

ABSTRACT
Cyclin-G-associated kinase (GAK), the ubiquitously expressed J-domain protein, is essential for the chaperoning and uncoating of clathrin that is mediated by Hsc70 (also known as HSPA8). Adjacent to the C-terminal J-domain that binds to Hsc70, GAK has a clathrin-binding domain that is linked to an N-terminal kinase domain through a PTEN-like domain. Knocking out GAK in fibroblasts caused inhibition of clathrin-dependent trafficking, which was rescued by expressing a 62-kDa fragment of GAK, comprising just the clathrin-binding and J-domains. Expressing this fragment as a transgene in mice rescued the lethality and the histological defects caused by knocking out GAK in the liver or in the brain. Furthermore, when both GAK and auxilin (also known as DNAJC6), the neuronal-specific homolog of GAK, were knocked out in the brain, mice expressing the 62-kDa GAK fragment were viable, lived a normal life-span and had no major behavior abnormalities. However, these mice were about half the size of wild-type mice. Therefore, the PTEN-like domains of GAK and auxilin are not essential for Hsc70-dependent chaperoning and uncoating of clathrin, but depending on the tissue, these domains appear to increase the efficiency of these co-chaperones.
KEY WORDS: GAK, Auxilin, Chaperone, Clathrin
Summary: Our studies on GAK- and auxilin-knockout mice show that the only essential domains of GAK and auxilin in the brain are the clathrin-binding domain and the J-domain that binds to Hsc70.
INTRODUCTION
A variety of physiological receptors are internalized through clathrin-mediated endocytosis (CME) (Doherty and McMahon, 2009; Kirchhausen et al., 2014). The endocytic cycle starts with the binding of ligands to receptors in clathrin coated pits (CCPs). Dynamin then causes constriction of the necks of the pits, and ultimately the scission of the CCPs releases clathrin-coated vesicles (CCVs) into the cytosol (Hinshaw, 2000). Finally, the clathrin coat is dissociated by Hsc70 (also known as HSPA8) before the fusion of the uncoated vesicles with the early endosome (Chappell et al., 1986). In addition to uncoating, biochemical studies have shown that Hsc70 chaperones the dissociated clathrin (Jiang et al., 2000).
The uncoating and chaperone activities of Hsc70 are dependent on a co-chaperone, either the ubiquitously expressed GAK (also known as auxilin 2) or the neuronally expressed auxilin (also known as DNAJC6) (Eisenberg and Greene, 2007). Both GAK and auxilin share the same multi-domain structure, except that GAK has an N-terminal kinase domain (Eisenberg and Greene, 2007; Lemmon, 2001). Auxilin has three domains; an N-terminal PTEN-like domain and a C-terminal J-domain are bridged by a clathrin-binding domain. In vitro uncoating studies have shown that the clathrin-binding domain and the J-domain, which binds to Hsc70, are both necessary and sufficient to support the uncoating of clathrin-coated baskets by Hsc70 (Holstein et al., 1996; Ma et al., 2002). In fact, even a C-terminal fragment of auxilin (Aux-C20), with just a single clathrin-binding motif upstream of the J-domain, efficiently supports the uncoating of clathrin baskets by Hsc70 in vitro (Ma et al., 2002).
GAK and auxilin have been targeted to examine the role of Hsc70 in clathrin-dependent trafficking both in tissue culture cells and in animal models. When these co-chaperones are knocked down or knocked out in tissue culture cells, clathrin-mediated endocytosis (CME) and other clathrin-dependent pathways are inhibited (Hirst et al., 2008; Kametaka et al., 2007; Lee et al., 2005; Zhang et al., 2005). Clathrin and clathrin adaptors are mislocalized at the plasma membrane and the trans-Golgi network (TGN) in cells that do not express GAK. In addition, instead of clathrin being readily diffusible in the cytosol, the cytosolic clathrin is immobilized into cages (Hirst et al., 2008; Lee et al., 2005). As expected, given the importance of these co-chaperones, their absence causes lethality in Caenorhabditis elegans, Drosophila and mice (Eun et al., 2008; Greener et al., 2001; Kandachar et al., 2008).
In contrast to the in vitro uncoating studies that show the kinase and the PTEN-like domains of GAK are not essential for activity, animal model studies have yielded confusing and sometimes conflicting results as to the function of these domains. Functional analysis of GAK in zebrafish has been performed by injecting morpholinos into the zygote (Bai et al., 2010). When 20 residues are excised from the kinase domain of GAK, there are defects in Notch signaling during early zebrafish development, whereas later in development, there is an increased level of apoptosis in neuronal tissues, resulting in neurodegeneration. Interestingly, the same phenotype is obtained when a nonsense mutation is introduced into the PTEN-like domain of GAK, which if expressed, produces a GAK fragment without the clathrin-binding and J-domains. Based on these results, it appears that the kinase domain might have an essential role in GAK function, but this has not been validated by expressing only the clathrin-binding and J-domains of GAK in the morpholino-injected fish. This is especially important in light of the finding that morpholinos frequently do not give the same phenotype as that obtained using gene editing to disrupt proteins in zebrafish (Kok et al., 2015).
The importance of the rescue experiment in defining domain function is evident from studies using the Drosophila model system. Drosophila expresses only one auxilin ortholog that is structurally similar to GAK as it contains an N-terminal kinase domain. A nonsense mutation in the auxilin gene causes larval lethality, whereas missense mutations in either the kinase or PTEN-like domains produce developmental defects in the eye and the wing owing to impaired Notch signaling (Eun et al., 2008, 2007; Hagedorn et al., 2006; Kandachar et al., 2008). These latter results suggest a role for the kinase and PTEN-like domains in Drosophila auxilin, but surprisingly, expression of a truncated molecule comprising only the clathrin-binding and J-domains of Drosophila auxilin rescues these defects, indicating that the missense mutations apparently cause partial misfolding of the full-length auxilin protein. Therefore, there is no known function for the kinase and PTEN-like domains in Drosophila.
A definitive role and highly specific role for the kinase domain of GAK has been found in higher organisms. Engineering of mice to express a kinase-dead mutant of GAK from the GAK chromosomal locus results in newborn mice dying shortly after birth (Tabara et al., 2011). The neonatal mortality is caused by a defect in lung development. Interestingly, mouse embryonic fibroblasts (MEFs) derived from the above mice show normal distribution of clathrin, indicating that trafficking is not defective in these cells. In addition, our own studies using the GAK conditional knockout mice raised the possibility that the kinase domain has a specialized function in the mouse brain (Lee et al., 2008). When GAK is knocked out in the brain by expressing Cre recombinase from the nestin promoter, the mice die within 4 days of birth, even though there is still expression of auxilin in neuronal tissue. Histological analysis of the brains from the embryonic and newborn GAK-knockout mice shows gross morphological defects. By contrast, auxilin-knockout mice, which still express GAK in neuronal tissue, are viable even though many mice die at birth (Yim et al., 2010). Because neuronal expression of GAK, but not auxilin, is essential for mice viability, this raises the possibility that the kinase domain of GAK has a specialized function in neurons as it does in the lung.
Although a function for the PTEN-like domain has not been established in animal models, this domain is necessary for a rapid wave of GAK and auxilin recruitment to newly budded CCVs, as determined by imaging green fluorescent protein (GFP)-labeled proteins in tissue culture cells (Lee et al., 2006; Massol et al., 2006). Similar to the PTEN protein, the PTEN-like domain binds to phospholipids, preferentially phosphatidylinositol monophosphates (Lee et al., 2006; Massol et al., 2006), but unlike PTEN, it does not have tyrosine-protein phosphatase activity (Haynie and Ponting, 1996). When mutations in the PTEN-like domain of auxilin are made to weaken its affinity for lipids, the mutated auxilins are not recruited to newly budded CCVs in tissue culture cells (Guan et al., 2010). Based on these results, the expression of GAK fragments that lack the PTEN-like domain should markedly reduce the rate of uncoating of CCVs in GAK-knockout MEFS, which in turn might affect the overall rate of CME. Therefore, if the rate of recruitment of GAK and/or auxilin is important in the endocytic cycle, this should be most apparent in neuronal tissue, a tissue which requires rapid endocytosis of synaptic vesicles during neuronal transmission.
In this study, the physiological functions of the kinase and PTEN-like domains of GAK were examined in the conditional GAK-knockout and conventional auxilin-knockout mice models, which were engineered in our laboratory, by expressing a C-terminal fragment of GAK, GAK-C62. First, the GAK-C62 fragment, which comprises only the clathrin-binding and J-domains, rescued all the defects in clathrin chaperoning and uncoating that were caused by knocking out GAK in MEFs derived from the GAK-knockout mouse. Second, by engineering a transgenic mouse that expresses the GAK-C62 fragment, we found that this fragment rescued the lethality and morphological defects caused by knocking out GAK either in the liver or the brain. Therefore, neither the kinase nor the PTEN-like domains were necessary for GAK function in these tissues. Third, when both auxilin and GAK were knocked out in the brain of the GAK-C62 transgenic mice, surprisingly, these mice were viable and lived a normal life-span, albeit they were significantly smaller than their siblings. Therefore, the PTEN-like domain is not essential for auxilin and GAK function in the brain, a tissue in which rapid endocytosis is required for fast recycling of synaptic vesicles during neuronal transmission, although it might increase the efficiency of endocytosis in the brain.
RESULTS
Rescue of clathrin localization and steady-state distribution of TfnR and M6PR by different GAK fragments
Using MEFs derived from the conditional GAK-knockout mouse, GAK was knocked out in these cells through infection with adenovirus expressing Cre recombinase, as described in the Materials and Methods. At 5 days post infection, western blot analysis of cell lysates showed a greater than 95% reduction in GAK levels, whereas the levels of clathrin, AP1 and AP2 were unaffected (Fig. 1A). As expected, MEFs showed no detectable expression of the co-chaperone auxilin, which is highly homologous to GAK, but unlike GAK, is neuron-specific.
Fig. 1.

Rescue of clathrin–AP2 puncta on the plasma membrane of GAK-knockout MEFs by expressing different GAK fragments and chimeras. (A) Western blot of lysates from control (Con) MEFs, GAK-knockout (GAK K/O) MEFs, and cow brain extract. Cell lysates were blotted for GAK, clathrin heavy chain (CHC), auxilin (Aux), α-adaptin, γ-adaptin and β-actin (loading control). Cow brain extract was used a positive control for auxilin. (B) Schematic of different constructs used to rescue the defects in MEFs after knocking out GAK. The chimeras AP180–C58J and GGA1J comprise either the 58-kDa C-terminus of AP180 or the full-length GGA1 fused with the J-domain of GAK. The indicated domains are as follows: CB, Clathrin-binding domain; J, J-domain; K, kinase domain; P, PTEN-like domain. (C) Localization of clathrin and AP2 on the plasma membrane in control and GAK-knockout (GAK KO) MEFs. Fixed cells were immunostained for AP2 and clathrin. Images at low (left) and higher magnification (inset showing magnification of boxed region). (D) Effect of localization of clathrin and AP2 on the plasma membranes in GAK-knockout MEFs expressing different GAK fragments or chimeras. Five days after treating cells with adenovirus expressing Cre recombinase, the cells were transfected with the indicated mCherry-labeled constructs (upper panel). Two days after transfection, the cells were fixed and stained for clathrin and AP2 (lower panels).
Because Cre recombinase excises GAK from its chromosomal locus, the GAK-knockout MEFs are an excellent tool to determine whether the kinase and PTEN-like domains of GAK are necessary to restore the clathrin uncoating and chaperoning functions of Hsc70. The GAK-knockout cells were transfected with different GAK fragments in order to determine which fragments rescue GAK function (Fig. 1B). First, we examined the rescue of the clathrin puncta on the plasma membrane, which was disrupted in the GAK-knockout cells (Fig. 1C). After transfecting the GAK-knockout MEFs with different fragments, cells were co-stained for clathrin and AP2. Fig. 1D shows that, as expected, full-length GAK rescued the clathrin localization. The clathrin–AP2 puncta were also rescued by expressing GAK-ΔK, GAK-C62 and even GAK-C20 (a fragment that contains only one clathrin-binding motif and the J-domain), but not by GAK-C11, which comprises only the J-domain of GAK. Therefore, the clathrin-binding and J-domains appear to be necessary and sufficient for GAK co-chaperone activity. This was confirmed by expressing two different chimeras, which were engineered from monomeric clathrin adaptors and the J-domain of GAK (Fig. 1B). The expression of either the AP180–C58J chimera, comprising the 58-kDa C-terminal clathrin-binding domain of AP180 fused to the J-domain of GAK, or the GGA1J chimera, comprising full-length GGA1 fused to the J-domain of GAK, restored the clathrin puncta on the plasma membrane in the GAK-knockout cells (Fig. 1D).
We next examined whether these same constructs were able to rescue the clathrin-dependent trafficking in the GAK-knockout MEFs by examining the localization of transferrin receptor (TfnR) and mannose 6-phosphate receptor (M6PR). These two cargos bud from the membrane in CCVs, but differ in that TfnR and M6PR bud from the plasma membrane and TGN, respectively. The M6PR traffics through the endo-lysosomal pathway (Hirst et al., 1998) and then is recycled back to the TGN through the retromer complex (Arighi et al., 2004). When budding of CCVs from the TGN is inhibited, the M6PR is transported to the plasma membrane through a default secretory pathway (Traub and Kornfeld, 1997). Therefore, immunostaining shows the presence of TfnR and M6PR on the plasma membrane in the GAK-knockout MEFs (Fig. 2A), whereas these receptors have, predominantly, a juxtanuclear location in control cells. As shown in Fig. 2B and C, the juxtanuclear localization of TfnR and M6PR was restored by expressing different GFP-labeled GAK fragments, including GAK-C62 and GAK-C20.
Fig. 2.

Localization of TfnR and M6PR in control and GAK-knockout MEFs expressing different GAK constructs and chimeras. (A) Localization of TnfR and M6PR in control (Con) and GAK-knockout (GAK KO) MEFs. Cells were immunostained for TfnR and M6PR. (B) Effect of expressing different GFP-labeled GAK constructs and chimeras on the localization of TfnR in GAK-knockout MEFs. GAK-knockout cells that had been transfected with the indicated constructs were stained for TfnR. (C) Effect of expressing different GFP-labeled constructs and chimers on the localization of M6PR in GAK-knockout MEFs. GAK-knockout cells that had been transfected with the indicated constructs were stained for M6PR. (D) Effect of expressing GAK-C62 and GAK-C20 in GAK-knockout cells that had been treated with RNA oligonucleotides to knock down AP2. Cells were immunostained for TfnR and M6PR. The outlined cells in B–D indicate the cells expressing the mCherry-labeled GAK fragments.
Because M6PR on the cell surface can be internalized through CME (Le Borgne and Hoflack, 1997), we wanted to ensure that the juxtanuclear rescue of M6PR localization was due to the budding of CCVs from the TGN and not from the plasma membrane. Therefore, we repeated this rescue experiment in GAK-knockout MEFS that had been treated with small interfering (si)RNA oligonucleotides against AP2 to inhibit CME. The cells were then transfected with the different GAK constructs, followed by staining for TfnR and M6PR. As shown in Fig. 2D, GAK-C62 and GAK-C20 rescued the juxtanuclear localization of M6PR (Fig. 2D), but did not rescue TfnR internalization, indicating efficient knockdown of AP2. Therefore, the clathrin-binding and J-domains of GAK are sufficient to rescue clathrin-dependent trafficking.
Rescue of clathrin flashing and clathrin exchange by GAK fragments
It has been previously shown that the PTEN-like domain of either GAK or auxilin is necessary for a burst of recruitment of these co-chaperones to the CCVs, which occurs shortly after the scission of the CCPs by dynamin (Lee et al., 2006; Massol et al., 2006). Because these studies were performed in cells expressing endogenous GAK (Lee et al., 2006; Massol et al., 2006), we have reexamined whether there is a burst of recruitment, which appears as a flash, of the different GFP-labeled GAK fragments in the GAK-knockout MEFs. After imaging the plasma membrane with total internal reflection fluorescence (TIRF) microscopy, Metamorph software was used to analyze the frequency of GFP flashing on the plasma membrane. As shown in Fig. 3A, the rate of flashing of the GAK-ΔK fragment and the GAK-C62 fragment was 70% and 40%, respectively, of the rate obtained with the full-length molecule. There was no detectable flashing when the GAK-C20 fragment was expressed in the GAK-knockout cells. These results show that although the PTEN-like domain is not essential for flashing, it greatly enhances the rate of flashing.
Fig. 3.
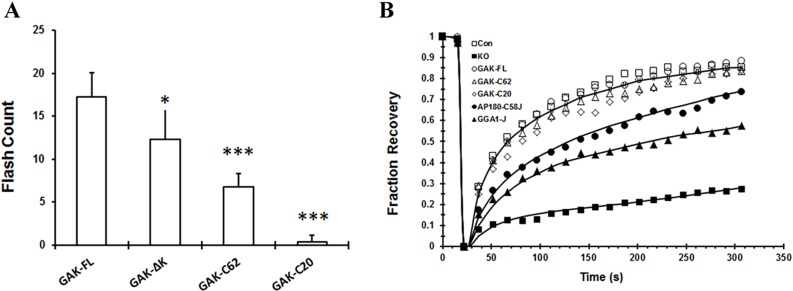
Recruitment of GAK fragments to the plasma membrane and rescue of clathrin exchange in GAK-knockout MEFs by the different constructs. (A) GAK-knockout cells were transfected with different GFP-labeled GAK fragments and then imaged using a TIRF microscope. GAK-FL, full-length GAK. The number of GAK flashes that occurred in a 25-µm2 area over a 6-s time period (a total of 60 images) was counted for each sample set on a minimum of 4 cells. *P<0.05 and ***P<0.001 are the statistical differences compared to the cells transfected with the GFP-labeled full-length GAK, determined using Student's t-test. (B) Rate of clathrin exchange in GAK-knockout (GAK KO) cells that had been transfected with different mCherry-labeled constructs and GFP-tagged clathrin light chain. Control (Con) cells were the MEFs derived from the GAK-knockout mice that still expressed endogenous GAK. At 48 h post transfection, a small region on the plasma membrane containing GFP-clathrin structures was photobleached, and imaging of the bleached area was performed to measure the fluorescence recovery. The fraction of initial fluorescence intensity was plotted versus time when data were analyzed, as described previously (Wu et al., 2001). For each construct, the plotted data were the average from 10 to 15 measurements. The data are representative of one of three sets of experiments that measured the fluorescence recovery of GFP–clathrin in cells that had been transfected with different mCherry-labeled GAK fragments.
Another function of GAK is that it is needed for the exchange of clathrin between CCPs and the cytosol (Hirst et al., 2008; Lee et al., 2008, 2005). To determine which domains of GAK are necessary for this function, GAK-knockout MEFs were co-transfected with GFP–clathrin-light-chain and mCherry-GAK fragments. Clathrin exchange was measured by photobleaching the GFP-labeled clathrin puncta, followed by determination of the rate of fluorescence recovery of the clathrin puncta. As shown in Fig. 3B, the rate of fluorescence recovery in cells that had been transfected with either GAK-C62 or GAK-C20 was not significantly different from that of control MEFs that still expressed endogenous GAK (Fig. 3B). In cells expressing the chimeras – either AP180–C58J or GGA1J – clathrin exchange occurred at a slower rate. Our previous study shows that the AP180–C58J construct stoichiometrically supports the uncoating of clathrin baskets by Hsc70, whereas the GAK fragments work catalytically (Ma et al., 2002). This might account for the slower rate of clathrin exchange in cells that express AP180–C58J than in cells that express the GAK fragments.
Rescue of transferrin internalization by GAK fragments
The rate of CME in the GAK-knockout cells that expressed different GFP-labeled GAK fragments was determined from the uptake of Alexa647-conjugated transferrin (Tfn) in 10 min, a time when Tfn internalization is still in the linear range. Cells were sorted based on the fluorescence intensities of both GFP and Alexa647 to determine the amount of Tfn internalized in the population of GFP-labeled cells. The Tfn uptake was then normalized to that of control MEFs that still expressed endogenous GAK. As shown in Fig. 4A, the extent of Tfn internalization in cells expressing GAK-ΔK and GAK-C62 was comparable to that in control cells, whereas less Tfn was internalized in cells expressing GAK-C20 as well as those expressing the AP180–C58J and GGA1J chimeras. These results suggest that neither the kinase nor the PTEN-like-domains of GAK affect the rate of Tfn internalization.
Fig. 4.
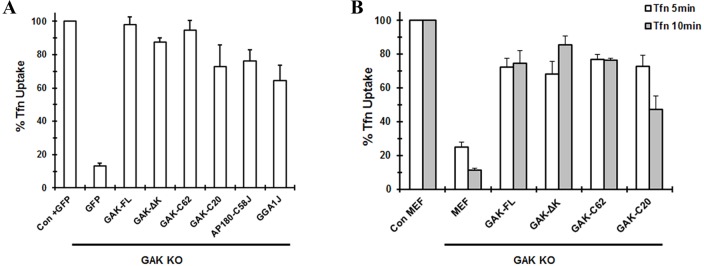
Histogram of Tfn uptake in GAK-knockout MEF cells transfected with different constructs. (A) Internalization of AlexaFluor647–Tfn was measured in GAK-knockout cells that had been transfected with GFP, different GFP-labeled GAK fragments or GFP-labeled chimeras. Control (Con) cells, the MEFs derived from the GAK-knockout mice that still expressed endogenous GAK, were transfected with GFP. Cells were sorted based on the intensity of GFP and AlexaFluor647 fluorescence. From the fluorescence-activated cell sorting (FACS) plot, the median fluorescence intensity of AlexaFluor674 for each population of GFP-expressing cells was calculated. Data are from four independent experiments (mean±s.d.). GAK-FL, full-length GAK. (B) Internalization of Tfn in GAK-knockout cells that had been stably transfected with different GAK fragments. After establishing the stable cell lines, GAK was knocked out by treating cells with adenovirus expressing Cre recombinase. Control MEFs were the stable MEF cell lines derived from the GAK-knockout mice that still expressed endogenous GAK. In control cells and in the adenovirus-treated stable cell lines, AlexaFluor488–Tfn was internalized for either 5 min (open bars) or 10 min (shaded bars). Cells were sorted based on the intensity of the AlexaFluor488 fluorescence to determine the internalization of Tfn. Data are from three independent experiments (mean±s.d.).
To confirm the above conclusion, instead of using transient transfection to express the different GAK constructs, stable MEF cell lines were made expressing Flag-tagged full length GAK, GAK-ΔK, GAK-C62 or GAK-C20. After deriving the stable cell lines, the expression levels of the Flag-tagged constructs were determined after knocking out the chromosomal GAK with Cre recombinase. Lysates from the stable cell lines were run on SDS-PAGE gels, followed by probing with antibodies against GAK and Flag. The expression of Flag-tagged GAK, GAK-ΔK and GAK-C62 was not significantly different from that of the endogenous GAK protein (the ratio of expressed GAK constructs to endogenous GAK was 1.1±0.2), whereas GAK-C20 was expressed at about half the level of that of the endogenous proteins (the ratio of GAK-C62 to endogenous GAK was 0.6±0.1).
Tfn uptake was measured in the different stable cell lines before (control) and after infection with adenovirus that encoded Cre recombinase. Flow cytometry was used to quantify the amount of Alexa488–Tfn internalized within 5 min (open bars) and 10 min (shaded bars). Fig. 4B shows that the Tfn uptake in cells that stably expressed GAK, GAK-ΔK or GAK-C62 was about 80% of the uptake of the controls both at 5 and 10 min. The GAK-C20-expressing cells showed reduced Tfn uptake compared to the control, especially at 10 min, which might be owing to the reduced expression level of this fragment. These results appear to show that GAK-C62 rescues CME as well as other clathrin-dependent pathways in GAK-knockout MEFs. Therefore, the kinase and PTEN-like domains do not appear to have an essential function for the co-chaperone activity of GAK in MEFs.
Transgenic GAK-C62 mice
Having established that GAK-C62 restored chaperoning of clathrin by Hsc70 in the GAK-knockout MEFS, we next examined whether this truncated fragment, which lacks both the kinase and PTEN-like domains, is able to rescue the lethality caused by knocking out GAK in the mouse. It has been previously shown that if the chromosomal GAK is mutated such that a kinase dead GAK is expressed, the mice all die shortly after birth owing to a defect in lung development (Tabara et al., 2011). Therefore, we decided to engineer a transgenic mouse to ubiquitously express the GAK-C62 fragment from the ROSA 26 promoter (Kisseberth et al., 1999). In all of our breeding schemes, we ensured that mice expressing GAK-C62 were heterozygous for this transgene (GAK-C62+/−). The expression of GAK-C62 in different tissues from the transgenic mice was confirmed by western blotting. As shown in Fig. 5A, GAK-C62 was expressed at comparable levels in the different tissues, which was expected based on the expression pattern of ROSA 26 (Giel-Moloney et al., 2007).
Fig. 5.
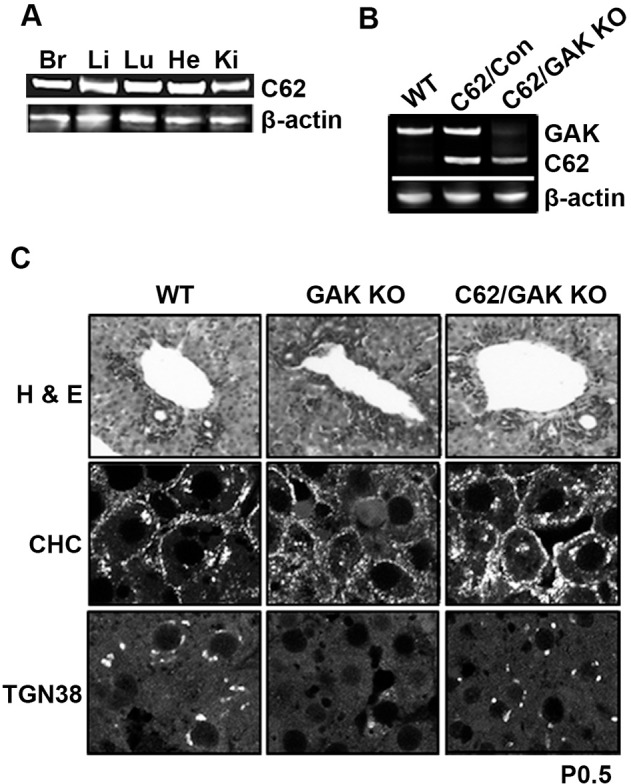
Transgenic expression of GAK-C62 rescues the lethality that is caused by knocking out GAK in the mouse liver. (A) Western blot of GAK-C62 (C62) in lysates prepared from different organs isolated from 1-week-old GAK-C62 control mouse (GAK-C62+/−). The following organs were used: Br, brain; Li, liver; Lu, lung, He, heart; Ki, kidney. GAK-C62 was probed using an antibody against GAK. Actin was loaded as an internal loading control. (B) Immunoblotting of GAK and GAK-C62 in liver lysates from 3-week-old wild-type (WT) mice, GAK-C62-expressing control mice (C62/Con) and GAK-C62-expressing GAK-knockout mice (C62/GAK KO). GAK and GAK-C62 (C62) were probed using an antibody against GAK. Actin was used as an internal loading control. (C) Histological analysis of livers from wild-type mice, GAK-knockout mice and GAK-C62/GAK-knockout mice. H&E and immunostaining of liver sections from newborn mice [postnatal day (P)0.5]. Cells were stained for clathrin heavy chain (CHC) and TGN38. The genotypes are as follows: WT (GAK+/+), C62/Con (GAK-C62+/−,GAKfl/fl), GAK KO (AlbCre+/−,GAKfl/fl), C62/GAK KO (GAK-C62+/−,AlbCre+/−,GAKfl/fl).
The GAK-C62 transgenic mice were crossed to the GAKfl/fl mice, and their offspring were then bred with mice expressing Cre recombinase from different tissue-specific promoters. First, we examined whether GAK-C62 rescues the lethality and liver dysfunction that is observed when GAK is knocked out in the liver (Lee et al., 2008). As previously reported, when GAK was knocked out in the liver by expressing Cre recombinase from the albumin promoter, the newborn mice were jaundiced, smaller than their littermates, and died several days after birth. When the 3-week-old offspring of mating GAK-C62+/−,GAKfl/fl mice with AlbCre+/−,GAKfl/− mice were genotyped, there were mice with the GAK-C62-expressing GAK-knockout (GAK-C62/GAK-knockout) genotype (GAK-C62+/−,AlbCre+/−,GAKfl/fl). In fact, genotyping many litters showed that the percentage of the GAK-C62/GAK-knockout mice was consistent with Mendelian genetics. Western blots of liver lysates were used to confirm that these mice were expressing GAK-C62, but not endogenous GAK (Fig. 5B). In addition, the western blot analysis provides a measure of the relative level of GAK-C62 relative to endogenous GAK. The expression level of GAK-C62 was, at most, twice (1.8±0.2, n=4) that of the full-length protein. As expected, the level of GAK-C62 was unaffected by knocking out endogenous GAK.
The mice that expressed GAK-C62, but not endogenous GAK, were healthy and lived a normal life span. To better characterize the phenotype of the rescued mice, histological liver sections were examined from wild-type, GAK-knockout and GAK-C62/GAK-knockout mice. As shown in Fig. 5C, the hematoxylin and eosin (H&E)-stained liver sections from the GAK-C62/GAK-knockout mice showed normal differentiation and biliary duct formation, unlike the liver from the GAK-knockout. Furthermore, immunostaining showed that the localization of clathrin and TGN38 at the Golgi in the GAK-C62/GAK-knockout mice appeared similar to that in the wild-type mice, instead of being mislocalized as observed in the GAK-knockout mouse (Lee et al., 2008).
Next, we examined whether GAK-C62 rescues the lethality that is caused by knocking out GAK in the brain by expressing Cre recombinase from the nestin promoter (Lee et al., 2008). Here again, we genotyped the mice at 3 weeks of age and found mice with the GAK-C62/GAK-knockout genotype (GAK-C62+/−,NesCre+/−,GAKfl/fl). Genotyping of many litters showed that the percentage of the GAK-C62/GAK-knockout mice was consistent with Mendelian genetics. Western blot analysis of brain lysates confirmed that these mice expressed GAK-C62 but not endogenous GAK (Fig. 6A). Before knocking out GAK, the relative expression level of GAK-C62 to endogenous GAK was 1.7±0.3 (n=5), which is similar to the ratio we obtained in the liver. There was no compensation in the amount of GAK-C62 or auxilin expressed when GAK was knocked out in neuronal tissue.
Fig. 6.

Expression of GAK-C62 rescues the lethality that is caused by knocking out GAK in mice both in the absence of auxilin expression. (A) Immunoblotting of GAK, GAK-C62 (C62) and auxilin in brain lysates from 3-week-old mice with the indicated genotypes. Actin was used as an internal loading control. (B) Ultrastructure of the brain of embryonic mice with altered expression of the co-chaperones GAK and auxilin. Brains were sectioned from mice [embryonic day (E)15.5] with the following genotypes: wild type (WT), GAK-knockout (GAK KO), GAK-C62/GAK-knockout (C62/GAK KO), GAK-C62/double knockout (C62/DKO). H&E staining of the coronal section of the cerebral cortex. CP, cortical plate; VZ, ventricular zone. (C) Body size comparison of GAK-C62 transgenic mice with the indicated genotypes at 3 weeks of age. Aux KO, auxilin knockout; DKO, GAK and auxilin double knockout; GAK knockout, GAK KO. Mice genotypes are given below. (D) Comparison of the relative body weights of GAK-C62 transgenic mice with the indicated genotypes. The body weight of 3-week-old mice was measured from 10 mice for each genotype. Error bars show mean±s.e.m. (E) Western blot of brain lysates from 3-week-old GAK-C62 transgenic mice with the indicated genotypes. The blots were probed with the indicated antibodies. Genotypes are as follows: wild type (GAK+/+); GAK-C62-expressing control mice, C62/Con (GAK-C62+/−,GAKfl/fl); GAK KO (NesCre+/−,GAKfl/fl); C62/GAK KO (GAK-C62+/−,NesCre+/−,GAKfl/fl); C62/Aux KO (GAK-C62+/−,Aux−/−,); C62/DKO (GAK-C62+/−,NesCre+/−,GAKfl/fl,Aux−/−).
The GAK-C62/GAK-knockout mice lived a normal life-span with no obvious behavioral defects. H&E staining of brain sections from the GAK-C62/GAK-knockout mice showed no prominent loss of cells in the cerebellar cortex (Fig. 6B), unlike the brains from the GAK-knockout mice that were not expressing GAK-C62. The GAK-C62/GAK-knockout mice were on average 25% smaller than control mice (Fig. 6D), similar to the reduction in size observed in the conventional auxilin-knockout mouse. These results show that the kinase and PTEN-like domains of GAK do not have a specialized function in the brain. However, the above results do not address whether the PTEN-like domain has an essential role in the brain because auxilin is still expressed in the GAK-C62/GAK-knockout mice. Therefore, to examine whether this domain has an essential function in neurons, we engineered a mouse that expressed GAK-C62 but neither GAK nor auxilin in neuronal cells by breeding the GAK conditional knockout mice and the auxilin conventional knockout mice with the GAK-C62 transgenic mouse and then crossing the offspring to mice expressing Cre recombinase from the nestin promoter. Genotyping the mice at 3 weeks of age showed that there were mice expressing GAK-C62 that were also double knockout for GAK and auxilin (GAK-C62/double knockout) (GAK-C62+/−,Aux−/−,NesCre+/−,GAKfl/fl). Because the conventional auxilin-knockout mouse already has multiple defects – including high neonatal mortality, infertility problems and low body weight (Yim et al., 2010) – we were surprised that the GAK-C62/double-knockout mice were viable. As expected, the GAK-C62/double-knockout mice had the same phenotypic defects as the auxilin-knockout mice, but in addition, the mice were distinguished by being much smaller than littermates with different genotypes (Fig. 6C). At 3 weeks of age, when the mice were weaned, the weight of the GAK-C62/double-knockout mice were about half the weight of the control mice that expressed both auxilin and full-length GAK, along with GAK-C62, in the brain (Fig. 6D). The small size of the transgenic GAK-C62/double-knockout mice persisted into adulthood, which shows that insufficient supply of mother's milk did not cause their small size.
In addition to genotyping the mice, western blot analysis of brain lysates from the GAK-C62/double-knockout mice confirmed the lack of GAK and auxilin expression. As noted above, before knocking out GAK, the level of GAK-C62 was, at most, twice the level of endogenous GAK. Knocking out GAK and/or auxilin did not affect the expression of GAK-C62. Western blot analysis was used to compare the levels of other endocytic proteins in brain lysates to determine whether there was a change in their expression. In particular, we were interested in whether synaptojanin or endophilin was upregulated in the GAK-C62/double-knockout mice. Both the synaptojanin- and endophilin-knockout mice had more CCVs (Cremona et al., 1999; Milosevic et al., 2011), which suggests that these proteins play a role in dissociating the clathrin coat. As shown in the western blot analysis, there was no detectable upregulation of synaptojanin 1 or endophilin 1 in the GAK-C62/double-knockout mouse (Fig. 6E). Therefore, GAK-C62 is sufficient to support the interaction of clathrin with Hsc70 in the brain, which shows that the kinase and PTEN-like domains are not essential for synaptic function in the mouse.
DISCUSSION
The numerous studies on the function of members of the auxilin family have shown that these proteins, which are conserved from yeast to man, have an essential role in the uncoating and chaperoning of clathrin by Hsc70. A further understanding of these co-chaperones has come from functional analysis of their individual domains. At the C-terminus of GAK and auxilin, there is a J-domain, which binds to Hsc70, and adjacent to the J-domain is the clathrin-binding domain. The clathrin-binding and J-domains of auxilin and GAK have been shown to be necessary and sufficient to support clathrin uncoating in biochemical studies (Holstein et al., 1996; Ma et al., 2002). The roles of the other domains – the PTEN-like domain, which is present in both GAK and auxilin, and the kinase domain, which is present only in GAK – are less well understood. In this study, we addressed whether the kinase and PTEN-like domains are important in mammalian cells and in mammals. Specifically, we examined the role of these domains in our GAK-knockout mouse model and MEFs derived from these mice.
By expressing different GAK fragments in the GAK-knockout MEFs, we showed that a fragment comprising the clathrin-binding and J-domains of GAK (GAK-C62) restored clathrin uncoating and chaperoning when GAK was knocked out in the MEFs, in agreement with other studies (Eun et al., 2008; Kandachar et al., 2008). Specifically, similar to the control MEFs, the GAK-knockout MEFs expressing GAK-C62 restored the localization of clathrin, the steady-state distribution of TfnR and M6PR, the rate of clathrin exchange, and the rate of Tfn uptake. Compared to full-length GAK, there was a marked reduction in the frequency of GAK-C62 flashing on the plasma membrane, which is a measure of its bulk recruitment onto newly budded CCVs, which occurs just before uncoating (Lee et al., 2006). Evidently, this reduction in recruitment, which is expected to decrease the rate of uncoating, does not manifest itself in a decreased rate of Tfn uptake. These results show that the kinase and PTEN-like domains are not required for clathrin uncoating or chaperoning in MEFs. However, they might modulate the function of GAK. For example, expression of a kinase-dead mutant of GAK in cells in which GAK levels were knocked down with RNA interference caused a reduction in the rate of maturation of cathepsin D (Kametaka et al., 2007), a process in which cathepsin D traffics in a clathrin-dependent pathway from the TGN to the lysosome.
Other studies using mouse models have provided mixed results as to the function of the kinase and PTEN-like domains of GAK and auxilin. Interestingly, auxilin from yeast and C. elegans does not contain a kinase domain or a PTEN-like domain, which shows that these domains are not necessary for the co-chaperone function of auxilin in these organisms (Lemmon, 2001). In Drosophila, only one member of the auxilin family is expressed, which contains a domain structure similar to that of GAK. Because missense mutations in the kinase and PTEN-like domain of Drosophila auxilin cause developmental defects through impaired notch signaling (Eun et al., 2008, 2007; Hagedorn et al., 2006; Kandachar et al., 2008), at first, it seems as if these domains are essential. However, because these missense mutations, as well as nonsense mutations that cause larval lethality, can be rescued by expressing a fragment comprising just the clathrin-binding and J-domains (Eun et al., 2008; Kandachar et al., 2008), it appears that the missense mutations cause overall misfolding of the full-length protein rather than simply disrupting the kinase and PTEN-like domains (Eun et al., 2008).
In higher organisms, the two members of the auxilin family differ in their expression patterns; GAK is expressed ubiquitously, and auxilin is expressed only in neurons. When morpholinos are used to excise a small region of the kinase domain of GAK in zebrafish, the brain has developmental defects due to impairment of notch signaling (Bai et al., 2010), just as observed in Drosophila (Eun et al., 2008; Hagedorn et al., 2006). Surprisingly, a similar phenotype in the zebrafish is obtained when morpholinos are used to generate a nonsense mutation in the PTEN-like domain of GAK (Bai et al., 2010). However, there has been no attempt to rescue the zebrafish defects with a fragment comprising just the clathrin-binding and J-domains of GAK so it is unclear whether, as in Drosophila (Eun et al., 2008; Kandachar et al., 2008), these domains are sufficient for GAK function in zebrafish. Nor it is clear why the effects of mutating GAK are not rescued by auxilin, which is also present in neurons; perhaps auxilin is not expressed early in development or maybe it does not localize to the same part of the neuron as GAK.
In the mouse, it has been well established that the kinase domain of GAK is essential for lung development (Tabara et al., 2011), but it is not clear whether it has an important function in other tissues or in the adult mouse. It is also unclear whether the PTEN-like domain of GAK and auxilin are important in the mouse to rapidly recruit these co-chaperones to the plasma membrane to promote fast uncoating of the CCVs. In a model of clathrin uncoating by Hsc70, proposed by the Kirchhausen laboratory (Massol et al., 2006), the hydrolysis of phospholipids on the membrane of the CCVs by synaptojanin regulates the recruitment of GAK and auxilin. This recruitment is dependent on the binding of the PTEN-like domains of these co-chaperones to phospholipids on the newly budded CCVs (Guan et al., 2010). Therefore, it might be expected that this domain is required to achieve rapid endocytosis.
In the present study, to better understand the function of the kinase and PTEN-like domains, we engineered a transgenic GAK-C62 mouse to determine whether this fragment rescues the defects caused by tissue-specific knockouts in the GAK- and auxilin-knockout mice models (Lee et al., 2008; Yim et al., 2010). First, we found that GAK-C62 rescued the lethality that is caused by knocking out GAK in the liver. The rescued mice were healthy with no apparent histological or physiological defects, which indicate a non-essential function for the kinase and PTEN-like domain in this tissue. Similar results were obtained when we knocked out GAK in the brain. This shows that not only is the kinase domain not required in the brain but, in addition, the lethality that we previously observed when GAK was knocked out was either due to lack of expression of auxilin during the early stages of brain development or else due to different subcellular localization of GAK and auxilin (Lee et al., 2008).
Knocking out the neuronally expressed auxilin in mice produces multiple defects, even though the neurons express GAK (Yim et al., 2010). The auxilin-knockout mice have a high incidence of perinatal mortality, low body weight and infertility (Yim et al., 2010). Furthermore, electron microscopy studies show that synapses from the auxilin-knockout mice have an increase in the number of clathrin-coated structures, whereas electrophysiology studies have shown an impairment of clathrin-mediated endocytosis in the synaptic vesicles of neurons from the knockout mice (Yim et al., 2010). In this study, we used the auxilin-knockout mice, along with the GAK-knockout mice, to generate a mouse that expresses only GAK-C62 in neuronal tissue in order to examine whether the PTEN-like domain has an essential function in the brain. Because the PTEN-like domain has been proposed to be necessary for the rapid recruitment of auxilin to the plasma membrane (Guan et al., 2010), we expected profound defects in the GAK-C62/double-knockout mice due to the lack of rapid endocytosis in order to recycle the synaptic vesicles during neurotransmission. Surprisingly, the major defect of the GAK-C62/double knockout was their small size, which suggests that a hypothalamus defect causes a reduction in the secretion of growth hormone from the pituitary gland in these animals (Slabaugh et al., 1981). Otherwise, these mice were viable with no apparent behavioral defects and lived a normal life span.
Although our data show that the PTEN-like domains are not essential in the mice brain and liver, missense mutations in the PTEN-like domain of auxilin has been reported to cause juvenile Parkinson's disease (Edvardson et al., 2012). In a possibly related effect, GAK, through its PTEN-like domain, forms a complex with leucine-rich kinase 2 (LRRK2), a kinase that when mutated is associated with a high risk of the development of Parkinson's disease, along with mutations in Bag5 and Rab7L1 (Beilina et al., 2014). This multi-protein complex is needed to maintain the integrity of the TGN. Therefore, these results suggest that the PTEN-like domains of GAK and auxilin have other functions that are not yet understood, aside from facilitating the recruitment of these co-chaperones to phospholipids.
MATERIALS AND METHODS
Cell culture and Ad-Cre infection
MEFs derived from mice with GAK fl/fl (Lee et al., 2008) were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 4.5 g/l glucose, 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM l-glutamine, and 10% fetal bovine serum (FBS; Invitrogen). For adenoviral infection, cells plated 1 day before infection were incubated in fresh DMEM culture medium with adenovirus expressing Cre recombinase (Ad-Cre; Vector Biolabs) at a multiplicity of infection (MOI) of 200 in the presence of 5 µg/ml polybrene (Sigma-Aldrich), as described previously (Olszewski et al., 2014). The stable cell lines were treated with adenovirus as described above to knockout the endogenous GAK. To knockdown AP2, oligonucleotide duplexes (Dharmacon) against α-adaptin were transfected using Lipofectamine RNAiMAX reagent (Invitrogen).
Stable cell lines expressing full-length and different GAK fragments were generated by infecting the GAK-knockout MEFs with different pBabe retroviral expression vectors. The stable cell lines were selected for 4–6 weeks with puromyocin. The stable cell lines were then treated with adenovirus expressing Cre recombinase to knockout GAK gene expression.
Immunofluorescence microscopy
MEFs plated onto Lab-Tek glass chamber slides (Nalge Nunc) were fixed in 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) and then immunostained with the following primary antibodies: rabbit polyclonal antibody against clathrin heavy chain (Abcam); rabbit polyclonal antibodies against TGN38 (AbD Serotec), cation-independent M6PR (from Dr Linton Traub, University of Pittsburgh, Pittsburgh, PA), mouse monoclonal antibodies against GM130 (clone 35; BD Biosciences), α-adaptin (clone AP6; Affinity BioReagents), and TfnR (Invitrogen). Cells were immunostained with fluorescently labeled secondary antibodies (Invitrogen and Jackson ImmunoResearch Laboratories).
Western blotting
Protein lysates from cells or tissues were run on 4–12% NuPAGE gels (Invitrogen) and transferred onto nitrocellulose membranes (Invitrogen) through Trans Blot SD system (Bio-Rad, Hercules, CA). The proteins were immunoblotted with the following antibodies: rabbit polyclonal antibodies against GAK (Greener et al., 2000), auxilin (Yim et al., 2010), synaptonin 1 (Dr Pietro de Camilli, Yale University, New Haven, CT), and endophilin 1 (Dr Pietro de Camilli); mouse monoclonal antibodies to β-actin (clone AC-15; Abcam), dynamin (clone Hudy1 from Invitrogen), FLAG antibody (Sigma-Aldrich), clathrin heavy chain (clone 23, BD Biosciences), α-adaptin (clone 8, BD Biosciences) and γ-adaptin (clone 88, BD Biosciences). Protein bands were detected by using a classic chemiluminescent method with the ECL blotting substrate (Thermo Fisher Scientific) and horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories), or with an imaging method using infrared secondary antibodies, such as IRDye 680 and 800CW (Li-Cor Bioscience, Lincoln, NE) followed by scanning in the Odyssey infrared detection system (Li-Cor Bioscience).
Plasmids and transfection
The construction of GFP-tagged full-length GAK, GFP-tagged GAK truncation constructs (GAK-ΔK, GAK-C62, GAK-C20, GAK-C11) and GFP-LCa (encoding GFP-tagged clathrin light chain A) has been described previously (Lee et al., 2005). The GAK fragments were also subcloned into pmCherry-C1 vector (Clontech). To generate chimeric constructs, referred to as AP180-C58J and GGA1J, DNA fragments of a 58-kDa C-terminal domain of AP180 and a full-length GGA1 protein were amplified from pQE30-AP180-C58J (Ma et al., 2002) and pEGFP-GGA1 (Puertollano et al., 2003), respectively. The J-domain of GAK was cloned in frame at the C-terminus of GGA1. The chimeras were cloned into either pEGFP or pmCherry vectors (Invitrogen). To make the stable cell lines, the pBabe retro viral expression vector (Cell Biolabs) was used to express the full-length and fragments of GAK. Transient transfections were performed using FuGENE HD reagent (Roche) as per the manufacturer's instructions. To transfect the cells, FuGENE HD reagent (Roche) in Opti-MEM (Invitrogen) was mixed with plasmid DNA at a 6:2 ratio.
Fluorescent recovery after photobleaching and TIRF imaging of cells
Cells were imaged using the Zeiss LSM 510 confocal microscope imaging system (Carl Zeiss MicroImaging) with a 40× or a 63×, 1.4 NA objectives.
GFP–clathrin in normal MEFs or GAK-knockout MEFs with or without expression of rescue constructs was imaged and photobleached using a 488-nm laser light with a 40×, 1.4 NA objective. A defined region was photobleached at high laser power resulting in 50–80% reduction in the fluorescence intensity. Scanning at low laser power monitored the fluorescence recovery after photobleaching (FRAP). Data were analyzed as described previously (Wu et al., 2001).
For TIRF imaging, the Olympus microscope was used with a 60×1.45 NA oil-immersion objective using the 488-nm line of an argon laser (Melles Griot, Carlsbad, CA). Images were collected using a Photometrics Cool Snap HQ CCD camera (Photometrics, Tucson, AZ). Streams of 120 frames were exposed and acquired at 10 frames/s for 120 s.
Tfn internalization
MEFs were incubated in DMEM containing 0.2% bovine serum albumin (DMEM-BSA) for 1 h to deplete serum. The cells were transferred to fresh DMEM-BSA medium containing 10 µg/ml AlexaFluor-conjugated Tfn and incubated for 10 min. The cells were then washed with cold DMEM-BSA, incubated with cold acid buffer (0.2 M acetic acid, 0.5 M NaCl) to dissociate surface Tfn and then fixed with 4% PFA. AlexaFluor-labeled Tfn was analyzed with flow cytometry using a FACS Calibur instrument.
Engineering and breeding of transgenic mouse
The GAK-C62 fragment was expressed from the ROSA 26 promoter. The fragment was cloned into the AgeI and EcoRI restriction sites that are present in the multiple-cloning site of the pBroad3 vector (InvivoGen). The vector was linearized using the PacI restriction enzyme (New England Biolabs) and then purified by using gel extraction. The vector was then injected into fertilized C57BL/6 mouse eggs, followed by their implantation into pseudo-pregnant recipient mice. The genotype of the transgenic mouse was determined by using PCR analysis with genomic DNA isolated from the tail of the mice.
To determine whether GAK-C62 rescued the GAK-knockout phenotypes, the GAK-C62 transgenic mouse was bred with a GAKfl/fl mouse to produce GAK-C62+/−,GAKfl/fl. These mice were then bred with AlbCre+/− and NesCre+/− mice to knockout GAK in the liver and brain, respectively. The transgenic Cre mice lines were purchased from Jackson Laboratory. To generate the GAK-C62 double knockout of neuronal GAK and auxilin, GAK-C62+/−,NesCre+/−,GAKfl/fl were bred to the Aux−/−,GAKfl/fl to generate Gak-C62+/−. GAKfl/fl,Aux−/−,NesCre+/− mice. Mice were genotyped by using PCR analysis with genomic DNA isolated from the mice tail. In all of the GAK-C62 rescue experiments, the mice were always heterozygous for GAK-62.
All mice were handled in accordance with National Institutes of Health guide for the care and use of laboratory animals.
Histological studies
The liver and brain were dissected from the mice followed by fixation in 10% phosphate-buffered formalin for a day at room temperature. The organs were paraffin embedded and sectioned (5 µm) by HistoServ (Germantown, MD). H&E staining and immunostaining were performed as described previously (Lee et al., 2008; Yim et al., 2010).
Acknowledgements
We would like to acknowledge the following National Heart, Lung, and Blood Institute core facilities – transgenic core, light microscopy core and flow cytometry core.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
B.-C.P., Y.-I.Y., X.Z. and M.B.O. performed experiments and analyzed data. L.E.G. and E.E. planned experiments and wrote the manuscript.
Funding
This work was supported by the Intramural Research Program in the National Heart, Lung, and Blood Institute (NHLBI) at the National Institutes of Health (NIH). Deposited in PMC for release after 12 months.
References
- Arighi C. N., Hartnell L. M., Aguilar R. C., Haft C. R. and Bonifacino J. S. (2004). Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 165, 123-133. 10.1083/jcb.200312055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai T., Seebald J. L., Kim K.-E., Ding H.-M., Szeto D. P. and Chang H. C. (2010). Disruption of zebrafish cyclin G-associated kinase (GAK) function impairs the expression of Notch-dependent genes during neurogenesis and causes defects in neuronal development. BMC Dev. Biol. 10, 7 10.1186/1471-213X-10-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A., Rudenko I. N., Kaganovich A., Civiero L., Chau H., Kalia S. K., Kalia L. V., Lobbestael E., Chia R., Ndukwe K. et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 111, 2626-2631. 10.1073/pnas.1318306111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell T. G., Welch W. J., Schlossman D. M., Palter K. B., Schlesinger M. J. and Rothman J. E. (1986). Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell 45, 3-13. 10.1016/0092-8674(86)90532-5 [DOI] [PubMed] [Google Scholar]
- Cremona O., Di Paolo G., Wenk M. R., Lüthi A., Kim W. T., Takei K., Daniell L., Nemoto Y., Shears S. B., Flavell R. A. et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179-188. 10.1016/S0092-8674(00)81649-9 [DOI] [PubMed] [Google Scholar]
- Doherty G. J. and McMahon H. T. (2009). Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857-902. 10.1146/annurev.biochem.78.081307.110540 [DOI] [PubMed] [Google Scholar]
- Edvardson S., Cinnamon Y., Ta-Shma A., Shaag A., Yim Y.-I., Zenvirt S., Jalas C., Lesage S., Brice A., Taraboulos A. et al. (2012). A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 7, e36458 10.1371/journal.pone.0036458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E. and Greene L. E. (2007). Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic 8, 640-646. 10.1111/j.1600-0854.2007.00568.x [DOI] [PubMed] [Google Scholar]
- Eun S. H., Lea K., Overstreet E., Stevens S., Lee J.-H. and Fischer J. A. (2007). Identification of genes that interact with Drosophila liquid facets. Genetics 175, 1163-1174. 10.1534/genetics.106.067959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun S. H., Banks S. M. L. and Fischer J. A. (2008). Auxilin is essential for Delta signaling. Development 135, 1089-1095. 10.1242/dev.009530 [DOI] [PubMed] [Google Scholar]
- Giel-Moloney M., Krause D. S., Chen G., Van Etten R. A. and Leiter A. B. (2007). Ubiquitous and uniform in vivo fluorescence in ROSA26-EGFP BAC transgenic mice. Genesis 45, 83-89. 10.1002/dvg.20269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greener T., Zhao X., Nojima H., Eisenberg E. and Greene L. E. (2000). Role of cyclin G-associated kinase in uncoating clathrin-coated vesicles from non-neuronal cells. J. Biol. Chem. 275, 1365-1370. 10.1074/jbc.275.2.1365 [DOI] [PubMed] [Google Scholar]
- Greener T., Grant B., Zhang Y., Wu X., Greene L. E., Hirsh D. and Eisenberg E. (2001). Caenorhabditis elegans auxilin: a J-domain protein essential for clathrin-mediated endocytosis in vivo. Nat. Cell Biol. 3, 215-219. 10.1038/35055137 [DOI] [PubMed] [Google Scholar]
- Guan R., Han D., Harrison S. C. and Kirchhausen T. (2010). Structure of the PTEN-like region of auxilin, a detector of clathrin-coated vesicle budding. Structure 18, 1191-1198. 10.1016/j.str.2010.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn E. J., Bayraktar J. L., Kandachar V. R., Bai T., Englert D. M. and Chang H. C. (2006). Drosophila melanogaster auxilin regulates the internalization of Delta to control activity of the Notch signaling pathway. J. Cell Biol. 173, 443-452. 10.1083/jcb.200602054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynie D. T. and Ponting C. P. (1996). The N-terminal domains of tensin and auxilin are phosphatase homologues. Protein Sci. 5, 2643-2646. 10.1002/pro.5560051227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw J. E. (2000). Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 16, 483-519. 10.1146/annurev.cellbio.16.1.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J., Futter C. E. and Hopkins C. R. (1998). The kinetics of mannose 6-phosphate receptor trafficking in the endocytic pathway in HEp-2 cells: the receptor enters and rapidly leaves multivesicular endosomes without accumulating in a prelysosomal compartment. Mol. Biol. Cell 9, 809-816. 10.1091/mbc.9.4.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst J., Sahlender D. A., Li S., Lubben N. B., Borner G. H. H. and Robinson M. S. (2008). Auxilin depletion causes self-assembly of clathrin into membraneless cages in vivo. Traffic 9, 1354-1371. 10.1111/j.1600-0854.2008.00764.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein S. E., Ungewickell H. and Ungewickell E. (1996). Mechanism of clathrin basket dissociation: separate functions of protein domains of the DnaJ homologue auxilin. J. Cell Biol. 135, 925-937. 10.1083/jcb.135.4.925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R., Gao B., Prasad K., Greene L. E. and Eisenberg E. (2000). Hsc70 chaperones clathrin and primes it to interact with vesicle membranes. J. Biol. Chem. 275, 8439-8447. 10.1074/jbc.275.12.8439 [DOI] [PubMed] [Google Scholar]
- Kametaka S., Moriyama K., Burgos P. V., Eisenberg E., Greene L. E., Mattera R. and Bonifacino J. S. (2007). Canonical interaction of cyclin G-associated kinase with Adaptor protein 1 regulates lysosomal enzyme sorting. Mol. Biol. Cell 18, 2991-3001. 10.1091/mbc.E06-12-1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandachar V., Bai T. and Chang H. C. (2008). The clathrin-binding motif and the J-domain of Drosophila Auxilin are essential for facilitating Notch ligand endocytosis. BMC Dev. Biol. 8, 50 10.1186/1471-213X-8-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T., Owen D. and Harrison S. C. (2014). Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 6, a016725 10.1101/cshperspect.a016725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseberth W. C., Brettingen N. T., Lohse J. K. and Sandgren E. P. (1999). Ubiquitous expression of marker transgenes in mice and rats. Dev. Biol. 214, 128-138. 10.1006/dbio.1999.9417 [DOI] [PubMed] [Google Scholar]
- Kok F. O., Shin M., Ni C.-W., Gupta A., Grosse A. S., van Impel A., Kirchmaier B. C., Peterson-Maduro J., Kourkoulis G., Male I. et al. (2015). Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 32, 97-108. 10.1016/j.devcel.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R. and Hoflack B. (1997). Mannose 6-phosphate receptors regulate the formation of clathrin-coated vesicles in the TGN. J. Cell Biol. 137, 335-345. 10.1083/jcb.137.2.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-w., Zhao X., Zhang F., Eisenberg E. and Greene L. E. (2005). Depletion of GAK/auxilin 2 inhibits receptor-mediated endocytosis and recruitment of both clathrin and clathrin adaptors. J. Cell Sci. 118, 4311-4321. 10.1242/jcs.02548 [DOI] [PubMed] [Google Scholar]
- Lee D. W., Wu X., Eisenberg E. and Greene L. E. (2006). Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J. Cell Sci. 119, 3502-3512. 10.1242/jcs.03092 [DOI] [PubMed] [Google Scholar]
- Lee D.-w., Zhao X., Yim Y.-I., Eisenberg E. and Greene L. E. (2008). Essential role of cyclin-G-associated kinase (Auxilin-2) in developing and mature mice. Mol. Biol. Cell 19, 2766-2776. 10.1091/mbc.E07-11-1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon S. K. (2001). Clathrin uncoating: auxilin comes to life. Curr. Biol. 11, R49-R52. 10.1016/S0960-9822(01)00010-0 [DOI] [PubMed] [Google Scholar]
- Ma Y., Greener T., Pacold M. E., Kaushal S., Greene L. E. and Eisenberg E. (2002). Identification of domain required for catalytic activity of auxilin in supporting clathrin uncoating by Hsc70. J. Biol. Chem. 277, 49267-49274. 10.1074/jbc.M203695200 [DOI] [PubMed] [Google Scholar]
- Massol R. H., Boll W., Griffin A. M. and Kirchhausen T. (2006). A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc. Natl. Acad. Sci. USA 103, 10265-10270. 10.1073/pnas.0603369103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I., Giovedi S., Lou X., Raimondi A., Collesi C., Shen H., Paradise S., O'Toole E., Ferguson S., Cremona O. et al. (2011). Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587-601. 10.1016/j.neuron.2011.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski M. B., Chandris P., Park B.-C., Eisenberg E. and Greene L. E. (2014). Disruption of clathrin-mediated trafficking causes centrosome overduplication and senescence. Traffic 15, 60-77. 10.1111/tra.12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R., van der Wel N. N., Greene L. E., Eisenberg E., Peters P. J. and Bonifacino J. S. (2003). Morphology and dynamics of clathrin/GGA1-coated carriers budding from the trans-Golgi network. Mol. Biol. Cell 14, 1545-1557. 10.1091/mbc.02-07-0109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabaugh M. B., Lieberman M. E., Rutledge J. J. and Gorski J. (1981). Growth hormone and prolactin synthesis in normal and homozygous Snell and Ames dwarf mice. Endocrinology 109, 1040-1046. 10.1210/endo-109-4-1040 [DOI] [PubMed] [Google Scholar]
- Tabara H., Naito Y., Ito A., Katsuma A., Sakurai M. A., Ohno S., Shimizu H., Yabuta N. and Nojima H. (2011). Neonatal lethality in knockout mice expressing the kinase-dead form of the gefitinib target GAK is caused by pulmonary dysfunction. PLoS ONE 6, e26034 10.1371/journal.pone.0026034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub L. M. and Kornfeld S. (1997). The trans-Golgi network: a late secretory sorting station. Curr. Opin. Cell Biol. 9, 527-533. 10.1016/S0955-0674(97)80029-4 [DOI] [PubMed] [Google Scholar]
- Wu X., Zhao X., Baylor L., Kaushal S., Eisenberg E. and Greene L. E. (2001). Clathrin exchange during clathrin-mediated endocytosis. J. Cell Biol. 155, 291-300. 10.1083/jcb.200104085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim Y.-I., Sun T., Wu L.-G., Raimondi A., De Camilli P., Eisenberg E. and Greene L. E. (2010). Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Natl. Acad. Sci. USA 107, 4412-4417. 10.1073/pnas.1000738107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. X., Engqvist-Goldstein A. E. Y., Carreno S., Owen D. J., Smythe E. and Drubin D. G. (2005). Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic 6, 1103-1113. 10.1111/j.1600-0854.2005.00346.x [DOI] [PubMed] [Google Scholar]