

Abstract
Enzymatic metabolism of the 20C polyunsaturated fatty acid (PUFA) arachidonic acid (AA) occurs via the cyclooxygenase (COX) and lipoxygenase (LOX) pathways, and leads to the production of various bioactive lipids termed eicosanoids. These eicosanoids have a variety of functions, including stimulation of homeostatic responses in the cardiovascular system, induction and resolution of inflammation, and modulation of immune responses against diseases associated with chronic inflammation, such as cancer. Because chronic inflammation is essential for the development of colorectal cancer (CRC), it is not surprising that many eicosanoids are implicated in CRC. Oftentimes, these autacoids work in an antagonistic and highly temporal manner in inflammation; therefore, inhibition of the pro-inflammatory COX-2 or 5-LOX enzymes may subsequently inhibit the formation of their essential products, or shunt substrates from one pathway to another, leading to undesirable side-effects. A better understanding of these different enzymes and their products is essential not only for understanding the importance of eicosanoids, but also for designing more effective drugs that solely target the inflammatory molecules found in both chronic inflammation and cancer. In this review, we have evaluated the cancer promoting and anti-cancer roles of different eicosanoids in CRC, and highlighted the most recent literature which describes how those molecules affect not only tumor tissue, but also the tumor microenvironment. Additionally, we have attempted to delineate the roles that eicosanoids with opposing functions play in neoplastic transformation in CRC through their effects on proliferation, apoptosis, motility, metastasis, and angiogenesis.
Keywords: Eicosanoids, Cyclooxygenase, Lipoxygenase, Inflammation, Colorectal cancer
Core tip: Eicosanoids are bioactive lipids generated from polyunsaturated fatty acids (usually arachidonic acid) through highly regulated enzymatic pathways in many different cell types. These molecules are effective in small amounts, and may act in an autocrine or paracrine manner to regulate some of the most important steps in the development of acute inflammation and its resolution. Aberrant expression of the enzymes that help synthesize these bioactive lipids is frequently seen in diseases associated with chronic inflammation, including cancer.
INFLAMMATION AND COLORECTAL CANCER
Tumors show characteristics of inflamed tissue, including the presence of immune cells within the tumor tissue[1]. Although the presence of leukocytes within tumors was initially thought to result from anti-tumor immune responses, a role for inflammation in tumorigenesis is now generally accepted. Epidemiologic and clinical studies indicate that in response to chronic inflammatory conditions, epithelial cells (transformed and/or normal) and tissue-resident immune cells locally secrete cytokines, chemokines, growth factors, and pro-inflammatory mediators that recruit inflammatory cells from the circulation into the tumor site[2]. Furthermore, immune cells that invade the local tumor microenvironment are phenotypically different from normal immune cells, and can maintain the inflammatory milieu and promote invasion and migration of the transformed epithelial cells[3].
Colorectal carcinoma (CRC) is one of the best examples of an inflammation-associated cancer[4]. During colorectal carcinogenesis, epithelial cells in the colon accumulate mutations, which lead to either inactivation or activation of certain target genes that provide a selective growth advantage. In turn, this results in the transformation of normal epithelium to an adenomatous polyp, and finally to invasive CRC. The transformed epithelial cells then acquire the ability to secrete inflammatory mediators that act on pro-inflammatory leukocytes, endothelial cells, and fibroblasts to establish a tumor-promoting reactive microenvironment. For example, when compared with the general population, epidemiological studies have shown a higher incidence of CRC in patients with a previous history of inflammatory bowel disease (IBD)[5]. It has also become evident that inflammation is a significant factor in the progression of tumors. The regular use of non-steroidal anti-inflammatory drugs (NSAIDs) lowers mortality from sporadic CRC and suppresses adenoma growth in patients with familial adenomatous polyposis (FAP) and who inherit a mutation in the tumor suppressor APC gene[6]. Similar to other solid malignancies, pathological examinations of CRC tissue reveal the presence of innate immune cells, including neutrophils, mast cells, natural killer cells, and dendritic cells that are recruited to the tumor and suppress tumor growth and angiogenesis[7]. This phenomenon is called immune-surveillance, and assists in the early detection and elimination of transformed cells and preneoplastic aberrant crypt foci (ACF), which may progress to adenomas and adenocarcinomas in CRC. On the contrary, colorectal and colitis-associated tumorigenesis are associated with the presence of an inflammatory microenvironment that favors the inhibition of immune-surveillance and promotes a tolerogenic environment with the release of growth factors, thereby supporting further tumor growth[8,9]. In addition to paracrine signaling by growth factors, cytokines, chemokines, and oxygen radicals[10], bioactive lipids derived from polyunsaturated fatty acids (PUFAs) are among the earliest signaling molecules released in response to an injury or inflammatory stimulus. The role played by these small mediators in inflammation and its resolution has garnered a great amount of recent interest[11,12].
POLYUNSATURATED FATTY ACIDS
Polyunsaturated fatty acids (PUFAs) that can be metabolized to bioactive lipids include arachidonic acid (AA), linoleic acid (LA), linolenic acid (LNA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). AA is a 20C polyunsaturated fatty acid (20:4n-6) that is usually esterified to the second carbon in membrane phospholipids, and gives rise to a wide variety of lipid products termed eicosanoids. AA is also known as an n-6 fatty acid, signifying the position of the carbon with the first double bond when considering the terminal methylene carbon group as the first carbon. AA is derived from linoleic acid (LA, 18:2n-6), an 18C essential fatty acid, through the subsequent actions of desaturases and elongases located primarily in the liver. The release of AA from phospholipids in the outer nuclear membrane is achieved through the activity of phospholipases such as the calcium-dependent cytosolic phospholipase A2 (cPLA2). After being released, the free fatty acid can be metabolized via enzymatic pathways including the cyclooxygenase (COX) and lipoxygenase (LOX) pathways to generate 2-series prostaglandins (PGs) and thromboxanes (Txs) (COX pathway) or 4-series leukotrienes (LTs) and hydroxyeicosatetraenoic acids (HETEs) (LOX pathway)[11,13] (Figure 1). The eicosanoids are highly potent, short-lived molecules that act locally, and have been strongly implicated in a variety of cancers, including CRC.
Figure 1.
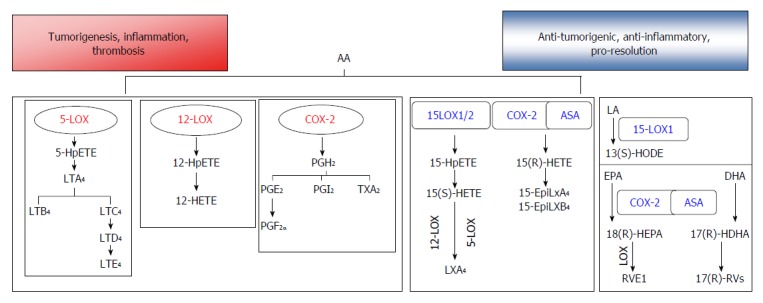
Enzymatic metabolism of polyunsaturated fatty acid can generate bioactive lipids that induce inflammation, tumorigenesis, and thrombosis, while also generating mediators with anti-tumorigenic, pro-resolution properties. In the pro-tumorigenic arm, arachidonic acid (AA) is metabolized via the cyclooxygenase (COX) pathway to generate prostaglandins (PGE2, PGI2) and thromboxanes (TxA2). Lipoxygenase (LOX) enzymes convert AA to hydroxyeicosatetraenoic acids (HETEs), which are active on their own, or can be further converted to leukotrienes (LTs). In the anti-tumorigenic, pro-resolution arm, metabolism of AA through 15-LOX1/2 or acetyl salicylic acid (ASA) acetylated COX-2 generates intermediates that can be converted to lipoxins (Lxs) through the transcellular activity of other LOXs (5- or 12-LOX). Conversion of linoleic acid (LA) to 13(S)-HODE may produce anti-inflammatory effects mediated through activation of PPARγ. The fish oils eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) may be converted by acetylated COX-2 to pro-resolution mediators E- and D- series resolvins (Rvs), respectively. PUFA: Polyunsaturated fatty acid.
Long-chain PUFAs such as EPA and DHA, commonly known as n-3 fatty acids, are extensively found in fatty fish, but are not efficiently synthesized in humans[14]. As these fatty acids are primarily obtained through the diet, increased consumption of fish oil can alter the fatty acid profiles of the plasma and cell membranes in a time and dose-dependent manner[15], primarily at the expense of AA. This implies a decreased production of inflammatory AA-derived eicosanoids, which has been verified in healthy human volunteers who showed decreased levels of PGs and LTs after consuming EPA and DHA supplements for varying lengths of time[16]. EPA, being a 20C highly unsaturated fatty acid and therefore classified as an eicosanoid, can also be metabolized by the COX and LOX pathways into 3-series PGs and 5-series LTs. However, these lipids are readily recognized by PG and LT receptors, and are therefore considerably less potent in inducing inflammation[17]. Both EPA and DHA are substrates for the production of newly identified autacoids that are essential for the resolution of inflammation[18].
EICOSANOID PATHWAYS AND COLORECTAL CANCER
Bioactive lipids may modulate the incidence of cancer via several different mechanisms that include, but are not limited to, induction of inflammation, regulation of cellular oxidative stress, activation of receptors involved in cellular signaling pathways, and the alteration of membrane dynamics[19].
COX-2-derived lipid mediators
AA is metabolized to prostaglandins either by constitutively expressed COX-1 or by COX-2, which is expressed when induced by inflammatory stimuli[20]. COX-2 is an immediate-early response gene that is not expressed in most cells, but is highly induced at sites of inflammation and in the tumor microenvironment[21]. The primary prostaglandin produced from AA (PGH2) can be further metabolized to a several other prostaglandins; among which, PGE2 has been strongly implicated in the development of gastrointestinal tumors[22]. This prostanoid acts via four G-protein coupled receptors (EP1-4) and can enhance tumorigenesis through a variety of mechanisms, including enhanced cell proliferation, suppression of apoptosis, and induction of angiogenesis[23].
Elevated levels of COX-2 and an accompanying elevation of PGE2 are often seen in CRC, and COX-2 expression is correlated with a lower survival rate among CRC patients[24]. It is well accepted that there are concerted interactions between carcinoma cells and other cells in the tumor microenvironment, and that these interactions contribute to cancer progression. PGE2 modulates cancer-associated immune suppression by recruiting T cells, CD8+ cytotoxic T cells, regulatory T cells, dendritic cells, and myeloid-derived suppressor cells (MDSCs)[20]. Additionally, secretion of PGE2 may enhance oxidative stress, leading to a state of low grade continuous inflammation characterized by the infiltration of neutrophils and macrophages, and eventually leading to mitogenic signaling[18]. PGE2 has been shown to stimulate macrophages to produce pro-inflammatory chemokines and cytokines, such as macrophage chemoattractant protein-1 (MCP-1), which recruits leukocytes from the circulation to local sites of inflammation[25]. A previous study[26] showed that MCP-1 levels were higher in intestinal epithelial cells, and that MCP-1 could stimulate COX-2 expression as well as the release of PGE2 and vascular endothelial growth factor (VEGF) in human macrophages.
In the adaptive response, PGE2 mediated signaling may affect cytokine production in antigen-presenting cells by influencing the functional phenotype of T cells [i.e., by switching the anti-tumor T helper 1 (Th1) responses to immunosuppressive Th2 responses)] during priming[27,28]. In trinitrobenzene sulfonic acid (TNBS)-induced colitis, a model of IBD, PGE2 was shown to worsen inflammation and disease severity by increasing neutrophil and Th17 cell infiltration into colonic tissue[29]. Furthermore, PGE2 was shown to amplify IL-23-mediated Th17 cell expansion by acting on its receptor (EP4) located on T cells and dendritic cells[30].
The pro-tumorigenic effects of PGE2 may also be mediated by Treg cells, which contribute to immune evasion by tumor cells in a variety of cancers. Increased COX-2 expression and elevated PGE2 production in adaptive FoxP3-positive Treg cells found within tumors and mesenteric lymph nodes have been shown to contribute to an immunosuppressive microenvironment in CRC. This type of microenvironment facilitates tumor growth by suppressing effector T cells and inducing resistance to antigen-specific cancer immunotherapy[31,32]. A role for COX-2 in tumor immunity was also exhibited in COX-2 expressing colon cancer cell lines, where expression of FasL and TRAIL was shown to cause a counter-attack against cytotoxic T cells[33].
COX-2 overexpression is frequently associated with the concomitant expression of microsomal PGE synthase-1 (mPGES-1), the terminal synthase that leads to the preferential production of PGE2[20,34]. Accordingly, in Apc-mutant mice, genetic deletion of mPGES-1 was reported to suppress intestinal cancer growth by reducing the size and number of aberrant crypt foci (ACF) in a carcinogen-induced colon cancer model[35]. Together, these findings suggest that mPGES-1 plays crucial roles during colon cancer progression, and that these roles are relevant to the promotion of inflammation. Thus targeting mPGES-1 may be a feasible option for cancer chemoprevention[36].
Interestingly, prostaglandins are also essential for the health of gastrointestinal mucosa by maintaining mucosal integrity, promoting wound healing, and limiting inflammation[36]. The absence of cPLA2 in mice was recently shown to globally reduce the formation of AA-derived bioactive lipids, increase mucosal ulceration, and increase the expression of pro-inflammatory cytokines[34]. Mice with a targeted deletion of COX-2 in their endothelial cells and myeloid cells and treated with dextran sulfate sodium (DSS) displayed greater weight loss and had worse clinical scores compared to their WT littermates. However, mice with a targeted deletion of COX-2 in their colonic epithelial cells were not susceptible to DSS[37]. Additionally, mPGES-1-/- mice were recently reported to show more extensive inflammation in the GI tract when compared with WT mice[34]. This could have resulted from a shift in the metabolism of prostaglandins; e.g., a loss of PGE2 was associated with a gain in PGD2, which has tumor suppressive functions[34,38].
Influx and efflux carriers such as the prostaglandin transporter (PGT) and multidrug resistance-associated protein 4 (MRP4), as well as the inactivation of prostaglandins (specifically PGE2) by hydroxyprostaglandin dehydrogenase 15-(NAD) (15-PGDH) can regulate the availability and efficacy of prostanoids[20]. In fact, 15-PGDH is frequently down-regulated in a variety of cancers, suggesting its tumor-suppressive role[11]. Overexpression of 15-PGDH in colon cancer cells was shown to strongly inhibit tumor growth in immune-deficient mice. It was also demonstrated that colonic 15-PGDH expression was directly controlled and strongly induced by the activation of TGF-β, which has tumor-suppressive functions in colon cancer[39,40]. Therefore, the combined induction of COX-2 and inactivation of 15-PGDH in colon cancer can markedly increase PGE2 levels, which may allow cancer cells to escape immune surveillance.
Randomized clinical trials and observational studies have indicated that long-term use (≥ 5 years) of acetyl salicylic acid (ASA, Aspirin®), which inhibits both COX-1 and COX-2, leads to significant reductions in the risks for developing colorectal, esophageal, gastric, biliary, and breast cancer, as well as their distant metastases[41]. Furthermore, the daily use of ASA has been reported to specifically prevent the development of colorectal polyps and reduce the risk for developing sporadic CRC or CRC from Lynch syndrome[42-44]. Moreover, the use of ASA after a diagnosis of CRC can also improve survival, and especially in patients with COX-2-overexpressing tumors[24]. A recent prospective, observational study of ASA and COX-2 inhibitor use either during or after chemotherapy as an adjuvant in stage III colon cancer patients reported reduced cancer recurrence and mortality[45]. Moreover, ASA use was associated with a greater reduction in the risk for developing colorectal tumors when the normal colonic mucosa showed a higher expression of 15-PGDH[45].
Other prostaglandins produced in the eicosanoid pathway, such as PGD2, have been shown to down-regulate granulocyte infiltration into colonic mucosa at the early stages of TNBS induced inflammation[38,46]. More recently, it has been shown that mast cell-derived PGD2 can function as an inhibitor of colitis and colitis-associated cancer (CAC) in mouse models[47]. Taken together, the findings suggest that different COX-2-derived prostaglandins can have opposing effects on inflammation, and that selective modulation of these mediators may prevent tumor growth in CRC.
LOX-derived lipid mediators
PUFAs are oxygenated via the enzymatic action of LOXs to form LTs and hydroxyeicosatetraenoic acids (HETEs), which both exert significant effects on the development and progression of human cancers[48]. Several different isoforms of LOX exist, including 5-LOX, 15-LOX-1, 15-LOX-2, and 12-LOX[13], and are named according to the position of the carbon atom in AA that these enzymes oxygenate. Except for ALOX5, which is located on chromosome 10, most of the other LOX genes are located within a few megabases of each other on the short arm of chromosome 17[13]. Although AA is the preferred substrate for oxygenation for most LOXs, some LOX isoforms are capable of oxygenating fatty acids esterified to phospholipids or cholesterol[49,50].
In humans, 5-LOX is highly expressed in cells of myeloid origin, and especially in leukocytes[51]. This enzyme catalyzes the conversion of AA to 5S-hydroperoxyeicosatetraenoic acid (5-HpETE), and its subsequent conversion to LTA4. LTA4 can be converted to LTB4 and then to cysteinyl leukotrienes by the actions of LTA4 hydrolase and LTC4, synthase, respectively[52] (Figure 1). 5-LOX activity is exquisitely sensitive to various stimuli, including the second messenger Ca2+. Ca2+ can bind to the N-terminus of 5-LOX, which contains a hydrophobic domain and facilitates the binding of 5-LOX to membrane phospholipids[53]. The 5-LOX enzyme is usually located in the cytoplasm as a soluble protein; however, in the presence of Ca2+, it may become phosphorylated and translocate to the nuclear or endoplasmic reticulum (ER) membrane, where with the help of 5-LOX activating protein (FLAP), it catalyzes the oxygenation of AA[53]. Therefore, many stimuli that increase the levels of intracellular calcium ions (e.g., antigens, microbes, cytokines, and toxins) can induce the production of LTs[53,54].
LTs are classified into two general categories: LTB4 and cysteinyl LTs (LTC4, LTD4, and LTE4)[55]. LTs play key roles in the pathogenesis of inflammatory disorders, including IBD, and typically stimulate quick and short-lasting events (e.g., smooth muscle contraction, phagocyte infiltration, increased vascular permeability), which are important in the pro-inflammatory context. These responses are mediated by G-protein coupled receptors: BLT1/2 for LTB4, and CysLT1/2 and GPR17 for the Cys-LTs[52,56].
5-LOX is overexpressed in tissues with chronic inflammation and also in transformed cells. 5-LOX was shown to be up-regulated in patients with polyps and colon cancer[57], whereas in the APCΔ468 mouse model of polyposis, the loss of 5-LOX was protective[58]. In the same model, 5-LOX metabolic products produced by hematopoietic cells were shown to promote tumorigenesis by enhancing both the proliferation of intestinal epithelial cells and recruitment of MDSCs to the spleen, mesenteric lymph nodes, and primary tumor[59]. Dietary administration of a 5-LOX inhibitor (Zileuton) to APCΔ468 mice resulted in overall reductions in systemic inflammation, polyp number, and inflammatory infiltration into lesions[59].
Overproduction of LTB4 in human colon cancer tissue is implicated in the pathogenesis of IBD. Additionally, high expression of the LTB4 receptor BLT1 has been detected in human colon tissues[60]. These findings indicate the importance of an inflammatory autocrine loop during the promotion and progression phases of colon tumors. The inflammatory mediators can cause intestinal epithelial cells to up-regulate their expression of enzymes needed for the biosynthesis of eicosanoids, including the CysLTs, and signal transducing CysLT receptors, to provide a self-sufficient signaling mechanism needed to maintain both inflammation and tumor progression[58]. Taken together, these studies show that pro-inflammatory LTs might facilitate tumor growth by establishing an inflammatory microenvironment.
Metabolism of AA by 12-lipoxygenase (12-LOX) leads to the production of 12-HETE, which has been shown to stimulate the growth of various cancers[61]. Additionally, a Gln261Arg polymorphism in the ALOX12 gene was shown to be associated with an enhanced susceptibility to several malignancies, which also indicates a potential oncogenic role for 12-LOX[62,63]. Although a recent meta-analysis of studies with 8379 subjects revealed that this specific polymorphism was not associated with an increased risk of colon cancer[64], other studies have reported that 12-LOX expression was associated with an oncogenic phenotype in CRC[62]. 12-LOX was also shown to be up-regulated in colon cancer specimens associated with inflammation[61]. Moreover, 12-LOX expressing colon cancer cell lines have demonstrated increased migration as a result of decreased E-cadherin and integrin-β1 expression[61], or enhanced production of reactive oxygen species (ROS) and activation of the catalytic subunit of the NADPH oxidase complex, Nox1[65].
Unlike 5-LOX and 12-LOX, 15-lipoxygenase-1 (15-LOX-1), which can oxygenate AA and LA as well as complex substrates such as biomembranes[66], may have an anti-inflammatory, tumor suppressive role in CRC. This enzyme can oxygenate AA to 15-HETE, or LA to 13(S)-hydroxyoctadecadienoic acid [13(S)-HODE]. Profiling of LOX metabolic products in CRC has shown that 13(S)-HODE was the only metabolite that significantly increased in the Caco-2 model of cellular differentiation[67,68]. Additionally, an assay of > 120 cancer cell lines from 20 different cancer types indicated an almost universal loss of 15-LOX-1 expression in de-differentiated cell lines when compared with well-differentiated cancer cells or non-transformed cells[68]. Moreover, levels of 13(S)-HODE were shown to be reduced in colorectal polyp samples obtained from patients suffering from FAP when compared to their levels in paired normal tissues[67]. A loss of 15-LOX-1 expression is primarily due to epigenetic factors, such as nucleosomal remodeling and the histone deacetylase (NuRD) complex[69]. Re-expression of 15-LOX-1 has been achieved via routes including histone methylation/demethylation, acetylation[70,71] or the activation of transcription factors such as STAT-6[72]. In a mouse model with gut targeted expression of human 15-LOX-1 exposed to azoxymethane, the number of tumors was lower in the animals with transgene expression, and 15-LOX-1 expression was lower in samples of tumor tissue compared to normal tissue[73].
An inverse link between 15-LOX-1 expression and secretion of pro-inflammatory cytokines has been indicated in recent years. Gut-targeted expression of 15-LOX-1 has resulted in lower levels of TNFα and inducible nitric oxide synthase (iNOS) in epithelial cells[73]. In human CRC, down-regulation of 15-LOX-1 was associated with increased IL-1β expression[74]. This was further substantiated by a loss of NF-κB (a key inflammatory transcription factor) signaling both in colon cancer cell lines and mouse models when 15-LOX-1 was re-expressed in the gut[73,75,76]. Additionally, there is evidence indicating that 15-LOX-1 expression can inhibit CAC. Chemical inhibition of 15-LOX-1 by PD146176 was shown to cause significant deterioration of intestinal functions in a murine model of experimental colitis[77]. While LA is efficiently oxygenated by 15-LOX-1 to produce 13(S)-HODE, AA can also be metabolized by both 15-LOX-1 and 15-LOX-2 to produce 15(S)-HETE[78]. 15(S)-HETE levels were reported to be significantly lower in the serum of colorectal cancer patients when compared to their levels in control subjects[78].
Thus, activation of acute inflammatory responses, anti-inflammatory and anti-tumorigenic pathways or a neoplastic transformation may occur as a result of the opposing effects of various metabolites formed downstream of the enzymatic action of different LOXs De-regulation of any of these pathways may lead to a loss of homeostasis.
EICOSANOIDS AND THE HALLMARKS OF CANCER
Various types of cancer cells and surrounding stromal cells produce high amounts of pro-inflammatory eicosanoids. These bioactive lipid metabolites can modulate tumor progression through several mechanisms, including directly activating receptors on tumor epithelial cells that help regulate cell proliferation, apoptosis, migration, and invasion, and inducing epithelial cells to secrete growth factors, pro-inflammatory mediators, and angiogenic factors. All of these molecules can facilitate tumor growth, and also support tumor-associated angiogenesis and evasion of the immune system[20].
Proliferation and apoptosis
It is already well documented that tumor growth relies on a deregulated balance between cellular proliferation and cell death. It is not surprising that various eicosanoids that activate/inhibit important signaling pathways in cells can also regulate cellular proliferation and apoptosis in colon cancer cells.
COX-2 pathway in cell proliferation and apoptosis
COX-2 is overexpressed in 50%-80% of all colorectal cancers[79]. At the cellular level, COX-2 overexpression was shown to increase cell-to-matrix adhesion and inhibit apoptosis in human CRC cells[80-82]. Furthermore, in the APCΔ716 mouse model, the number and size of the polyps were shown to be reduced dramatically when the COX-2 gene was knocked out[83]. In accordance with this finding, ASA and sulindac have been shown to reduce the number and size of adenomatous colonic polyps in patients with FAP[84]. Additionally, conventional NSAIDs are known to inhibit chemically-induced colon cancer in rodent models by inhibiting COX-2 activity[85]. In the human colon cancer cell line HCT-116, COX-2 was induced through wild-type p53-mediated activation of the Ras/Raf/ERK cascade, which subsequently blocked p53 or genotoxic stress-mediated apoptosis. This anti-apoptotic effect may represent a mechanism for diminishing cellular stress associated with p53 induction[86]. On the other hand, NSAIDs inhibited expression of the anti-apoptotic protein Bcl-XL, resulting in an altered BAX to Bcl-XL ratio and enhanced apoptosis[87]. Increased expression of the anti-apoptotic protein Bcl-2 and reduced expression of the pro-apoptotic protein Bim caused by the COX-2-derived eicosanoid PGE2 have also been reported[21,88].
A considerable amount of crosstalk has been reported between the COX-2 and EGFR pathways. For instance, PGE2 treatment was shown to significantly increase cellular proliferation and reduce apoptosis in a rodent CAC model[89], and also induce COX-2 expression in intestinal adenomas by activating the MAPK signaling pathway[90]. PGE2 was shown to induce ERK2 signaling in colon cancer cell lines by stimulating the rapid phosphorylation of EGFR[91]. Inhibition of both EGFR and COX-2 achieved by using a targeted liposome carrying the COX-2 inhibitor celecoxib and a monoclonal antibody against EGFR (Cetuximab) has been shown to additively inhibit the proliferation of colon cancer cell lines expressing both EGFR and COX-2[92].
Roberts et al[93] reported that during glucose deprivation, PGE2 can promote tumor cell survival in the colon by activating the PI3K/AKT pathway, which in turn may up-regulate COX-2 and down-regulate 15-PGDH. Moreover, glucose deprivation was also demonstrated to activate the unfolded protein response (UPR), resulting in elevated expression of the C/EBP-homologous protein (CHOP), which was positively correlated with 15-PGDH expression. These data suggest that stress conditions can regulate PGE2 as a common and crucial mediator of cell survival during adaptation to the tumor microenvironment.
In the colorectal adenocarcinoma cell line DLD-1, PGE2 was shown to bind to EP2, which stimulated tumor growth by activating PI-3K/AKT signaling, followed by activation of the β-catenin signaling pathway[94]. PGE2 can also induce cell proliferation in colorectal tumors through the EP4 receptor by inducing phosphorylation of ERK[95]. Additionally, Park et al[96] have proposed that COX-2 inhibition may produce significant anti-tumorigenic effects by blocking stroma-derived PGs. These authors used a co-culture model to evaluate cancer cell-stromal cell relationships and reported that use of an EP4 antagonist resulted in decreased proliferation in the COX-2 non-expressing LS174T colon adenocarcinoma cell line.
In contrast to PGE2, 15d-PGJ2 was shown to induce apoptosis[97] and cell cycle arrest in CRC cells[98] by inhibiting activity of the inflammatory transcription factor NF-κB, reducing the levels of anti-apoptotic genes[97], down-regulating c-Myc expression, and upregulating c-Jun and GADD153[99]. When 15d-PGJ2 and histone deacetylase (HDAC) inhibitors were added in combination to colon cancer cell lines, they exerted a synergistic effect on caspase-dependent apoptosis, leading to ROS generation, ER stress, decreased expression of anti-apoptotic proteins Bcl-XL and XIAP, and increased expression of CHOP and DR5 (Death receptor 5, TRAIL-R2). Furthermore, the same effects of this co-treatment were also seen in vivo, with an inhibition in tumor growth in a nude mouse xenograft model inoculated with DLD-1 cells[100]. Shin et al[101] suggested that the growth inhibition and 15d-PGJ2-induced apoptosis seen in human and murine CRC cell lines were caused by ROS dependent down-regulation of AKT and p-AKT.
LOX pathways in proliferation and apoptosis
The 5-LOX protein is overexpressed in the early stages of colon cancer, where its expression is significantly correlated with patient age, polyp size, and the presence of intraepithelial neoplasia and villous and tubulovillous adenoma, all of which are considered to be typical markers of transformed adenomatous polyps[102]. Inhibition of 5-LOX with Zileuton was shown to significantly decrease proliferation in a colon cancer cell line and reduce the size of xenografted tumors[57]. LTB4 demonstrated pro-carcinogenic effects in CRC by activating the ERK pathway[103]. Induction and/or accumulation of COX-2, β-catenin, and Bcl-2, as well as PGE2 production in non-transformed epithelial linings in the colon have also been reported in the presence of LTB4[104]. Furthermore, LTD4, a cysteinyl leukotriene, was reported to inhibit caspase 3, and thereby increase resistance to NSAID-induced cell death[105].
In several different cancer types, COX-2 and 5-LOX signaling can converge to enhance cell proliferation[106]. For example, knock-out of 5-LOX or FLAP was shown to increase the amount of COX-2 metabolites produced by inflammatory cells, indicating that inhibition of one pathway could shunt metabolism of AA towards the other pathway[107-109]. Dual inhibition of 5-LOX and COX-2 may produce additive or synergistic effects on reducing cellular proliferation in colon cancer, as shown by the combination of AA861 (5-LOX inhibitor) and celecoxib[110], the dual COX/5-LOX inhibitor licofelone[111], and the combination of celecoxib and MK886 (5-LOX inhibitor)[112]. Gaining a better understanding of these pathways will have important implications for cancer chemoprevention and treatment[18].
The role of 15-LOX-1 in proliferation and apoptosis of colon cancer was initially considered to be controversial, although well-controlled in vitro and in vivo studies conducted in the past several years have revealed an unequivocal tumor suppressive role for 15-LOX-1 in CRC[113]. While initial studies indicated an anti-apoptotic role for the enzyme, those investigations were primarily conducted using inhibitors such as NDGA (nordihydroguaiaretic acid), which may have pleiotropic effects in cells[114]. Yoshinaga et al[115] reported that 15-LOX-1 over-expression in colon cancer cell lines increased cell proliferation via activation of ERK, followed by a decrease in p21(Cip/WAF1) expression. However, numerous subsequent studies have shown that the main product of 15-LOX-1, 13(S)-HODE, can inhibit cell proliferation and induce apoptosis in CRC[116,117]. Moreover, both 15-LOX-1 expression and levels of 13(S)-HODE were reduced in the polyps when compared to paired normal tissues obtained from patients with FAP[67]. Mice express 12/15-LOX, an enzyme that can simultaneously metabolize AA to 12-HETE and LA to 13(S)-HODE, which have opposing effects on tumorigenesis. Therefore a transgenic mouse model was established that can express human 15-LOX-1 specifically in gut epithelial cells[74]. These mice showed a decreased incidence of tumors[73]. Interestingly, an inverse correlation between 15-LOX-1 and COX-2 expression has been proposed to occur during the adenoma to carcinoma sequelae[118], leading to the accumulation of pro-tumorigenic PGs and the loss of apoptotic 13(S)-HODE. It has been suggested that 15-LOX-1-mediated inhibition of NF-κB, which can transcriptionally up-regulate COX-2, leads to a loss of expression of the latter. Epigenetic silencing of 15-LOX-1 in the later stages of progressive CRC may lead to an increase in COX-2 expression, and thus exacerbate the inflammatory milieu[119].
However, focusing on the effects of 15-LOX-1 expression only in epithelial intestinal cells may not provide sufficient information about how its expression contributes to CRC development. Additional knowledge concerning the effects of 15-LOX-1 and its metabolites on tumor-associated stromal cells and endothelial cells is also required to understand the underlying mechanisms of CRC development that lie beyond 15-LOX-1 signaling.
NF-κB and PPAR signaling pathways driven by eicosanoids in CRC
In colon cancer, the activity of NF-κB in intestinal epithelial cells and myeloid cells in the tumor environment plays an essential role in tumor formation[76]. Therefore, one may suggest that specific inactivation of the NF-κB pathway in cancer cells and surrounding myeloid cells may attenuate formation of inflammation-associated tumors[120].
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of a nuclear hormone receptor superfamily that includes PPARα, PPARγ, and PPARβ/δ. Each of these receptors can mediate the physiological actions of numerous fatty acids and fatty acid-derived molecules that serve as ligands for these transcription factors (Figure 2). Activated PPARs can also function as transcriptional repressors of NF-κB, STAT-1, and AP-1 signaling[121]. PPARγ is known to be expressed in normal colon tissue, and show reduced expression in colon tumors[122]; however, mutations of PPARγ in CRC are rare[123]. Agonists of PPARα and PPARγ were shown to inhibit DSS-induced colitis and formation of ACF in rats[124]. On the other hand, PPARβ/δ is associated with pro-inflammatory pathways and progression of CRC[121].
Figure 2.
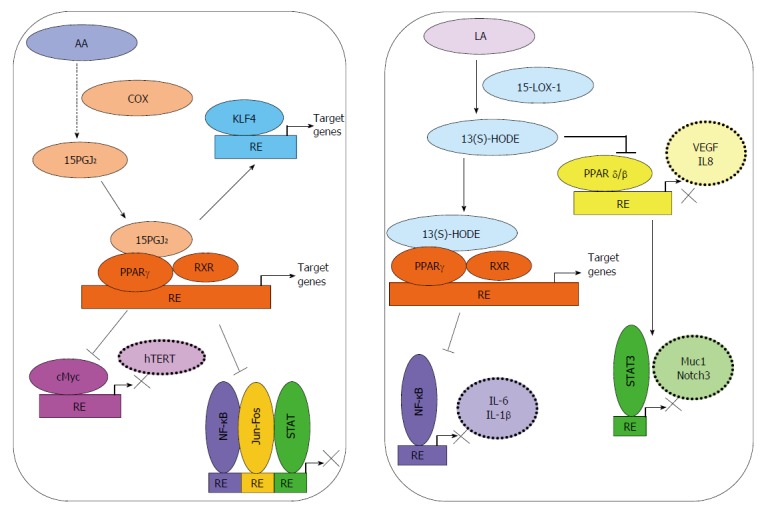
Activation of PPARγ by bioactive lipids can modulate signaling in progressive colorectal cancer. 15-deoxy-delta(12,14)-prostaglandin J2 (15-PGJ2), generated from arachidonic acid (AA) by the enzymatic action of COX-2, acts as a ligand for PPARγ. Co-activators such as RXR activation of tumor suppressive signaling through Kruppel-like factor 4 (KLF4) in colorectal cancer (CRC) have also been reported. Binding to co-repressors may lead to repression of various transcription factors such as nuclear factor kappa B (NF-κB), AP1 (Activator Protein-1, c-Jun and c-Fos), c-Myc or STAT. 13(S)-hydroxyoctadecadienoic acid [13(S)-HODE], generated by oxygenation of linoleic acid (LA) by 15-LOX-1, can act as a ligand for PPARγ and lead to inhibition of NF-κB activity. 13(S)-HODE may also inhibit the transcriptional activities of PPARβ/δ and STAT3, and thereby reduce inflammation and angiogenesis in CRC.
15d-PGJ2, a natural agonist of PPARγ, was shown to inhibit the proliferation of HT-29 human colon cancer cells by up-regulating tumor suppressive transcription factor, Kruppel-like factor 4 (Klf-4)[125]. 15d-PGJ2 and rosiglitazone, a synthetic ligand of PPARγ, were found to suppress proliferation of Caco-2 CRC cells by repressing telomerase activity and telomerase reverse transcriptase (hTERT) expression by down-regulating c-Myc and up-regulating Mad1[126].
13(S)-HODE produced in the 15-LOX-1 pathway can act as a ligand for PPARγ[127]. Re-expression of 15-LOX-1 in colon cancer cells was shown to down-regulate PPARδ, and thereby promote induction of endogenous PPARγ target genes related to induction of apoptosis[128]. In support of this finding, over-expression of 15-LOX-1 was associated with decreased proliferation and increased apoptosis, as well as reduced cellular motility, anchorage-independent growth, migration, and cell invasion in colon cancer cells[118]. Moreover, increased 13(S)-HODE-mediated PPARγ activation has been suggested to inhibit activation of NF-κB, which is associated with decreased cell viability[75]. In colitis and CAC[124], 15-LOX-1 activity was also shown to activate PPAR-γ[128,129], which suppressed the expression of key inflammatory genes; most likely by inhibiting NF-κB[130,131].
13-(S)HODE has been shown to suppress PPAR-δ; a receptor that can transcriptionally upregulate IL-6 expression, and thereby promote colitis and CAC[128,132,133]. Very recently, 15-LOX-1-induced inhibition of PPARδ during promotion of CAC was shown to be mediated by suppression of IL-6 expression, STAT3 phosphorylation, and Notch3 expression[134]. Moreover, elevated expression of PPARδ has been implicated in the pathogenesis of CRC[135,136], and a positive correlation between PPARδ expression and the late stages of CRC has also been observed[137]. PGI2 was shown to activate PPARδ, which may lead to a loss of apoptosis through sequestering of the pro-apoptotic protein BAD by 14-3-3 epsilon and reduced mitochondrial damage[138]. Similarly, stromal PGI2 generation was claimed to promote cell survival in colonocytes by activating PPARδ[139]. More recently, PPARδ activation was also associated with an increased expression of VEGF and IL-18 in colon cancer cells, through p300 and the PI3K/AKT pathway. Furthermore, hypoxia stimulated PPARδ activation enhanced angiogenesis, macrophage recruitment, and macrophage proliferation in the tumor microenvironment[140].
Metastasis
Although surgery is the most curative approach for CRC, approximately 40% of treated patients eventually show either local recurrence or distant metastases[141,142], primarily to the liver and lungs[143]. Both experimental and clinical studies have shown that daily use of ASA was associated with a reduced risk of metastasis[144], and inhibited the spread of primary tumor cells to other organs post-diagnosis[145]. These findings suggest a role for eicosanoids and eicosanoid-mediated signaling in CRC metastasis.
COX pathway and metastasis
Metastasis is a well-regulated cascade of events that requires the coordinated activation of several factors expressed/released not only by tumor cells, but also by stromal cells. PGE2 is claimed to promote a more metastatic CRC phenotype[146]. An analysis of sporadic colorectal adenocarcinoma tissue samples revealed a significant relationship between COX-2-derived PGE2 levels and tumor stage: higher PGE2 levels were reported in metastatic tumor specimens when compared to tumor specimens without metastases. Thus, it can be concluded that PGE2 levels may be correlated with tumor aggressiveness, its ability to metastasize, and patient prognosis[147].
Epidemiological, clinical, and animal studies have demonstrated that COX-2 and epidermal growth factor (EGF) signaling pathways coordinate their activities to play key roles in promoting CRC growth and metastasis[148]. For example, EGFR expression was directly correlated with the potential of human CRC cells to metastasize to the liver[149]. Moreover, Buchanan et al[150] suggested that in early stage CRC, the initial effects of PGE2 were mediated by EGFR transactivation and subsequent phosphorylation, which was also responsible for down-stream effects, including cell migration and invasion. In their following reports, the same group showed that PGE2 activated an EP4/β-arrestin1/c-Src signaling complex, resulting in EGFR transactivation and downstream Akt signaling, which subsequently stimulated migration of CRC cells in vitro as well as their metastatic spread to the liver in vivo[151]. Additionally, in the presence of functional EGFR, PGE2 was shown to transactivate hepatocyte growth factor receptor (c-Met-R), and thereby increase phosphorylation and accumulation of the oncogene β-catenin. This sequence of events induced expression of urokinase-type plasminogen activator receptor (uPAR), resulting in increased CRC cell invasiveness[152]. A significant decrease in liver metastasis with the use of selective EP4 receptor antagonists has also been reported[153]. In another report, PGE2 treatment was shown to activate JNK1/2 kinase. This activation was followed by an increase in the levels of migration-related factors uPA and MMP-9, which further promoted cellular motility in the human colon cancer cell line LoVo. However, pretreatment with 17β-estradiol down-regulated uPA and MMP-9 expression via deactivation of JNK1/2, and inhibited PGE2-induced LoVo cell motility. Based on these findings, the authors suggested that the incidence and mortality rates of CRC in women were lower than those in men because estrogen helps protect against development of fatal colon cancer, and thus reduces mortality from this disease[154].
In contrast to PGE2, PGI2 is known for its anti-metastatic effects in CRC. PGI2 analogues have been suggested to protect against metastasis by inhibiting CAM (Cell Adhesion Molecule) -mediated adherence of colon carcinoma to endothelial cells in metastatic target organs[155].
LOX pathway and metastasis
Data concerning the roles of LOX enzymes in colon cancer migration and invasion have recently been reported[156]. In one study, decreased levels of the selective LOX inhibitor, NDGA, were found in mobile human colon cancer cells; this was partly explained by inhibition of MMP-2 and MMP-9[157]. Loss of 15-LOX-1 expression was found in the lymph node and liver metastases of pancreatic cancer[158], and 15-LOX-1 re-expression in CRC cell lines inhibited their invasiveness, motility and migration[117]. More recently, Wu et al[159] showed that 15-LOX-1 re-expression in HCT116, HT29, and LoVo colon cancer cells inhibited cell survival, as well as angiogenesis, cancer cell migration, and invasion.
Angiogenesis
For tumors to grow and metastasize, they must generate their own blood supply; a process defined as neo-angiogenesis. Many cells in the tumor microenvironment, including tumor epithelial cells, stromal cells, and immune cells, secrete various pro-angiogenic factors needed for proliferation, migration, capillary tube formation, and recruitment of endothelial cells[160]. Numerous in vitro and in vivo studies have shown that eicosanoids can modulate angiogenesis at different levels[20].
VEGF is a major regulator of angiogenesis, and its expression is up-regulated in response to multiple micro-environmental “stress” factors such as hypoxia, acidosis, and starvation; which are all related to poor blood supply. In tumors, hypoxia can lead to stabilization of transcription factor HIF-1α, which activates genes which have the hypoxia-responsive element (HRE) in their promoter region, such as VEGF. VEGF exerts its effects on target cells through tyrosine kinase receptors, including VEGF receptors 1 (VEGFR1, Flt1) and 2 (VEGFR-2, Flk-1/KDR)[161]. Ligand binding induces receptor dimerization and activation of downstream signaling pathways including the MAPK family, PI3K/AKT and protein kinase C (PKC)[161]. Besides hypoxia, other factors that have been shown to stimulate VEGF expression include ROS[162], growth factors[163], cytokines[164], and various lipid mediators such as PGE2[165-168].
COX pathway in angiogenesis
PGE2 stimulation has been shown to induce HIF-1α stabilization[163] and VEGF expression in vitro[169]. Additionally, VEGF and COX-2 expression and tumor angiogenesis were shown to be highly correlated in samples of colon cancer tissue[147,170]. Through its receptor EP2, PGE2 was shown to stimulate the nuclear translocation of β-catenin[94], whereby it activated T cell factor 4 (TCF-4) and HIF-1α to trigger cell survival, proliferation, and angiogenesis in colon cancer[171,172]. Homozygous knock-out of EP2 completely abrogated induction of VEGF in the intestinal polyp stroma of APCΔ716 mice. It also decreased the number and size of intestinal polyps, showing that PGE2-directed induction of VEGF was an important factor for tumor growth[173]. Moreover, PGE2 was shown to induce expression and release of the pro-angiogenic chemokine CXCL1 in CRC, which in turn stimulated microvascular endothelial cell migration and tube formation both in vitro and in vivo[174]. Hypoxia was shown to induce EP1 expression in colon cancer cells, while inactivation of EP1 inhibited PGE2-dependent and hypoxia-inducible expression of angiopoietin-like protein 4 (ANGPTL4), whose lipid metabolizing functions are exerted via inhibition of lipoprotein lipase (LPL)[175].
In addition to inducing several angiogenic factors in epithelial cells, PG signaling in surrounding stromal cells also supports angiogenesis in colon cancer. For example, PGE2 and TXA2 were reported to regulate the adhesion and spreading of human umbilical vein endothelial cells (HUVEC) through cAMP-dependent activation of protein kinase A (PKA) and cAMP- and PKA-dependent activation of Rac, respectively[176]. Besides VEGF, PGE2 may also mediate the angiogenic effects of basic fibroblast growth factor (bFGF) by up-regulating expression of C-X-C chemokine receptor type 4 (CXCR4) in human microvascular endothelial cells (HMECs), and enhancing cellular response to stromal-derived factor 1 (SDF-1), a unique ligand for CXCR4[177]. TXA2 has been shown to enhance endothelial cell migration and angiogenesis[178]. An increase in TXA2 levels, as a result of overexpression of TXA2 synthase in C-26 colon adenocarcinoma cells allografted to BALB/c mice, was reported to stimulate accelerated tumor growth and tumor-associated angiogenesis[179].
The process of angiogenesis may require not only crosstalk between tumor epithelial and endothelial cells, but also the involvement of immune cells that produce pro-angiogenic factors. PGE2 has been shown to induce mast cells to release VEGF and the chemokine CCL2[180,181], which can induce tumor-associated angiogenesis by directly recruiting CCR2 expressing endothelial cells and inducing VEGF release from macrophages[26]. Contrary to the pro-angiogenic roles described above, PGE2, through its receptor EP2, was shown to inhibit secretion of VEGF in Caco-2 colon cancer cells exposed to hyperosmotic stress[182]. Additionally, 15d-PGJ2 was shown to down-regulate COX-2 and VEGF expression in colon carcinoma cells by inhibiting the transcription factor AP-1[183]. In an in vivo study in which PGI2 synthase was retrovirally transduced into C-26 colon adenocarcinoma cells and subsequently grafted to syngeneic BALB/c mice, the increased production of PGI2 resulted in slower tumor growth and a decreased amount of vasculature[179].
When viewed in total, these findings suggest that relative levels of pro- and anti-angiogenic prostanoids in the tumor microenvironment might be strong determinants of the degree of angiogenesis that occurs in colorectal tumors.
LOX pathway in angiogenesis
A growing body of evidence indicates that LOX-catalyzed products (LTs and HETEs) also exhibit important biological effects on the angiogenic process in colon tumors. Ye et al[184] implicated 5-LOX in the promotion of colon cancer growth by nicotine through up-regulation of VEGF, MMP-2, and MMP-9, resulting in stimulation of angiogenesis in the colon. The same group also reported that cigarette smoke extract indirectly stimulated endothelial cell proliferation, a biological phenomenon that may enhance neo-angiogenesis[185,186]. CysLT1R antagonists were shown to impair angiogenesis in colon cancer xenografts[187], while LTB4 was reported to induce neutrophil-mediated vascular permeability[188]. Additionally, LTB4 was shown to enhance hypoxia-induced microvascular alterations in vivo[189]. Expression of the LTB4 receptor BLT2 was found to be highly inducible by VEGF, suggesting interplay among VEGF, BLT2, and BLT2 ligands during vascular angiogenesis[190]. Similarly, LTC4 and LTD4 also promoted angiogenesis via receptor-mediated interactions[191]. Moreover, reduced vascular permeability was observed in LTC4 synthase knock-out mice which had impaired synthesis of cysteinyl LTs[192].
Very few reports have addressed the role of 15-LOX-1 in neo-angiogenesis in colorectal cancer. Recently, our group and others have shown that re-expression of 15-LOX-1 in colon cancer cell lines could reduce the expression and secretion of VEGF-A, and that treatment of HUVECs with conditioned medium from colon cancer cell lines ectopically expressing 15-LOX-1 resulted in reduced tube formation[159,193]. However, the signaling mechanism that medicates this angiostatic effect has not yet been reported.
Therefore, as with the prostanoids, it is likely that different bioactive lipids produced by the LOX pathway may have contrasting effects on angiogenesis, and their ultimate functional effects may be decided by the balance between pro- and anti-angiogenic products.
EICOSANOIDS IN THE RESOLUTION OF INFLAMMATION
The resolution of acute inflammation, rather than being a passive process of diluting out pro-inflammatory mediators, was shown to be actively conducted by several different bioactive lipid mediators[194]. The timely resolution of inflammation prevents the development of chronic inflammation and fibrosis, and enables an organism regain a state of homeostasis[194].
The primary drivers of resolution include cessation of neutrophil infiltration and the nonphlogistic recruitment of macrophages to clear debris at the site of inflammation[194]. Lipoxins (Lx) were the first bioactive lipids to be identified as mediators of these processes. Lx’s can be synthesized from AA in neutrophils through the enzymatic action of 5-LOX, and LTA4 can be converted to LXA4 and LXB4 by 12-LOX in platelets upon the latter’s adherence to neutrophils[195] (Figure 1). Additionally, AA can be metabolized by 15-LOX-1; after which, the oxygenated product can be converted to an epoxytetraene, and then to LXA4 or LXB4 by the action of hydrolases. ASA stimulates acetylation of COX-2, and thus shifts the activity of that enzyme from production of pro-inflammatory prostanoids to production 15(R)-HETE, which is subsequently metabolized by 5-LOX to 15-epi-Lx or aspirin triggered lipoxins (ATLs)[18]. Many of these bioactive lipids act through G-protein coupled receptors such as the lipoxin receptor/formyl peptide receptor (ALX/FPR2), which binds to LXA4 and ATLs[18]. Although most autacoids involved in resolution are known to be synthesized in a transcellular manner involving at least two cell types, a recent study indicates that lipoxins may also be generated from a single immune cell[196].
LXA4 expression or administration of LXA4 analogs has been shown to reduce DSS-induced colitis[197]. Inflammatory stimuli in intestinal epithelial cells have been shown to initiate a feedback reaction which up-regulates expression of the LXA4 receptor in intestinal epithelial cells[198]. Additionally, co-culture of Caco-2 cells with macrophages, where the cells were also treated with LXA4, resulted in decreased secretion of pro-inflammatory cytokines[199], most likely due to inhibition of NF-κB. In a recent study which used colonic biopsies obtained from patients experiencing a remission of ulcerative colitis, significantly increased levels of LXA4, along with enhanced expression of FPR2/ALX receptor mRNA and increased levels of macrophage infiltration were observed, suggesting that LXA4 levels may play an important role in restoring mucosal homeostasis[200]. FPR2 expression was also shown to be increased in the colons of patients with Crohn’s disease; again indicating that signaling through lipoxin was enhanced in the same inflammatory environments that were most likely to have enhanced clearance of debris or bacteria by macrophages[201].
Resolvins (Rvs) are derived from the n-3 fatty acids EPA (E-series Rvs) and DHA (D-series Rvs), and formed by the concerted actions of acetylated COX-2, 5-LOX or 15-LOX[18] (Figure 1). Rvs have demonstrated potency at very low concentrations when administered orally or intravenously[18], and are known to signal through ChemR23 and chemokine-like receptor 1 (CMKLR)[18]. RvD1 and RvD2 in the CACs of mice are known to be chemopreventive, and help reduce tumor growth[202]. RvE1 was reported to induce the clearance of neutrophils into the lumen of the gastrointestinal tract[203]. Moreover, RvE1 was shown to inhibit phosphorylation and activation of p65 NF-κB in the distal colons of mice in a DSS-colitis mouse model[199], suggesting its roles in both pro-resolution and anti-inflammatory pathways. Interestingly, the enzyme intestinal alkaline phosphatase (ALP1), a marker of differentiation, was shown to be induced in epithelial cells in the presence of RvE1, and also have a role in protecting against colitis[204]. Many of these potent bioactive molecules are currently being studied in large-scale clinical trials[205].
Mareisins (MaR) are generated in macrophages from DHA through the action of macrophage 12-LOX[202]. Intermediates formed during the conversion of DHA to MaR1 were shown to inhibit formation of LTB4 and oxygenation of AA by 12-LOX, but enhance conversion of M1 inflammatory macrophages to the M2 phenotype[202]. Another recently identified intermediate is 13,14-dihydroxydocosahexaenoic acid (13,14-diHDHA or MaR2), which is synthesized when macrophages are co-incubated with 12-LOX and soluble epoxide hydrolase (sEH). This compound has been shown to reduce neutrophil migration and enhance macrophage phagocytosis at nanogram concentrations[202]. MaR1 was recently used in a DSS and TNBS-induced mouse model of colitis. In that model, the disease activity index, amount of body weight loss, and tissue damage in the colon were all reduced. Additionally, there were significant decreases in the levels of inflammatory cytokines; most likely resulting from inhibition of the NF-κB pathway[206]. Moreover, in the same study, reduced migration of neutrophils, reduced ROS production, and reduced levels of inflammatory cytokines in LPS-stimulated bone marrow-derived macrophage cultures incubated with MaR1 were also reported[206].
CONCLUSION
There is no doubt that eicosanoids are an important family of immunoregulatory bioactive lipids with involvement in both the promotion and prevention of colon cancer. During inflammation, many of these autacoids act antagonistically or synergistically, and often in a temporal manner with the aid of different cell types in order to induce homeostasis. Many of these bioactive lipids are also essential for various cellular functions. Despite its importance, very few therapeutic agents are available that can modulate the aberrant production of these molecules specifically in the context of colorectal or other cancers. ASA is undoubtedly one of the best known drugs that interferes with the COX pathway; however, ASA needs to be consumed long term (at least 5 years) to provide any protective effect against cancer. Furthermore, use of ASA is associated with significant bleeding events, and is therefore not suitable for all patients. COX-2 inhibitors that specifically target the inflammatory arm of the COX metabolic pathway are approved primarily for pain relief rather than for cancer chemotherapy, and are also associated with significant cardiovascular side effects. On the other hand, CysLTR antagonists that were originally designed for asthma have also not demonstrated high levels of efficacy[207]. Because inhibition of one pathway leads to activation of another due to the shunting of substrates, combined COX/LOX inhibitors have proved to be more effective than signal pathway inhibitors, and should be further studied in the context of CRC.
The scientific community needs to develop drugs that are specifically effective in cancer, and perhaps the most promising candidates include newly discovered resolution mediators such as lipoxins, resolvins, and mareisins. The results of early studies have indicated that these mediators are effective at very low concentrations, and therefore may be viable chemopreventive/therapeutic agents for use in CRC. It is also interesting to note that COX-2 and 5-LOX, that are associated with pro-carcinogenic events, and 15LOX-1, which is associated with anti-carcinogenic events in CRC, rarely show any mutations. Deregulation in their activity results from their overexpression, enhanced enzymatic activity or epigenetic silencing. Therefore, one may envisage the design and development of chromatin modifiers capable of reducing the expression of pro-inflammatory enzymes such as COX-2 and 5-LOX, while enhancing the expression of the anti-inflammatory enzymes such as 15-LOX-1.
There is no dearth of information in the literature highlighting the importance of eicosanoids in cancer. Delving into the details of how eicosanoids function in both tumor and stromal cells will be essential for understanding the pathways involved. Such knowledge will aid in the design of novel cancer therapies.
ACKNOWLEDGMENTS
The members of the Banerjee Lab are gratefully acknowledged for reading and commenting on the manuscript. Given the large amount of literature covering the field, we apologize to those whose articles were not cited here.
Footnotes
Supported by The TÜBİTAK project, No. 113S935 (to Banerjee S).
Conflict-of-interest statement: No conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 6, 2015
First decision: June 2, 2015
Article in press: August 31, 2015
P- Reviewer: Koc S, Okamoto I S- Editor: Yu J L- Editor: Filipodia E- Editor: Zhang DN
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A. Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev. 2008;27:31–40. doi: 10.1007/s10555-007-9108-5. [DOI] [PubMed] [Google Scholar]
- 4.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 5.Yarur AJ, Strobel SG, Deshpande AR, Abreu MT. Predictors of aggressive inflammatory bowel disease. Gastroenterol Hepatol (N Y) 2011;7:652–659. [PMC free article] [PubMed] [Google Scholar]
- 6.Oshima M, Taketo MM. COX selectivity and animal models for colon cancer. Curr Pharm Des. 2002;8:1021–1034. doi: 10.2174/1381612023394953. [DOI] [PubMed] [Google Scholar]
- 7.Atreya I, Neurath MF. Immune cells in colorectal cancer: prognostic relevance and therapeutic strategies. Expert Rev Anticancer Ther. 2008;8:561–572. doi: 10.1586/14737140.8.4.561. [DOI] [PubMed] [Google Scholar]
- 8.Sung SY, Hsieh CL, Wu D, Chung LW, Johnstone PA. Tumor microenvironment promotes cancer progression, metastasis, and therapeutic resistance. Curr Probl Cancer. 2007;31:36–100. doi: 10.1016/j.currproblcancer.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008;99:1501–1506. doi: 10.1111/j.1349-7006.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011;96:27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariani F, Sena P, Roncucci L. Inflammatory pathways in the early steps of colorectal cancer development. World J Gastroenterol. 2014;20:9716–9731. doi: 10.3748/wjg.v20.i29.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta. 2015;1851:308–330. doi: 10.1016/j.bbalip.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calder PC. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim Biophys Acta. 2015;1851:469–484. doi: 10.1016/j.bbalip.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Kew S, Mesa MD, Tricon S, Buckley R, Minihane AM, Yaqoob P. Effects of oils rich in eicosapentaenoic and docosahexaenoic acids on immune cell composition and function in healthy humans. Am J Clin Nutr. 2004;79:674–681. doi: 10.1093/ajcn/79.4.674. [DOI] [PubMed] [Google Scholar]
- 16.Rees D, Miles EA, Banerjee T, Wells SJ, Roynette CE, Wahle KW, Calder PC. Dose-related effects of eicosapentaenoic acid on innate immune function in healthy humans: a comparison of young and older men. Am J Clin Nutr. 2006;83:331–342. doi: 10.1093/ajcn/83.2.331. [DOI] [PubMed] [Google Scholar]
- 17.Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, Yuan C, Warnock M, Schmaier AH, Yokoyama C, et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem. 2007;282:22254–22266. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- 18.Janakiram NB, Mohammed A, Rao CV. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011;30:507–523. doi: 10.1007/s10555-011-9311-2. [DOI] [PubMed] [Google Scholar]
- 19.Cockbain AJ, Toogood GJ, Hull MA. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2012;61:135–149. doi: 10.1136/gut.2010.233718. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheng H, Shao J, Williams CS, Pereira MA, Taketo MM, Oshima M, Reynolds AB, Washington MK, DuBois RN, Beauchamp RD. Nuclear translocation of beta-catenin in hereditary and carcinogen-induced intestinal adenomas. Carcinogenesis. 1998;19:543–549. doi: 10.1093/carcin/19.4.543. [DOI] [PubMed] [Google Scholar]
- 22.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69 Suppl 1:28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 23.Jiang J, Dingledine R. Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J Pharmacol Exp Ther. 2013;344:360–367. doi: 10.1124/jpet.112.200444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302:649–658. doi: 10.1001/jama.2009.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Zhu J, Lyu F, Panigrahy D, Ferrara KW, Hammock B, Zhang G. ω-3 polyunsaturated fatty acids-derived lipid metabolites on angiogenesis, inflammation and cancer. Prostaglandins Other Lipid Mediat. 2014;113-115:13–20. doi: 10.1016/j.prostaglandins.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka S, Tatsuguchi A, Futagami S, Gudis K, Wada K, Seo T, Mitsui K, Yonezawa M, Nagata K, Fujimori S, et al. Monocyte chemoattractant protein 1 and macrophage cyclooxygenase 2 expression in colonic adenoma. Gut. 2006;55:54–61. doi: 10.1136/gut.2004.059824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaliński P, Vieira PL, Schuitemaker JH, de Jong EC, Kapsenberg ML. Prostaglandin E(2) is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood. 2001;97:3466–3469. doi: 10.1182/blood.v97.11.3466. [DOI] [PubMed] [Google Scholar]
- 28.Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 2004;18:1318–1320. doi: 10.1096/fj.03-1367fje. [DOI] [PubMed] [Google Scholar]
- 29.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23--& gt; IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 30.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 31.Yaqub S, Henjum K, Mahic M, Jahnsen FL, Aandahl EM, Bjørnbeth BA, Taskén K. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother. 2008;57:813–821. doi: 10.1007/s00262-007-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Göbel C, Breitenbuecher F, Kalkavan H, Hähnel PS, Kasper S, Hoffarth S, Merches K, Schild H, Lang KS, Schuler M. Functional expression cloning identifies COX-2 as a suppressor of antigen-specific cancer immunity. Cell Death Dis. 2014;5:e1568. doi: 10.1038/cddis.2014.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, Takei Y, Kobayashi O, Osada T, Watanabe S. Cyclooxygenase 2 modulates killing of cytotoxic T lymphocytes by colon cancer cells. J Clin Biochem Nutr. 2009;45:163–170. doi: 10.3164/jcbn.09-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montrose DC, Nakanishi M, Murphy RC, Zarini S, McAleer JP, Vella AT, Rosenberg DW. The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat. 2014;116-117:26–36. doi: 10.1016/j.prostaglandins.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68:3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 36.Dey I, Lejeune M, Chadee K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol. 2006;149:611–623. doi: 10.1038/sj.bjp.0706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishikawa TO, Oshima M, Herschman HR. Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Carcinogenesis. 2011;32:417–426. doi: 10.1093/carcin/bgq268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JM, Kanaoka Y, Eguchi N, Aritake K, Grujic S, Materi AM, Buslon VS, Tippin BL, Kwong AM, Salido E, et al. Hematopoietic prostaglandin D synthase suppresses intestinal adenomas in ApcMin/+ mice. Cancer Res. 2007;67:881–889. doi: 10.1158/0008-5472.CAN-05-3767. [DOI] [PubMed] [Google Scholar]
- 39.Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, Lawrence E, Lutterbaugh J, Lu S, Willson JK, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci USA. 2004;101:17468–17473. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–527. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 42.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 43.Sostres C, Gargallo CJ, Lanas A. Interaction between Helicobacter pylori infection, nonsteroidal anti-inflammatory drugs and/or low-dose aspirin use: old question new insights. World J Gastroenterol. 2014;20:9439–9450. doi: 10.3748/wjg.v20.i28.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan AT, Arber N, Burn J, Chia WK, Elwood P, Hull MA, Logan RF, Rothwell PM, Schrör K, Baron JA. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res (Phila) 2012;5:164–178. doi: 10.1158/1940-6207.CAPR-11-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ng K, Meyerhardt JA, Chan AT, Sato K, Chan JA, Niedzwiecki D, Saltz LB, Mayer RJ, Benson AB, Schaefer PL, et al. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J Natl Cancer Inst. 2015;107:345. doi: 10.1093/jnci/dju345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ajuebor MN, Singh A, Wallace JL. Cyclooxygenase-2-derived prostaglandin D(2) is an early anti-inflammatory signal in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G238–G244. doi: 10.1152/ajpgi.2000.279.1.G238. [DOI] [PubMed] [Google Scholar]
- 47.Iwanaga K, Nakamura T, Maeda S, Aritake K, Hori M, Urade Y, Ozaki H, Murata T. Mast cell-derived prostaglandin D2 inhibits colitis and colitis-associated colon cancer in mice. Cancer Res. 2014;74:3011–3019. doi: 10.1158/0008-5472.CAN-13-2792. [DOI] [PubMed] [Google Scholar]
- 48.Steele VE, Holmes CA, Hawk ET, Kopelovich L, Lubet RA, Crowell JA, Sigman CC, Kelloff GJ. Lipoxygenase inhibitors as potential cancer chemopreventives. Cancer Epidemiol Biomarkers Prev. 1999;8:467–483. [PubMed] [Google Scholar]
- 49.Schewe T, Halangk W, Hiebsch C, Rapoport SM. A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett. 1975;60:149–152. doi: 10.1016/0014-5793(75)80439-x. [DOI] [PubMed] [Google Scholar]
- 50.Belkner J, Wiesner R, Kühn H, Lankin VZ. The oxygenation of cholesterol esters by the reticulocyte lipoxygenase. FEBS Lett. 1991;279:110–114. doi: 10.1016/0014-5793(91)80263-3. [DOI] [PubMed] [Google Scholar]
- 51.Rådmark O, Samuelsson B. 5-Lipoxygenase: mechanisms of regulation. J Lipid Res. 2009;50 Suppl:S40–S45. doi: 10.1194/jlr.R800062-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savari S, Vinnakota K, Zhang Y, Sjölander A. Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J Gastroenterol. 2014;20:968–977. doi: 10.3748/wjg.v20.i4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rådmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta. 2015;1851:331–339. doi: 10.1016/j.bbalip.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Peters-Golden M. Molecular mechanisms of leukotriene synthesis: the changing paradigm. Clin Exp Allergy. 1998;28:1059–1065. doi: 10.1046/j.1365-2222.1998.00333.x. [DOI] [PubMed] [Google Scholar]
- 55.Sharma JN, Mohammed LA. The role of leukotrienes in the pathophysiology of inflammatory disorders: is there a case for revisiting leukotrienes as therapeutic targets? Inflammopharmacology. 2006;14:10–16. doi: 10.1007/s10787-006-1496-6. [DOI] [PubMed] [Google Scholar]
- 56.Brock TG, Peters-Golden M. Activation and regulation of cellular eicosanoid biosynthesis. ScientificWorldJournal. 2007;7:1273–1284. doi: 10.1100/tsw.2007.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melstrom LG, Bentrem DJ, Salabat MR, Kennedy TJ, Ding XZ, Strouch M, Rao SM, Witt RC, Ternent CA, Talamonti MS, et al. Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clin Cancer Res. 2008;14:6525–6530. doi: 10.1158/1078-0432.CCR-07-4631. [DOI] [PubMed] [Google Scholar]
- 58.Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, Blatner NR, Hix LM, Zhang M, Dennis KL, et al. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCDelta468 mice. Cancer Res. 2011;71:1627–1636. doi: 10.1158/0008-5472.CAN-10-1923. [DOI] [PubMed] [Google Scholar]
- 59.Gounaris E, Heiferman MJ, Heiferman JR, Shrivastav M, Vitello D, Blatner NR, Knab LM, Phillips JD, Cheon EC, Grippo PJ, et al. Zileuton, 5-lipoxygenase inhibitor, acts as a chemopreventive agent in intestinal polyposis, by modulating polyp and systemic inflammation. PLoS One. 2015;10:e0121402. doi: 10.1371/journal.pone.0121402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soumaoro LT, Iida S, Uetake H, Ishiguro M, Takagi Y, Higuchi T, Yasuno M, Enomoto M, Sugihara K. Expression of 5-lipoxygenase in human colorectal cancer. World J Gastroenterol. 2006;12:6355–6360. doi: 10.3748/wjg.v12.i39.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klampfl T, Bogner E, Bednar W, Mager L, Massudom D, Kalny I, Heinzle C, Berger W, Stättner S, Karner J, et al. Up-regulation of 12(S)-lipoxygenase induces a migratory phenotype in colorectal cancer cells. Exp Cell Res. 2012;318:768–778. doi: 10.1016/j.yexcr.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prasad VV, Kolli P, Moganti D. Association of a functional polymorphism (Gln261Arg) in 12-lipoxygenase with breast cancer. Exp Ther Med. 2011;2:317–323. doi: 10.3892/etm.2011.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prasad VV, Padma K. Non-synonymous polymorphism (Gln261Arg) of 12-lipoxygenase in colorectal and thyroid cancers. Fam Cancer. 2012;11:615–621. doi: 10.1007/s10689-012-9559-x. [DOI] [PubMed] [Google Scholar]
- 64.Shan D, Shen K, Zhu J, Feng M, Wu Y, Wan C, Shen Y, Xu L. The polymorphism (Gln261Arg) of 12-lipoxygenase and cancer risk: a meta-analysis. Int J Clin Exp Med. 2015;8:488–495. [PMC free article] [PubMed] [Google Scholar]
- 65.de Carvalho DD, Sadok A, Bourgarel-Rey V, Gattacceca F, Penel C, Lehmann M, Kovacic H. Nox1 downstream of 12-lipoxygenase controls cell proliferation but not cell spreading of colon cancer cells. Int J Cancer. 2008;122:1757–1764. doi: 10.1002/ijc.23300. [DOI] [PubMed] [Google Scholar]
- 66.Kuhn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002;68-69:263–290. doi: 10.1016/s0090-6980(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 67.Shureiqi I, Wu Y, Chen D, Yang XL, Guan B, Morris JS, Yang P, Newman RA, Broaddus R, Hamilton SR, et al. The critical role of 15-lipoxygenase-1 in colorectal epithelial cell terminal differentiation and tumorigenesis. Cancer Res. 2005;65:11486–11492. doi: 10.1158/0008-5472.CAN-05-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moussalli MJ, Wu Y, Zuo X, Yang XL, Wistuba II, Raso MG, Morris JS, Bowser JL, Minna JD, Lotan R, et al. Mechanistic contribution of ubiquitous 15-lipoxygenase-1 expression loss in cancer cells to terminal cell differentiation evasion. Cancer Prev Res (Phila) 2011;4:1961–1972. doi: 10.1158/1940-6207.CAPR-10-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zuo X, Morris JS, Broaddus R, Shureiqi I. 15-LOX-1 transcription suppression through the NuRD complex in colon cancer cells. Oncogene. 2009;28:1496–1505. doi: 10.1038/onc.2008.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu C, Xu D, Sjöberg J, Forsell P, Björkholm M, Claesson HE. Transcriptional regulation of 15-lipoxygenase expression by promoter methylation. Exp Cell Res. 2004;297:61–67. doi: 10.1016/j.yexcr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 71.Zuo X, Shen L, Issa JP, Moy O, Morris JS, Lippman SM, Shureiqi I. 15-Lipoxygenase-1 transcriptional silencing by DNA methyltransferase-1 independently of DNA methylation. FASEB J. 2008;22:1981–1992. doi: 10.1096/fj.07-098301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shankaranarayanan P, Chaitidis P, Kühn H, Nigam S. Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J Biol Chem. 2001;276:42753–42760. doi: 10.1074/jbc.M102626200. [DOI] [PubMed] [Google Scholar]
- 73.Zuo X, Peng Z, Wu Y, Moussalli MJ, Yang XL, Wang Y, Parker-Thornburg J, Morris JS, Broaddus RR, Fischer SM, et al. Effects of gut-targeted 15-LOX-1 transgene expression on colonic tumorigenesis in mice. J Natl Cancer Inst. 2012;104:709–716. doi: 10.1093/jnci/djs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shureiqi I, Chen D, Day RS, Zuo X, Hochman FL, Ross WA, Cole RA, Moy O, Morris JS, Xiao L, et al. Profiling lipoxygenase metabolism in specific steps of colorectal tumorigenesis. Cancer Prev Res (Phila) 2010;3:829–838. doi: 10.1158/1940-6207.CAPR-09-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cimen I, Astarci E, Banerjee S. 15-lipoxygenase-1 exerts its tumor suppressive role by inhibiting nuclear factor-kappa B via activation of PPAR gamma. J Cell Biochem. 2011;112:2490–2501. doi: 10.1002/jcb.23174. [DOI] [PubMed] [Google Scholar]
- 76.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mangino MJ, Brounts L, Harms B, Heise C. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2006;79:84–92. doi: 10.1016/j.prostaglandins.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 78.Chen GG, Xu H, Lee JF, Subramaniam M, Leung KL, Wang SH, Chan UP, Spelsberg TC. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor gamma-dependent pathway. Int J Cancer. 2003;107:837–843. doi: 10.1002/ijc.11447. [DOI] [PubMed] [Google Scholar]
- 79.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 80.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 81.Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58:362–366. [PubMed] [Google Scholar]
- 82.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 83.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 84.Rigau J, Piqué JM, Rubio E, Planas R, Tarrech JM, Bordas JM. Effects of long-term sulindac therapy on colonic polyposis. Ann Intern Med. 1991;115:952–954. doi: 10.7326/0003-4819-115-12-952. [DOI] [PubMed] [Google Scholar]
- 85.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 86.Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, Aaronson SA, Lee SW. P53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 2002;21:5635–5644. doi: 10.1093/emboj/cdf591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 88.Greenhough A, Wallam CA, Hicks DJ, Moorghen M, Williams AC, Paraskeva C. The proapoptotic BH3-only protein Bim is downregulated in a subset of colorectal cancers and is repressed by antiapoptotic COX-2/PGE(2) signalling in colorectal adenoma cells. Oncogene. 2010;29:3398–3410. doi: 10.1038/onc.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawamori T, Uchiya N, Sugimura T, Wakabayashi K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis. 2003;24:985–990. doi: 10.1093/carcin/bgg033. [DOI] [PubMed] [Google Scholar]
- 90.Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, Tengowski MW, Wang F, Gualberto A, Beebe JS, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res. 2005;11:2063–2073. doi: 10.1158/1078-0432.CCR-04-1070. [DOI] [PubMed] [Google Scholar]
- 91.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 92.Limasale YD, Tezcaner A, Özen C, Keskin D, Banerjee S. Epidermal growth factor receptor-targeted immunoliposomes for delivery of celecoxib to cancer cells. Int J Pharm. 2015;479:364–373. doi: 10.1016/j.ijpharm.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 93.Roberts HR, Smartt HJ, Greenhough A, Moore AE, Williams AC, Paraskeva C. Colon tumour cells increase PGE(2) by regulating COX-2 and 15-PGDH to promote survival during the microenvironmental stress of glucose deprivation. Carcinogenesis. 2011;32:1741–1747. doi: 10.1093/carcin/bgr210. [DOI] [PubMed] [Google Scholar]
- 94.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 95.Cherukuri DP, Chen XB, Goulet AC, Young RN, Han Y, Heimark RL, Regan JW, Meuillet E, Nelson MA. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp Cell Res. 2007;313:2969–2979. doi: 10.1016/j.yexcr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park SW, Kim HS, Choi MS, Jeong WJ, Heo DS, Kim KH, Sung MW. The effects of the stromal cell-derived cyclooxygenase-2 metabolite prostaglandin E2 on the proliferation of colon cancer cells. J Pharmacol Exp Ther. 2011;336:516–523. doi: 10.1124/jpet.110.173278. [DOI] [PubMed] [Google Scholar]
- 97.Chen GG, Lee JF, Wang SH, Chan UP, Ip PC, Lau WY. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-kappaB in human colon cancer. Life Sci. 2002;70:2631–2646. doi: 10.1016/s0024-3205(02)01510-2. [DOI] [PubMed] [Google Scholar]
- 98.Lin MS, Chen WC, Bai X, Wang YD. Activation of peroxisome proliferator-activated receptor gamma inhibits cell growth via apoptosis and arrest of the cell cycle in human colorectal cancer. J Dig Dis. 2007;8:82–88. doi: 10.1111/j.1443-9573.2007.00290.x. [DOI] [PubMed] [Google Scholar]
- 99.Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut. 2002;50:658–664. doi: 10.1136/gut.50.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koyama M, Izutani Y, Goda AE, Matsui TA, Horinaka M, Tomosugi M, Fujiwara J, Nakamura Y, Wakada M, Yogosawa S, et al. Histone deacetylase inhibitors and 15-deoxy-Delta12,14-prostaglandin J2 synergistically induce apoptosis. Clin Cancer Res. 2010;16:2320–2332. doi: 10.1158/1078-0432.CCR-09-2301. [DOI] [PubMed] [Google Scholar]
- 101.Shin SW, Seo CY, Han H, Han JY, Jeong JS, Kwak JY, Park JI. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin Cancer Res. 2009;15:5414–5425. doi: 10.1158/1078-0432.CCR-08-3101. [DOI] [PubMed] [Google Scholar]
- 102.Wasilewicz MP, Kołodziej B, Bojułko T, Kaczmarczyk M, Sulzyc-Bielicka V, Bielicki D, Ciepiela K. Overexpression of 5-lipoxygenase in sporadic colonic adenomas and a possible new aspect of colon carcinogenesis. Int J Colorectal Dis. 2010;25:1079–1085. doi: 10.1007/s00384-010-0980-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci. 2007;103:24–32. doi: 10.1254/jphs.fp0060651. [DOI] [PubMed] [Google Scholar]
- 104.Ohd JF, Wikström K, Sjölander A. Leukotrienes induce cell-survival signaling in intestinal epithelial cells. Gastroenterology. 2000;119:1007–1018. doi: 10.1053/gast.2000.18141. [DOI] [PubMed] [Google Scholar]
- 105.Ohd JF, Nielsen CK, Campbell J, Landberg G, Löfberg H, Sjölander A. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology. 2003;124:57–70. doi: 10.1053/gast.2003.50011. [DOI] [PubMed] [Google Scholar]
- 106.Romano M, Claria J. Cyclooxygenase-2 and 5-lipoxygenase converging functions on cell proliferation and tumor angiogenesis: implications for cancer therapy. FASEB J. 2003;17:1986–1995. doi: 10.1096/fj.03-0053rev. [DOI] [PubMed] [Google Scholar]
- 107.Byrum RS, Goulet JL, Griffiths RJ, Koller BH. Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses. J Exp Med. 1997;185:1065–1075. doi: 10.1084/jem.185.6.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peters-Golden M, Bailie M, Marshall T, Wilke C, Phan SH, Toews GB, Moore BB. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am J Respir Crit Care Med. 2002;165:229–235. doi: 10.1164/ajrccm.165.2.2104050. [DOI] [PubMed] [Google Scholar]
- 109.Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Langenbach R, Roggli VL, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 1999;104:721–732. doi: 10.1172/JCI6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li N, Sood S, Wang S, Fang M, Wang P, Sun Z, Yang CS, Chen X. Overexpression of 5-lipoxygenase and cyclooxygenase 2 in hamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clin Cancer Res. 2005;11:2089–2096. doi: 10.1158/1078-0432.CCR-04-1684. [DOI] [PubMed] [Google Scholar]
- 111.Tavolari S, Bonafè M, Marini M, Ferreri C, Bartolini G, Brighenti E, Manara S, Tomasi V, Laufer S, Guarnieri T. Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade. Carcinogenesis. 2008;29:371–380. doi: 10.1093/carcin/bgm265. [DOI] [PubMed] [Google Scholar]
- 112.Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, Schiavone N, Messerini L, Vannacci A, Capaccioli S, Perna F, et al. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther. 2006;5:2716–2726. doi: 10.1158/1535-7163.MCT-06-0318. [DOI] [PubMed] [Google Scholar]
- 113.Bhattacharya S, Mathew G, Jayne DG, Pelengaris S, Khan M. 15-lipoxygenase-1 in colorectal cancer: a review. Tumour Biol. 2009;30:185–199. doi: 10.1159/000236864. [DOI] [PubMed] [Google Scholar]
- 114.Kamitani H, Geller M, Eling T. Expression of 15-lipoxygenase by human colorectal carcinoma Caco-2 cells during apoptosis and cell differentiation. J Biol Chem. 1998;273:21569–21577. doi: 10.1074/jbc.273.34.21569. [DOI] [PubMed] [Google Scholar]
- 115.Yoshinaga M, Buchanan FG, DuBois RN. 15-LOX-1 inhibits p21 (Cip/WAF 1) expression by enhancing MEK-ERK 1/2 signaling in colon carcinoma cells. Prostaglandins Other Lipid Mediat. 2004;73:111–122. doi: 10.1016/j.prostaglandins.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 116.Shureiqi I, Wojno KJ, Poore JA, Reddy RG, Moussalli MJ, Spindler SA, Greenson JK, Normolle D, Hasan AA, Lawrence TS, et al. Decreased 13-S-hydroxyoctadecadienoic acid levels and 15-lipoxygenase-1 expression in human colon cancers. Carcinogenesis. 1999;20:1985–1995. doi: 10.1093/carcin/20.10.1985. [DOI] [PubMed] [Google Scholar]
- 117.Cimen I, Tunçay S, Banerjee S. 15-Lipoxygenase-1 expression suppresses the invasive properties of colorectal carcinoma cell lines HCT-116 and HT-29. Cancer Sci. 2009;100:2283–2291. doi: 10.1111/j.1349-7006.2009.01313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yuri M, Sasahira T, Nakai K, Ishimaru S, Ohmori H, Kuniyasu H. Reversal of expression of 15-lipoxygenase-1 to cyclooxygenase-2 is associated with development of colonic cancer. Histopathology. 2007;51:520–527. doi: 10.1111/j.1365-2559.2007.02799.x. [DOI] [PubMed] [Google Scholar]
- 119.Zuo X, Shureiqi I. Eicosanoid profiling in colon cancer: emergence of a pattern. Prostaglandins Other Lipid Mediat. 2013;104-105:139–143. doi: 10.1016/j.prostaglandins.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 121.Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 123.Ikezoe T, Miller CW, Kawano S, Heaney A, Williamson EA, Hisatake J, Green E, Hofmann W, Taguchi H, Koeffler HP. Mutational analysis of the peroxisome proliferator-activated receptor gamma gene in human malignancies. Cancer Res. 2001;61:5307–5310. [PubMed] [Google Scholar]
- 124.Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, Hosokawa M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001;61:2424–2428. [PubMed] [Google Scholar]
- 125.Chen ZY, Tseng CC. 15-deoxy-Delta12,14 prostaglandin J2 up-regulates Kruppel-like factor 4 expression independently of peroxisome proliferator-activated receptor gamma by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Mol Pharmacol. 2005;68:1203–1213. doi: 10.1124/mol.105.014944. [DOI] [PubMed] [Google Scholar]
- 126.Toaldo C, Pizzimenti S, Cerbone A, Pettazzoni P, Menegatti E, Daniela B, Minelli R, Giglioni B, Dianzani MU, Ferretti C, et al. PPARgamma ligands inhibit telomerase activity and hTERT expression through modulation of the Myc/Mad/Max network in colon cancer cells. J Cell Mol Med. 2010;14:1347–1357. doi: 10.1111/j.1582-4934.2009.00966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 128.Zuo X, Wu Y, Morris JS, Stimmel JB, Leesnitzer LM, Fischer SM, Lippman SM, Shureiqi I. Oxidative metabolism of linoleic acid modulates PPAR-beta/delta suppression of PPAR-gamma activity. Oncogene. 2006;25:1225–1241. doi: 10.1038/sj.onc.1209160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sasaki T, Fujii K, Yoshida K, Shimura H, Sasahira T, Ohmori H, Kuniyasu H. Peritoneal metastasis inhibition by linoleic acid with activation of PPARgamma in human gastrointestinal cancer cells. Virchows Arch. 2006;448:422–427. doi: 10.1007/s00428-005-0110-4. [DOI] [PubMed] [Google Scholar]
- 130.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 131.Dubuquoy L, Rousseaux C, Thuru X, Peyrin-Biroulet L, Romano O, Chavatte P, Chamaillard M, Desreumaux P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55:1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM, Lippman SM. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci USA. 2003;100:9968–9973. doi: 10.1073/pnas.1631086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhong H, Wang R, Kelavkar U, Wang CY, Simons J. Enzyme 15-lipoxygenase 1 promotes hypoxia-inducible factor 1α turnover and reduces vascular endothelial growth factor expression: implications for angiogenesis. Cancer Med. 2014;3:514–525. doi: 10.1002/cam4.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mao F, Xu M, Zuo X, Yu J, Xu W, Moussalli MJ, Elias E, Li HS, Watowich SS, Shureiqi I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015;29:2359–2370. doi: 10.1096/fj.14-264515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat Med. 2004;10:245–247. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- 136.Gupta RA, Tan J, Krause WF, Geraci MW, Willson TM, Dey SK, DuBois RN. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci USA. 2000;97:13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Takayama O, Yamamoto H, Damdinsuren B, Sugita Y, Ngan CY, Xu X, Tsujino T, Takemasa I, Ikeda M, Sekimoto M, et al. Expression of PPARdelta in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–895. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu KK, Liou JY. Cyclooxygenase inhibitors induce colon cancer cell apoptosis Via PPARdelta --& gt; 14-3-3epsilon pathway. Methods Mol Biol. 2009;512:295–307. doi: 10.1007/978-1-60327-530-9_16. [DOI] [PubMed] [Google Scholar]
- 139.Cutler NS, Graves-Deal R, LaFleur BJ, Gao Z, Boman BM, Whitehead RH, Terry E, Morrow JD, Coffey RJ. Stromal production of prostacyclin confers an antiapoptotic effect to colonic epithelial cells. Cancer Res. 2003;63:1748–1751. [PubMed] [Google Scholar]
- 140.Jeong E, Koo JE, Yeon SH, Kwak MK, Hwang DH, Lee JY. PPARδ deficiency disrupts hypoxia-mediated tumorigenic potential of colon cancer cells. Mol Carcinog. 2014;53:926–937. doi: 10.1002/mc.22144. [DOI] [PubMed] [Google Scholar]
- 141.Davies RJ, Miller R, Coleman N. Colorectal cancer screening: prospects for molecular stool analysis. Nat Rev Cancer. 2005;5:199–209. doi: 10.1038/nrc1569. [DOI] [PubMed] [Google Scholar]
- 142.Lurje G, Zhang W, Lenz HJ. Molecular prognostic markers in locally advanced colon cancer. Clin Colorectal Cancer. 2007;6:683–690. doi: 10.3816/CCC.2007.n.037. [DOI] [PubMed] [Google Scholar]
- 143.Dubois RN. Role of inflammation and inflammatory mediators in colorectal cancer. Trans Am Clin Climatol Assoc. 2014;125:358–72; discussion 372-3. [PMC free article] [PubMed] [Google Scholar]
- 144.Usman MW, Luo F, Cheng H, Zhao JJ, Liu P. Chemopreventive effects of aspirin at a glance. Biochim Biophys Acta. 2015;1855:254–263. doi: 10.1016/j.bbcan.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 145.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 146.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 147.Cianchi F, Cortesini C, Bechi P, Fantappiè O, Messerini L, Vannacci A, Sardi I, Baroni G, Boddi V, Mazzanti R, et al. Up-regulation of cyclooxygenase 2 gene expression correlates with tumor angiogenesis in human colorectal cancer. Gastroenterology. 2001;121:1339–1347. doi: 10.1053/gast.2001.29691. [DOI] [PubMed] [Google Scholar]
- 148.Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett. 2010;295:7–16. doi: 10.1016/j.canlet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 149.Radinsky R. Modulation of tumor cell gene expression and phenotype by the organ-specific metastatic environment. Cancer Metastasis Rev. 1995;14:323–338. doi: 10.1007/BF00690601. [DOI] [PubMed] [Google Scholar]
- 150.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:35451–35457. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 151.Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–1497. doi: 10.1073/pnas.0510562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17:1640–1647. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 153.Fujino H, Toyomura K, Chen XB, Regan JW, Murayama T. Prostaglandin E₂ regulates cellular migration via induction of vascular endothelial growth factor receptor-1 in HCA-7 human colon cancer cells. Biochem Pharmacol. 2011;81:379–387. doi: 10.1016/j.bcp.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 154.Hsu HH, Hu WS, Lin YM, Kuo WW, Chen LM, Chen WK, Hwang JM, Tsai FJ, Liu CJ, Huang CY. JNK suppression is essential for 17β-Estradiol inhibits prostaglandin E2-Induced uPA and MMP-9 expressions and cell migration in human LoVo colon cancer cells. J Biomed Sci. 2011;18:61. doi: 10.1186/1423-0127-18-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Daneker GW, Lund SA, Caughman SW, Staley CA, Wood WC. Anti-metastatic prostacyclins inhibit the adhesion of colon carcinoma to endothelial cells by blocking E-selectin expression. Clin Exp Metastasis. 1996;14:230–238. doi: 10.1007/BF00053896. [DOI] [PubMed] [Google Scholar]
- 156.Cathcart MC, Lysaght J, Pidgeon GP. Eicosanoid signalling pathways in the development and progression of colorectal cancer: novel approaches for prevention/intervention. Cancer Metastasis Rev. 2011;30:363–385. doi: 10.1007/s10555-011-9324-x. [DOI] [PubMed] [Google Scholar]
- 157.Koontongkaew S, Monthanapisut P, Saensuk T. Inhibition of arachidonic acid metabolism decreases tumor cell invasion and matrix metalloproteinase expression. Prostaglandins Other Lipid Mediat. 2010;93:100–108. doi: 10.1016/j.prostaglandins.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 158.Hennig R, Kehl T, Noor S, Ding XZ, Rao SM, Bergmann F, Fürstenberger G, Büchler MW, Friess H, Krieg P, et al. 15-lipoxygenase-1 production is lost in pancreatic cancer and overexpression of the gene inhibits tumor cell growth. Neoplasia. 2007;9:917–926. doi: 10.1593/neo.07565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wu Y, Mao F, Zuo X, Moussalli MJ, Elias E, Xu W, Shureiqi I. 15-LOX-1 suppression of hypoxia-induced metastatic phenotype and HIF-1α expression in human colon cancer cells. Cancer Med. 2014;3:472–484. doi: 10.1002/cam4.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. Genes Cancer. 2011;2:1085–1096. doi: 10.1177/1947601911432334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kliche S, Waltenberger J. VEGF receptor signaling and endothelial function. IUBMB Life. 2001;52:61–66. doi: 10.1080/15216540252774784. [DOI] [PubMed] [Google Scholar]
- 162.Brown NS, Streeter EH, Jones A, Harris AL, Bicknell R. Cooperative stimulation of vascular endothelial growth factor expression by hypoxia and reactive oxygen species: the effect of targeting vascular endothelial growth factor and oxidative stress in an orthotopic xenograft model of bladder carcinoma. Br J Cancer. 2005;92:1696–1701. doi: 10.1038/sj.bjc.6602522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 164.Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996;271:736–741. doi: 10.1074/jbc.271.2.736. [DOI] [PubMed] [Google Scholar]
- 165.Fukuda R, Kelly B, Semenza GL. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res. 2003;63:2330–2334. [PubMed] [Google Scholar]
- 166.Jurek D, Udilova N, Jozkowicz A, Nohl H, Marian B, Schulte-Hermann R. Dietary lipid hydroperoxides induce expression of vascular endothelial growth factor (VEGF) in human colorectal tumor cells. FASEB J. 2005;19:97–99. doi: 10.1096/fj.04-2111fje. [DOI] [PubMed] [Google Scholar]
- 167.Ko HM, Seo KH, Han SJ, Ahn KY, Choi IH, Koh GY, Lee HK, Ra MS, Im SY. Nuclear factor kappaB dependency of platelet-activating factor-induced angiogenesis. Cancer Res. 2002;62:1809–1814. [PubMed] [Google Scholar]
- 168.Mezentsev A, Seta F, Dunn MW, Ono N, Falck JR, Laniado-Schwartzman M. Eicosanoid regulation of vascular endothelial growth factor expression and angiogenesis in microvessel endothelial cells. J Biol Chem. 2002;277:18670–18676. doi: 10.1074/jbc.M201143200. [DOI] [PubMed] [Google Scholar]
- 169.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 170.Ochs AM, Wong L, Kakani V, Neerukonda S, Gorske J, Rao A, Riggs M, Ward H, Keville L. Expression of vascular endothelial growth factor and HER2/neu in stage II colon cancer and correlation with survival. Clin Colorectal Cancer. 2004;4:262–267. doi: 10.3816/ccc.2004.n.025. [DOI] [PubMed] [Google Scholar]
- 171.Buchanan FG, DuBois RN. Connecting COX-2 and Wnt in cancer. Cancer Cell. 2006;9:6–8. doi: 10.1016/j.ccr.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 172.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 173.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 174.Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, Richmond A, Strieter R, Dey SK, DuBois RN. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203:941–951. doi: 10.1084/jem.20052124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kim SH, Park YY, Kim SW, Lee JS, Wang D, DuBois RN. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res. 2011;71:7010–7020. doi: 10.1158/0008-5472.CAN-11-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dormond O, Bezzi M, Mariotti A, Ruegg C. Prostaglandin E2 promotes integrin alpha Vbeta 3-dependent endothelial cell adhesion, rac-activation, and spreading through cAMP/PKA-dependent signaling. J Biol Chem. 2002;277:45838–45846. doi: 10.1074/jbc.M209213200. [DOI] [PubMed] [Google Scholar]
- 177.Salcedo R, Zhang X, Young HA, Michael N, Wasserman K, Ma WH, Martins-Green M, Murphy WJ, Oppenheim JJ. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood. 2003;102:1966–1977. doi: 10.1182/blood-2002-11-3400. [DOI] [PubMed] [Google Scholar]
- 178.Daniel TO, Liu H, Morrow JD, Crews BC, Marnett LJ. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res. 1999;59:4574–4577. [PubMed] [Google Scholar]
- 179.Pradono P, Tazawa R, Maemondo M, Tanaka M, Usui K, Saijo Y, Hagiwara K, Nukiwa T. Gene transfer of thromboxane A(2) synthase and prostaglandin I(2) synthase antithetically altered tumor angiogenesis and tumor growth. Cancer Res. 2002;62:63–66. [PubMed] [Google Scholar]
- 180.Abdel-Majid RM, Marshall JS. Prostaglandin E2 induces degranulation-independent production of vascular endothelial growth factor by human mast cells. J Immunol. 2004;172:1227–1236. doi: 10.4049/jimmunol.172.2.1227. [DOI] [PubMed] [Google Scholar]
- 181.Nakayama T, Mutsuga N, Yao L, Tosato G. Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J Leukoc Biol. 2006;79:95–104. doi: 10.1189/jlb.0405226. [DOI] [PubMed] [Google Scholar]
- 182.Gentile LB, Piva B, Diaz BL. Hypertonic stress induces VEGF production in human colon cancer cell line Caco-2: inhibitory role of autocrine PGE₂. PLoS One. 2011;6:e25193. doi: 10.1371/journal.pone.0025193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Grau R, Iñiguez MA, Fresno M. Inhibition of activator protein 1 activation, vascular endothelial growth factor, and cyclooxygenase-2 expression by 15-deoxy-Delta12,14-prostaglandin J2 in colon carcinoma cells: evidence for a redox-sensitive peroxisome proliferator-activated receptor-gamma-independent mechanism. Cancer Res. 2004;64:5162–5171. doi: 10.1158/0008-5472.CAN-04-0849. [DOI] [PubMed] [Google Scholar]
- 184.Ye YN, Liu ES, Shin VY, Wu WK, Luo JC, Cho CH. Nicotine promoted colon cancer growth via epidermal growth factor receptor, c-Src, and 5-lipoxygenase-mediated signal pathway. J Pharmacol Exp Ther. 2004;308:66–72. doi: 10.1124/jpet.103.058321. [DOI] [PubMed] [Google Scholar]
- 185.Ye YN, Liu ES, Shin VY, Wu WK, Cho CH. Contributory role of 5-lipoxygenase and its association with angiogenesis in the promotion of inflammation-associated colonic tumorigenesis by cigarette smoking. Toxicology. 2004;203:179–188. doi: 10.1016/j.tox.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 186.Ye YN, Wu WK, Shin VY, Cho CH. A mechanistic study of colon cancer growth promoted by cigarette smoke extract. Eur J Pharmacol. 2005;519:52–57. doi: 10.1016/j.ejphar.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 187.Savari S, Liu M, Zhang Y, Sime W, Sjölander A. CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One. 2013;8:e73466. doi: 10.1371/journal.pone.0073466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rosengren S, Olofsson AM, von Andrian UH, Lundgren-Akerlund E, Arfors KE. Leukotriene B4-induced neutrophil-mediated endothelial leakage in vitro and in vivo. J Appl Physiol (1985) 1991;71:1322–1330. doi: 10.1152/jappl.1991.71.4.1322. [DOI] [PubMed] [Google Scholar]
- 189.Steiner DR, Gonzalez NC, Wood JG. Leukotriene B(4) promotes reactive oxidant generation and leukocyte adherence during acute hypoxia. J Appl Physiol (1985) 2001;91:1160–1167. doi: 10.1152/jappl.2001.91.3.1160. [DOI] [PubMed] [Google Scholar]
- 190.Kim GY, Lee JW, Cho SH, Seo JM, Kim JH. Role of the low-affinity leukotriene B4 receptor BLT2 in VEGF-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:915–920. doi: 10.1161/ATVBAHA.109.185793. [DOI] [PubMed] [Google Scholar]
- 191.Tsopanoglou NE, Pipili-Synetos E, Maragoudakis ME. Leukotrienes C4 and D4 promote angiogenesis via a receptor-mediated interaction. Eur J Pharmacol. 1994;258:151–154. doi: 10.1016/0014-2999(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 192.Kanaoka Y, Maekawa A, Penrose JF, Austen KF, Lam BK. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J Biol Chem. 2001;276:22608–22613. doi: 10.1074/jbc.M103562200. [DOI] [PubMed] [Google Scholar]
- 193.Cimen I. The mechanism of antitumorigenic effect of 15-Lipoxygenase-1 in colorectal cancer. PhD Thesis, Department of Biological Sciences: Middle East Technical University; 2012. Available from: http://library.metu.edu.tr/search~S15?/a{u00C7}imen/ac~aimen/1,9,10,B/l856~b2013263&FF=ac~aimen ismail&1,1,,1,0. [Google Scholar]
- 194.Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2015;7:a016311. doi: 10.1101/cshperspect.a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids. 2005;73:163–177. doi: 10.1016/j.plefa.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 196.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Natl Acad Sci USA. 2014;111:12746–12751. doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gewirtz AT, Collier-Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF, Williams IR, Neish AS, Madara JL. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J Immunol. 2002;168:5260–5267. doi: 10.4049/jimmunol.168.10.5260. [DOI] [PubMed] [Google Scholar]
- 198.Gronert K, Gewirtz A, Madara JL, Serhan CN. Identification of a human enterocyte lipoxin A4 receptor that is regulated by interleukin (IL)-13 and interferon gamma and inhibits tumor necrosis factor alpha-induced IL-8 release. J Exp Med. 1998;187:1285–1294. doi: 10.1084/jem.187.8.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kure I, Nishiumi S, Nishitani Y, Tanoue T, Ishida T, Mizuno M, Fujita T, Kutsumi H, Arita M, Azuma T, et al. Lipoxin A(4) reduces lipopolysaccharide-induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor-kappaB activation. J Pharmacol Exp Ther. 2010;332:541–548. doi: 10.1124/jpet.109.159046. [DOI] [PubMed] [Google Scholar]
- 200.Vong L, Ferraz JG, Dufton N, Panaccione R, Beck PL, Sherman PM, Perretti M, Wallace JL. Up-regulation of Annexin-A1 and lipoxin A(4) in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS One. 2012;7:e39244. doi: 10.1371/journal.pone.0039244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Prescott D, McKay DM. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am J Physiol Gastrointest Liver Physiol. 2011;301:G487–G497. doi: 10.1152/ajpgi.00042.2011. [DOI] [PubMed] [Google Scholar]
- 202.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin Immunol. 2015;27:200–215. doi: 10.1016/j.smim.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Campbell EL, Louis NA, Tomassetti SE, Canny GO, Arita M, Serhan CN, Colgan SP. Resolvin E1 promotes mucosal surface clearance of neutrophils: a new paradigm for inflammatory resolution. FASEB J. 2007;21:3162–3170. doi: 10.1096/fj.07-8473com. [DOI] [PubMed] [Google Scholar]
- 204.Campbell EL, MacManus CF, Kominsky DJ, Keely S, Glover LE, Bowers BE, Scully M, Bruyninckx WJ, Colgan SP. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc Natl Acad Sci USA. 2010;107:14298–14303. doi: 10.1073/pnas.0914730107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hull MA, Sandell AC, Montgomery AA, Logan RF, Clifford GM, Rees CJ, Loadman PM, Whitham D. A randomized controlled trial of eicosapentaenoic acid and/or aspirin for colorectal adenoma prevention during colonoscopic surveillance in the NHS Bowel Cancer Screening Programme (The seAFOod Polyp Prevention Trial): study protocol for a randomized controlled trial. Trials. 2013;14:237. doi: 10.1186/1745-6215-14-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Marcon R, Bento AF, Dutra RC, Bicca MA, Leite DF, Calixto JB. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J Immunol. 2013;191:4288–4298. doi: 10.4049/jimmunol.1202743. [DOI] [PubMed] [Google Scholar]
- 207.Castro-Rodriguez JA, Rodrigo GJ. The role of inhaled corticosteroids and montelukast in children with mild-moderate asthma: results of a systematic review with meta-analysis. Arch Dis Child. 2010;95:365–370. doi: 10.1136/adc.2009.169177. [DOI] [PubMed] [Google Scholar]