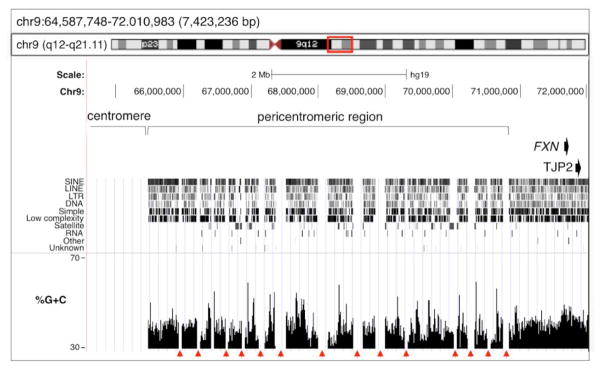
The ~7,4 Mb of DNA centromeric to the FXN gene was analyzed using the USCS database (http://genome.ucsc.edu/cgi-bin/hgBlat) browsing tools and the Feb .2009 (GRCh37/hg19) assembly of the human genome. The red arrowheads indicate sequence gaps.
A number of other repeated sequences were seen in this region. There is a segmental duplication involving a 2284 bp region of the FXN 3′ UTR and part of the last intron of the suppressor of cancer cell invasion (SCAI) gene at 9q33.3. There is also a 1058 bp sequence located in the intergenic region between FXN and TJP2 that is 94% homologous to a region of 9p24.1, and >88% homologous to sequences on chromosomes 3, 7, 8, 10 and 12. The region of homology on 9p24.1 corresponds to the last 3 exons of the adenylate kinase 3 (AK3) gene suggesting that the other loci represent integration sites of a partial AK3 pseudogene. The last intron of the FXN gene also contains a 226 bp region homologous to part of the repeat unit of the SATR2 minisatellite.
Comparison of the region corresponding to the RP11-876N18 probe with 10 randomly selected genomic intervals of the same size showed that the probe region is not significantly different from any of these intervals with respect to many of the factors thought to be associated with fragile sites (Table 1). Specifically, it has a %GC of 44.3% compared to the average for the randomly chosen intervals of 45%, an average number of CpG islands (n=5) and an average number of SINEs per 2 kb window (n=7). The overall SINE density was modestly higher than normal (1.4-fold). However, the ORI density was 1.2-fold lower than average and the average flexibility was also lower than average (5 peaks compared to an average of 12).
Table 1.
Comparison of RP11-876N18 probe region with 10 randomly chosen regions of the human genome with respect to density of SINEs, ORI prevalence, CpG islands and %GC.
| Target | Position | TwistFlex Peaks | Total SINEs | Max SINEs in 2Kb | No. of ORIs (HeLa) | CpG islands | GC% |
|---|---|---|---|---|---|---|---|
| RP11-876N18 | chr9:71641920–71846939 | 5 | 259 | 7 | 13 | 5 | 44.3 |
| Random_00 | chr14:103503541–103708561 | 4 | 165 | 7 | 23 | 11 | 52.4 |
| Random_01 | chr1:203992795–204197815 | 22 | 212 | 8 | 27 | 3 | 49.8 |
| Random_02 | chr7:35566971–35771991 | 27 | 120 | 6 | 17 | 2 | 41.2 |
| Random_03 | chr11:93684735–93889755 | 18 | 104 | 5 | 15 | 2 | 41.2 |
| Random_04 | chr16:89808205–90013225 | 0 | 305 | 8 | 24 | 16 | 53.4 |
| Random_05 | chr19:48508081–48713101 | 15 | 305 | 9 | 18 | 7 | 48.5 |
| Random_06 | ch15:52553923–52758943 | 8 | 154 | 7 | 11 | 1 | 41.0 |
| Random_07 | chr4:106430981–106636001 | 7 | 94 | 5 | 0 | 1 | 36.5 |
| Random_08 | chr10:11880565–12085585 | 4 | 237 | 8 | 11 | 3 | 43.9 |
| Random_09 | chr11:124779335–124984355 | 15 | 85 | 7 | 12 | 4 | 41.7 |
3.3 The FXN locus is very close to the pericentric heterochromatic region of chromosome 9
The FXN gene is less than 850kb from the pericentromeric region of chromosome 9. This region is the longest stretch of pericentric heterochromatin of any chromosome. It has many unsequenced gaps that are often flanked by long segmental duplications (indicated by the arrowheads in Fig. 3; [31]). It is also enriched for satellite repeats including α, β and satellite III sequences. The unusual length of this region and the high density of these repeats is thought to account for the very high frequency of structural variation of chromosome 9 seen in the general population [32].
While late replication has been suggested to be a common feature of fragile sites, high resolution time-sensitive maps of replication patterns shows that the FXN region actually replicates early in many cell types including normal lymphoblastoid cells where replication is initiated in G1 (Fig. 4; [33]). In contrast, replication of the FMR1 locus that forms a fragile site in individuals with FXS is initiated late in S phase and only finishes in G2 even in normal cells.
Fig. 4. Replication timing of the FXN and FMR1 loci.
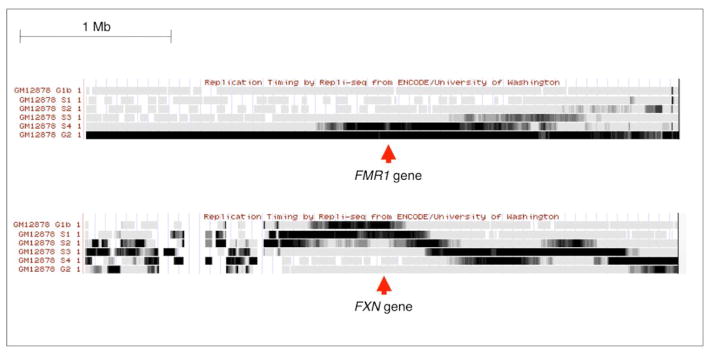
Information as to the replication timing of the two loci in the EBV-transformed normal female lymphoblastoid cell line, GM12878, was extracted from the appropriate ENCODE track in the USCS database. This data was obtained using the “Repli-seq” method [33].
3.4 Inhibition of the ATM kinase increases the frequency of chromosomal abnormalities in the FXN region only in FRDA patient cells
Since ATM inhibition has been shown to increase the frequency of common fragile sites as well as the FRAXA fragile site, we treated control and FRDA patient cell lines with ATM inhibitor (ATMi) KU55933 and scored the frequency of chromosomal aberrations. No significant changes in the number of chromosomal abnormalities in the FXN region were seen in control cell lines treated with the ATMi when normalized to the number of abnormalities seen in the absence of inhibitor (Fig. 5).
Fig. 5. FRDA cells show elevated levels of chromosomal abnormalities in response to ATM inhibition.

The percentage of ATM-sensitive chromosome abnormalities was obtained by subtracting the abnormalities seen in mock treated cells from the abnormalities seen in ATMi treated cells. P-values were calculated using the Fisher’s test. No significant change was seen in the proportion of duplications to deletions on inhibition of ATM.
In contrast, treatment of FRDA patient cells with the ATMi resulted in a significant increase in the number of abnormal chromosomes observed. In the case of GM15850 the frequency of ATM-dependent chromosomal abnormalities was 9.3%, while in GM16209 it was 7.5%. This would suggest that the expanded GAA/TTC repeat that causes FRDA is prone to chromosome breakage that is ATM-sensitive. Inhibition of ATM did not have a significant effect on the proportion of duplications and deletions observed.
4. Discussion
We show here that the 205 kb region that spans the FXN gene is prone to chromosomal rearrangements in both control and FRDA patient cells (Fig. 1). Our analysis of the FXN region for features thought to be characteristic of fragile sites did not reveal any clear factor that would make it recombination prone other than its proximity to the pericentric heterochromatic region of chromosome 9 (Fig. 2 and 3). The pericentromeric heterochromatic region of chromosome 9 is the largest of any autosome. It contains very little unique DNA being comprised primarily of repeats including α, β and satellite III DNA. There are also large blocks of duplications within and between the two pericentromeric regions along with matches to pericentromeric and non-pericentromeric sequences on other chromosomes [34]. This highly repetitive region has been suggested to be responsible for the very high frequency of structural variations seen on this chromosome: this region is heteromorphic in up to 50% of Caucasians and pericentric inversions are also common [32]. Many of these structural variations are not associated with any deleterious consequences. Thus not only is this region recombination prone, but without a penalty for these events, they may well accumulate in cells in tissue culture. It may be that some events triggered in this region also involve the FXN-TJP2 region thus accounting for the high frequency of rearrangements that were seen with our probe.
In addition to evidence of previous chromosome breakage and rearrangement seen in both normal and FRDA cells (Fig. 1), inhibition of ATM, a treatment known to increase the frequency of expression of a number of known fragile sites, significantly increased the number of chromosomal abnormalities observed in FRDA cells but not in normal ones (Fig. 5). This suggests that there may be two sources of the chromosomal rearrangements involving the FXN locus, one that is common to all cells and presumably reflects sequences other than the expanded repeat and one that is specific to cells with the expanded repeat. The spontaneously occurring chromosomal rearrangements may have accumulated in the culture over time with those rearrangements compatible with cell viability persisting. Thus it is difficult to estimate the true frequency with which such events occur. However, it is possible to do so for the ATM-sensitive rearrangements. In the case of the two FRDA cell lines tested, these rearrangements occurred at a frequency of 7.5–9.3% per cell division (Fig. 5). While the frequency with which fragile sites are expressed varies widely depending on the specific site as well as the growth conditions, the level of ATM-dependent chromosomal abnormalities we observe at the FXN locus is comparable to the frequency of expression of many common fragile sites [35–37].
The chromosome abnormalities that we observed are consistent with a high frequency of breaks in the region. The fact that duplications predominate might indicate that repair occurs by homologous recombination rather than non-homologous end-joining. While the 9q21.1 region has not previously been shown to contain a fragile site, it has been implicated in a number of chromosome aberrations including triplications [38], partial tetrasomy of chromosome 9 [39–43] and a paracentric inversion/duplication [44]. A 269 kb inverted duplication associated with progressive non-syndromic hearing loss (DFNA51) has also been described in which one breakpoint occurs upstream of TJP2 in the region covered by the probe [45].
While the chromosome rearrangements we observe are consistent with chromosome fragility they lack the constrictions, gaps or breaks in chromosomes or chromatids that are typically seen in metaphase spreads of cells containing “classic” fragile sites. This may reflect the fact that the FXN locus is replicated in late G1 or early S phase, much earlier in the cell cycle than fragile sites like the Fragile X fragile site, FRAXA. Even the normal FMR1 allele completes replication late in S phase or in G2 even in the absence of replication inhibitors ([46] and Fig. 4) and the FX alleles are known to complete replication even later [47–49]. Initiation of replication of the FXN region early in the cell cycle may give the cell more time to “repair” the gaps or breaks before mitosis begins.
The repeats responsible for FRAXA and those responsible for FRDA share many common features including the ability to form secondary structures [50–58] and to block DNA synthesis and cause chromosome breakage in yeast[15, 16, 18, 59]. The fact that long GAA/TTC-repeats are also prone to breakage in mammalian cells raises the possibility that a structural block to DNA synthesis is responsible for the chromosome fragility at both the FMR1 and the FXN loci. Since CTG/CAG-repeats which are also associated with other Repeat Expansion Diseases also impede DNA synthesis and show chromosome fragility in yeast [60], they too may be prone to chromosome rearrangements in humans, along with other disease-associated repeats that form similar structures.
Highlights.
The region of chromosome 9 that contains the Frataxin (FXN) locus is prone to breakage and rearrangements.
Friedreich ataxia (FRDA) is caused by a GAA/TTC-repeat expansion in the FXN gene
FRDA alleles are prone to chromosome rearrangements induced by an ATM inhibitor.
Acknowledgments
Funding:
This work was supported by a grant from the Intramural program of the National Institute of Diabetes, Digestive and Kidney Diseases to KU (DK057808 08).
Footnotes
Conflict of interest:
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pandolfo M. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich’s ataxia patients and in a mouse model. PLoS One. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soragni E, Herman D, Dent SY, Gottesfeld JM, Wells RD, Napierala M. Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res. 2008;36:6056–6065. doi: 10.1093/nar/gkn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 4.Kumari D, Biacsi RE, Usdin K. Repeat expansion affects both transcription initiation and elongation in Friedreich ataxia cells. J Biol Chem. 2011;286:4209–4215. doi: 10.1074/jbc.M110.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35:3383–3390. doi: 10.1093/nar/gkm271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol. 2009;256(Suppl 1):3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- 7.Usdin K, House NC, Freudenreich CH. Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol. 2015;50:142–167. doi: 10.3109/10409238.2014.999192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proops R, Webb T. The ‘fragile’ X chromosome in the Martin-Bell-Renpenning syndrome and in males with other forms of familial mental retardation. J Med Genet. 1981;18:366–373. doi: 10.1136/jmg.18.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritchie RJ, Knight SJ, Hirst MC, Grewal PK, Bobrow M, Cross GS, Davies KE. The cloning of FRAXF: trinucleotide repeat expansion and methylation at a third fragile site in distal Xqter. Hum Mol Genet. 1994;3:2115–2121. doi: 10.1093/hmg/3.12.2115. [DOI] [PubMed] [Google Scholar]
- 10.Parrish JE, Oostra BA, Verkerk AJ, Richards CS, Reynolds J, Spikes AS, Shaffer LG, Nelson DL. Isolation of a GCC repeat showing expansion in FRAXF, a fragile site distal to FRAXA and FRAXE. Nat Genet. 1994;8:229–235. doi: 10.1038/ng1194-229. [DOI] [PubMed] [Google Scholar]
- 11.Winnepenninckx B, Debacker K, Ramsay J, Smeets D, Smits A, FitzPatrick DR, Kooy RF. CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am J Hum Genet. 2007;80:221–231. doi: 10.1086/510800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metsu S, Rooms L, Rainger J, Taylor MS, Bengani H, Wilson DI, Chilamakuri CS, Morrison H, Vandeweyer G, Reyniers E, Douglas E, Thompson G, Haan E, Gecz J, Fitzpatrick DR, Kooy RF. FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet. 2014;10:e1004242. doi: 10.1371/journal.pgen.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukusa T, Fryns JP. Human chromosome fragility. Biochim Biophys Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Yu S, Mangelsdorf M, Hewett D, Hobson L, Baker E, Eyre HJ, Lapsys N, Le Paslier D, Doggett NA, Sutherland GR, Richards RI. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell. 1997;88:367–374. doi: 10.1016/s0092-8674(00)81875-9. [DOI] [PubMed] [Google Scholar]
- 15.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16:226–228. doi: 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balakumaran BS, Freudenreich CH, Zakian VA. CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum Mol Genet. 2000;9:93–100. doi: 10.1093/hmg/9.1.93. [DOI] [PubMed] [Google Scholar]
- 17.Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol Cell Biol. 2004;24:2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, Lobachev KS. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, Lobachev KS. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell. 2012;48:254–265. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumari D, Somma V, Nakamura AJ, Bonner WM, D’Ambrosio E, Usdin K. The role of DNA damage response pathways in chromosome fragility in Fragile X syndrome. Nucleic Acids Res. 2009;37:4385–4392. doi: 10.1093/nar/gkp391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozeri-Galai E, Schwartz M, Rahat A, Kerem B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene. 2008;27:2109–2117. doi: 10.1038/sj.onc.1210849. [DOI] [PubMed] [Google Scholar]
- 22.Punga T, Buhler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med. 2010;2:120–129. doi: 10.1002/emmm.201000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, Kerem B. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol. 2012;19:837–844. doi: 10.1038/nsmb.2339. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 2012;22:993–1005. doi: 10.1101/gr.134395.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palakodeti A, Lucas I, Jiang Y, Young DJ, Fernald AA, Karrison T, Le Beau MM. Impaired replication dynamics at the FRA3B common fragile site. Hum Mol Genet. 2010;19:99–110. doi: 10.1093/hmg/ddp470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell. 2011;43:122–131. doi: 10.1016/j.molcel.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 29.Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 30.Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, Gorgoulis VG. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- 31.Eichler EE, Clark RA, She X. An assessment of the sequence gaps: unfinished business in a finished human genome. Nature reviews Genetics. 2004;5:345–354. doi: 10.1038/nrg1322. [DOI] [PubMed] [Google Scholar]
- 32.Verma RS, Dosik H, Lubs HA. Size and pericentric inversion heteromorphisms of secondary constriction regions (h) of chromosomes 1, 9, and 16 as detected by CBG technique in Caucasians: classification, frequencies, and incidence. Am J Med Genet. 1978;2:331–339. doi: 10.1002/ajmg.1320020403. [DOI] [PubMed] [Google Scholar]
- 33.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphray SJ, Oliver K, Hunt AR, Plumb RW, Loveland JE, Howe KL, Andrews TD, Searle S, Hunt SE, Scott CE, Jones MC, Ainscough R, Almeida JP, Ambrose KD, Ashwell RI, Babbage AK, Babbage S, Bagguley CL, Bailey J, Banerjee R, Barker DJ, Barlow KF, Bates K, Beasley H, Beasley O, Bird CP, Bray-Allen S, Brown AJ, Brown JY, Burford D, Burrill W, Burton J, Carder C, Carter NP, Chapman JC, Chen Y, Clarke G, Clark SY, Clee CM, Clegg S, Collier RE, Corby N, Crosier M, Cummings AT, Davies J, Dhami P, Dunn M, Dutta I, Dyer LW, Earthrowl ME, Faulkner L, Fleming CJ, Frankish A, Frankland JA, French L, Fricker DG, Garner P, Garnett J, Ghori J, Gilbert JG, Glison C, Grafham DV, Gribble S, Griffiths C, Griffiths-Jones S, Grocock R, Guy J, Hall RE, Hammond S, Harley JL, Harrison ES, Hart EA, Heath PD, Henderson CD, Hopkins BL, Howard PJ, Howden PJ, Huckle E, Johnson C, Johnson D, Joy AA, Kay M, Keenan S, Kershaw JK, Kimberley AM, King A, Knights A, Laird GK, Langford C, Lawlor S, Leongamornlert DA, Leversha M, Lloyd C, Lloyd DM, Lovell J, Martin S, Mashreghi-Mohammadi M, Matthews L, McLaren S, McLay KE, McMurray A, Milne S, Nickerson T, Nisbett J, Nordsiek G, Pearce AV, Peck AI, Porter KM, Pandian R, Pelan S, Phillimore B, Povey S, Ramsey Y, Rand V, Scharfe M, Sehra HK, Shownkeen R, Sims SK, Skuce CD, Smith M, Steward CA, Swarbreck D, Sycamore N, Tester J, Thorpe A, Tracey A, Tromans A, Thomas DW, Wall M, Wallis JM, West AP, Whitehead SL, Willey DL, Williams SA, Wilming L, Wray PW, Young L, Ashurst JL, Coulson A, Blocker H, Durbin R, Sulston JE, Hubbard T, Jackson MJ, Bentley DR, Beck S, Rogers J, Dunham I. DNA sequence and analysis of human chromosome 9. Nature. 2004;429:369–374. doi: 10.1038/nature02465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramos FJ, Emanuel BS, Spinner NB. Frequency of the common fragile site at Xq27.2 under conditions of thymidylate stress: implications for cytogenetic diagnosis of the fragile-X syndrome. Am J Med Genet. 1992;42:835–838. doi: 10.1002/ajmg.1320420618. [DOI] [PubMed] [Google Scholar]
- 36.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, le Sage C, Chen BP, Chen DJ, Agami R, Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Gene Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahoo T, Wang JC, Elnaggar MM, Sanchez-Lara P, Ross LP, Mahon LW, Hafezi K, Deming A, Hinman L, Bruno Y, Bartley JA, Liehr T, Anguiano A, Jones M. Concurrent triplication and uniparental isodisomy: evidence for microhomology-mediated break-induced replication model for genomic rearrangements. Eur J Hum Genet. 2015:61–66. doi: 10.1038/ejhg.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvieri F, Tozzi C, Benincori C, De Merulis MV, Bellussi A, Genuardi M, Neri G. Partial tetrasomy 9 in an infant with clinical and radiological evidence of multiple joint dislocations. Eur J Pediatr. 1988;147:645–648. doi: 10.1007/BF00442483. [DOI] [PubMed] [Google Scholar]
- 40.Abe T, Morita M, Kawai K, Misawa S, Takino T, Hashimoto H, Nakagome Y. Partial tetrasomy 9(9pter to 9q2101) due to an extra iso-dicentric chromosome. Ann Genet. 1977;20:111–114. [PubMed] [Google Scholar]
- 41.Wisniewski L, Politis GD, Higgins JV. Partial tetrasomy 9 in a liveborn infant. Clin Genet. 1978;14:147–153. doi: 10.1111/j.1399-0004.1978.tb02120.x. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura Y, Sato E, Sakai K, Sakuma S, Hashimoto T, Sindou S. Abnormal chromosome 9 in a neonate program. Report of three cases. Arch Pathol Lab Med. 1990;114:185–187. [PubMed] [Google Scholar]
- 43.Andou R, Mimaki T, Ogihara T, Tamai H, Mino M. A case of tetrasomy 9p. Acta Paediatr Jpn. 1994;36:724–726. doi: 10.1111/j.1442-200x.1994.tb03280.x. [DOI] [PubMed] [Google Scholar]
- 44.Luke S, Verma RS, PeBenito R, Macera MJ. Inversion-duplication of bands q13----q21 of human chromosome 9. Am J Med Genet. 1991;40:57–60. doi: 10.1002/ajmg.1320400111. [DOI] [PubMed] [Google Scholar]
- 45.Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W, McCarthy S, Nord AS, Gordon CR, Ben-Neriah Z, Sebat J, Kanaan M, Lee MK, Frydman M, King MC, Avraham KB. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet. 2010;87:101–109. doi: 10.1016/j.ajhg.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brylawski BP, Chastain PD, 2nd, Cohen SM, Cordeiro-Stone M, Kaufman DG. Mapping of an origin of DNA replication in the promoter of fragile X gene FMR1. Exp Mol Pathol. 2007;82:190–196. doi: 10.1016/j.yexmp.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian PS, Nelson DL, Chinault AC. Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am J Hum Genet. 1996;59:407–416. [PMC free article] [PubMed] [Google Scholar]
- 48.Webb T. Delayed replication of Xq27 in individuals with the fragile X syndrome. Am J Med Genet. 1992;43:1057–1062. doi: 10.1002/ajmg.1320430633. [DOI] [PubMed] [Google Scholar]
- 49.Yudkin D, Hayward BE, Aladjem MI, Kumari D, Usdin K. Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum Mol Genet. 2014;23:2940–2952. doi: 10.1093/hmg/ddu006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Usdin K, Woodford KJ. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mitas M, Yu A, Dill J, Haworth IS. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry. 1995;34:12803–12811. doi: 10.1021/bi00039a041. [DOI] [PubMed] [Google Scholar]
- 53.Yu A, Barron MD, Romero RM, Christy M, Gold B, Dai J, Gray DM, Haworth IS, Mitas M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry. 1997;36:3687–3699. doi: 10.1021/bi9625410. [DOI] [PubMed] [Google Scholar]
- 54.Fry M, Loeb LA. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc Natl Acad Sci U S A. 1994;91:4950–4954. doi: 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nadel Y, Weisman-Shomer P, Fry M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J Biol Chem. 1995;270:28970–28977. doi: 10.1074/jbc.270.48.28970. [DOI] [PubMed] [Google Scholar]
- 56.Mariappan SV, Catasti P, Silks LA, 3rd, Bradbury EM, Gupta G. The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich’s ataxia. J Mol Biol. 1999;285:2035–2052. doi: 10.1006/jmbi.1998.2435. [DOI] [PubMed] [Google Scholar]
- 57.Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- 58.Potaman VN, Oussatcheva EA, Lyubchenko YL, Shlyakhtenko LS, Bidichandani SI, Ashizawa T, Sinden RR. Length-dependent structure formation in Friedreich ataxia (GAA)n*(TTC)n repeats at neutral pH. Nucleic Acids Res. 2004;32:1224–1231. doi: 10.1093/nar/gkh274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol. 2003;23:1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]