

Abstract
Objectives
Peroxisome proliferator-activated receptor γ coactivator 1 (PPARGCA1, PGC-1) transcriptional coactivators control gene programs important for nutrient metabolism. Islets of type 2 diabetic subjects have reduced PGC-1α expression and this is associated with decreased insulin secretion, yet little is known about why this occurs or what role it plays in the development of diabetes. Our goal was to delineate the role and importance of PGC-1 proteins to β-cell function and energy homeostasis.
Methods
We investigated how nutrient signals regulate coactivator expression in islets and the metabolic consequences of reduced PGC-1α and PGC-1β in primary and cultured β-cells. Mice with inducible β-cell specific double knockout of Pgc-1α/Pgc-1β (βPgc-1 KO) were created to determine the physiological impact of reduced Pgc1 expression on glucose homeostasis.
Results
Pgc-1α and Pgc-1β expression was increased in primary mouse and human islets by acute glucose and palmitate exposure. Surprisingly, PGC-1 proteins were dispensable for the maintenance of mitochondrial mass, gene expression, and oxygen consumption in response to glucose in adult β-cells. However, islets and mice with an inducible, β-cell-specific PGC-1 knockout had decreased insulin secretion due in large part to loss of the potentiating effect of fatty acids. Consistent with an essential role for PGC-1 in lipid metabolism, β-cells with reduced PGC-1s accumulated acyl-glycerols and PGC-1s controlled expression of key enzymes in lipolysis and the glycerolipid/free fatty acid cycle.
Conclusions
These data highlight the importance of PGC-1s in coupling β-cell lipid metabolism to promote efficient insulin secretion.
Keywords: Beta cell, PGC-1, Insulin secretion, Lipid metabolism, Mitochondria, Lipolysis
Graphical abstract
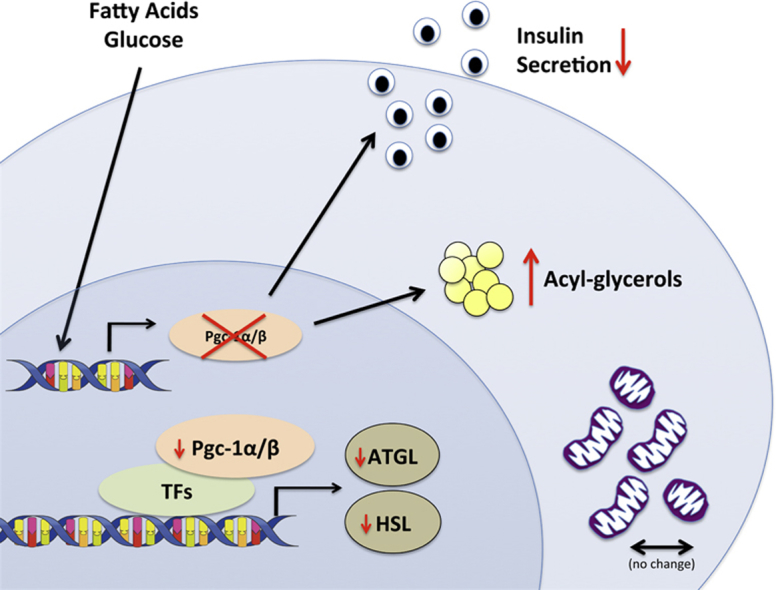
Highlights
-
•
Loss of Pgc-1s in adult β-cells decreases insulin secretion in response to glucose/palmitate.
-
•
Pgc-1α/β is not required to maintain basal mitochondrial mass or oxidative capacity in mature β-cells.
-
•
Pgc-1α/β regulates expression of the lipolytic enzymes HSL and ATGL in β-cells.
-
•
Reduced β-cell Pgc-1 causes accumulation of intracellular acyl-glycerols and cholesterol esters.
1. Introduction
The peroxisome proliferator-activated receptor γ coactivators-1α and β (PGC-1α and PGC-1β) have gained notoriety as important players in the development of diabetes due to altered expression in highly oxidative tissues (i.e. muscle, brown fat, liver) in patients [1], [2], [3], [4]. Decreased Pgc-1α mRNA expression in peripheral organs like liver and muscle is associated with insulin resistance and glucose intolerance [2], [5], and a gene variant of PGC-1α (Gly482Ser) correlates with increased diabetes risk in various human populations [6], [7]. PGC-1α and PGC-1β are transcriptional co-activators that regulate activity of multiple transcription factors including nuclear respiratory factor 1 (NRF1), estrogen related receptor α (ERRα) and peroxisome proliferator-activated receptors (PPAR) and are known to amplify expression of an extensive gene program controlling mitochondrial function and integrity [4], [8]. Reducing Pgc-1α specifically in rodent liver, muscle or adipose tissue can cause insulin resistance and glucose intolerance [8], [9], [10], [11], [12]. Pgc-1α expression is significantly decreased in islets of type 2 diabetic subjects, correlating with decreased insulin secretory capacity [13]. However, it is not known whether low levels of PGC-1s directly contribute to β-cell dysfunction and loss, two hallmarks of diabetes.
A number of studies implicate PGC-1α as an important mediator of β-cell function. Pgc1α expression in β-cells is induced by extracellular signals including both facilitators of glucose-stimulated insulin secretion (GSIS), such as glucagon-like peptide-1 and cAMP [14], and stressors that impair β-cell function including glucocorticoids, streptozotocin, cold exposure, obesity, and glucolipotoxic conditions [14], [15], [16], [17]. Pgc-1α levels are decreased in islets or cultured β-cells exposed to high glucose concentrations and knockdown of PGC-1α in human islets decreases GSIS [13], suggesting a mechanistic link between low PGC-1α expression and efficient insulin secretion. Paradoxically, overexpression of Pgc-1α in primary rat islets [15], [18] has also been shown to blunt insulin secretion; yet in vivo studies demonstrate this may only be the case when the coactivator is increased in β-cells during pancreatic development [17]. Very little is known about the role of the structurally related PGC-1β in β-cells, although it has also been suggested to play an inhibitory role on insulin secretion [16]. Taken together, these data demonstrate a clear importance for PGC-1 co-activators in β-cell function; yet, molecular pathways linking PGC-1 activity to GSIS are not understood and it remains unclear whether decreased PGC-1 expression in adult β-cells, as seen in diabetic subjects, augments the development and/or progression of β-cell dysfunction towards diabetes.
β-cell mitochondrial dysfunction is thought to play a key role in the pathogenesis of diabetes, as ATP production is necessary for optimal fuel-stimulated insulin secretion [19], [20]. β-cells of human diabetic islets exhibit decreased hyperpolarization of mitochondrial membranes and altered internal mitochondrial structure [21], but it is still not clear whether mitochondrial alterations are directly linked to β-cell failure. Given the extensive characterization of PGC-1α and -1β as master regulators of mitochondrial function, we hypothesized that decreased PGC-1 expression in adult β-cells would reduce mitochondrial oxidative capacity leading to impaired GSIS. Using an inducible β-cell specific PGC-1 knockout mouse model, we reveal an unexpectedly minor role for PGC-1s in maintaining β-cells mitochondrial mass and function. Also, we identify PGC-1s as essential for the potentiating action of fatty acids on glucose-stimulated insulin release and for maintaining expression of key lipolytic enzymes linked to insulin secretion within mature β-cells.
2. Research design and methods
2.1. Generation of MIP-CreERT:mT/mG and β-cell-specific Pgc-1α/β knockout mice
Hemizygote MIP-CreERT [22] and mT/mG mice [23] were crossed to generate MIP-CreERT:mT/mG mice. Mice carrying floxed Pgc-1α [9] and Pgc-1β alleles [24], interbred to generate Pgc-1α(fl/fl)/β(fl/fl) (C57BL/6N:129), were bred with MIP-CreERT mice (C57BL/6J) to generate homozygous Pgc-1α(fl/fl)/β(fl/fl) (WT fl/fl, littermate controls) and β-cell specific Pgc-1α/β−/− knock-out mice (βPgc-1 KO). For MIP-CreERT controls, Pgc-1α(fl/fl)/β(fl/fl):MIP-CreERT males were bred once with C57Bl/6J and interbred to eliminate floxed alleles. Unless specified, all mice were gavaged at six weeks of age for ten days with 100 mg/kg of tamoxifen (Sigma) in 0.5% methylcellulose/H2O. Mice were maintained on a 12 h dark/light cycle and given free access to water and standard laboratory chow (Teklad diets 2018). All animal procedures were approved by Animal Care Committee of the IRCM.
2.2. Histology of mT/mG:MIP-CreERT mice
All organs were frozen in OCT using isopentane, and frozen sections were visualized by fluorescence microscopy.
2.3. Islet isolation
Mouse pancreas was digested following perfusion with 0.4 u/ml Liberase TL (Roche) in Hank's Balanced Salt Solution (HBSS) buffer (Ca2+/Mg2+) and dispersed by gently shaking in buffer without Ca2+/Mg2+ containing 0.1% BSA and 20 mM HEPES pH 7.4. Pellet was washed in HBSS buffer and resuspended in histopaque 1077 (Sigma) overlaid with RPMI 1640 without glucose prior to separation by centrifugation. Handpicked islets were cultured overnight in 11 mM glucose RPMI 1640 (10% FBS and penicillin/streptomycin). Human islets obtained the day of isolation (Human Islet Transplant Laboratory, McGill University) were cultured overnight in similar media (protocols approved by the Human Ethics Committee (IRCM)).
2.4. Gene expression analysis
RNA extracted by Trizol (Invitrogen) or RNeasy Mini Kit (Qiagen) from INS1 cells and islets (∼120), respectively, was reverse transcribed (Life technologies) and mRNA measured by qPCR using SYBR green (Life Technologies). Relative expression was calculated by ΔΔct-method and normalized to hypoxanthine-guanine phosphoribosyltransferase (Hprt).
2.5. Immunoblotting
Islet proteins (∼150) lysed in RIPA buffer with protease inhibitors (Millipore) were separated by SDS-PAGE and blotted with anti-PGC-1α (1:1000, Calbiochem 4Cl.3), anti-ATGL (1:1000, Cell Signaling #2138) and anti-HSL (1:1000, Cell Signaling #4107).
2.6. Glucose/insulin tolerance tests and serum insulin measurements
Mice were maintained on standard chow or provided a high-fat/high-sucrose diet (HF/HS, Research Diets D12451i) starting at 8 weeks old. Physiological tests were performed at 25–30 weeks of age (4–5 months following initiation of HF/HS diet). Oral glucose tolerance tests were performed (1.5 g/kg d-glucose) in 16 h-fasted mice. Insulin tolerance tests were with human insulin (Lilly, 1 U/kg for chow-fed and 1.5 U/kg for HF/HS-fed mice) injected i.p. in 4 h-fasted mice. Fasted and refed glucose and insulin levels were measured following a 16 h-fast and 2 h of chow re-feeding. Glucose and serum insulin were measured in blood or media by standard glucometer or mouse ultrasensitive insulin and C-peptide ELISAs (Alpco).
2.7. Hyperglycemic clamp
Clamps were performed as described [25] on conscious, 20-week-old mice. Insulin clearance was calculated as C-peptide level divided by insulin level (30–60 min). The insulin sensitivity index (M/I) was calculated as the glucose infusion rate (M) divided by the average insulinemia (I) during the steady state hyperglycemia (30–60 min).
2.8. Palmitate oxidation
Palmitate oxidation was measured as described [26]. Briefly, islets (∼60) were incubated in Krebs–Ringer bicarbonate HEPES buffer (KRBH)/0.25% BSA, 0.1 mM palmitate, 1 mM carnitine, 2 μCi/ml[9,10(n)-3H] palmitate (51 Ci/mmol, Perkin Elmer), and 2.8 or 16.7 mM glucose for 2 h. Medium was acidified with HCl, 3H2O counted and normalized by protein.
2.9. Lipid measurements
Thin layer chromatography (TLC): Islets (∼250) were cultured in RPMI 1640 media containing lipoprotein deficient serum (Sigma) for 16 h with 1 μCi of [1–14C] oleic acid (17 pmol; Perkin Elmer). Lipids solubilized in chloroform were resolved by TLC as described [27]. Total Acyl-Glycerols: Measured in primary islets, as previously described [28]. Briefly, batches of 130–200 freshly isolated primary islets were subject to chloroform:methanol (2:1 vol:vol) extraction, dried material resuspended in isopropanol, and acyl-glycerol content measured by colorimetric assay (Sigma).
2.10. Mitochondrial mass and ATP measurements in dispersed islets
Mass: dispersed islets were incubated in RPMI 1640 containing 100 nM mitotracker green (Cell signaling) for 15 min and quantified by FACS (BD LSRFortessa™). ATP: dispersed islets were cultured for 15 min in Krebs–Ringer bicarbonate HEPES (KRBH) buffer containing 0.04 mM fatty-acid free BSA and 2.8 mM glucose, 20 mM glucose, or 20 mM glucose plus 0.2 mM palmitate pre-complexed to BSA (6:1 molar ratio). ATP was quantified by CellTiter Glo (Promega).
2.11. Static islet glucose-stimulated insulin secretion
Ten islets of similar size were equilibrated for 2 × 20 min in KRBH buffer containing 0.01 mM fatty-acid free BSA and 2.8 mM glucose. Islets were sequentially cultured for 1 h each in KRBH buffer containing 0.04 mM fatty-acid-free BSA and 2.8 mM glucose, 16 mM glucose, 16 mM glucose plus 0.2 mM palmitate pre-complexed to BSA (6:1 molar ratio), and 16 mM glucose plus 20 mM KCl or 100 μM TAK-875 (Selleckchem). Insulin content was determined following lysis in 1.5%/70% HCl/EtOH.
2.12. INS1 culture and adenovirus infection
Parental INS1 cells [29] cultured in RPMI 1640 were infected with adenoviruses expressing shPgc-1α (all variants) or shPgc-1β and cultured for 72 h or adenoviruses overexpressing mouse Pgc-1α1, Pgc-1α4, and/or Pgc-1β and cultured for 48 h.
2.13. Oxygen consumption rate analysis
Islets were cultured in XF media (DMEM containing 1% FBS, 4 mM glucose, 30 mM NaCl, 1 mM sodium pyruvate and 2 mM glutamine, pH 7.4) for 1 h without CO2 prior to measurement of O2 consumption by XFe24 (Seahorse Bioscience) with sequential addition of 11 mM glucose or 11 mM glucose and 0.2 mM palmitate (pre-complexed to BSA, 6:1 molar ratio), 5 μM oligomycin, 1 μM FCCP and 5 μM rotenone.
2.14. Islet area, insulin and glucagon immunohistochemistry, and mitochondrial morphology
Islet area: 10–12 pancreatic sections (5 μm) separated by 200 μm were stained with H&E and area of individual circled islets calculated using Matlab. β- and α-cells were stained using anti-insulin (1:100, Dako) or anti-glucagon (1:50, Sigma). EM: whole islets were fixed and embedded in Epon. Thin sections (60 nm) were viewed by Tecnai 12 transmission electron microscope at 120 kV. Mitochondrial surface area was calculated from 250 single mitochondria (∼30 β-cells).
2.15. Statistical analysis
Data comparing only two groups were analyzed by student t-tests, corrected for multiple comparisons (Sidak–Bonforroni) (*p < 0.05, **p < 0.01, ***p < 0.001). Normality was determined by Shapiro–Wilk test. For data sets with two variables or multiple measurements, two-way ANOVA followed by post-hoc analysis (Holm–Sidak or Fisher's LSD test) determined significance of individual points. Analysis performed using GraphPad Prism. Unless indicated, values are mean ± SEM.
3. Resultss
3.1. PGC-1s are increased in islets following short-term exposure to glucose or palmitate
Pgc-1α expression is altered in islets and β-cells by chronic exposure to glucotoxic or lipotoxic conditions [14]; however, it is unknown whether physiological changes in nutrients also regulate Pgc-1s. We found that acute (1 h) treatment of human (Figure 1A) or mouse islets (Figure S1A) with glucose or palmitate caused significant increases in Pgc-1α and/or Pgc-1β mRNA, while mRNA levels of PGC-1 related co-activator 1 (Prc) [30] remained stable (Figure 1A, S1A). We and others have shown that Pgc-1α exists as multiple variants due to alternative promoter usage and splicing [31]. Subgroups of known variants were similarly increased in response to high glucose and glucose/palmitate in human (Figure S1B) and mouse (data not shown) islets. We also noted that activation of adenylyl cyclase activity by forskolin significantly increased total PGC-1α and all PGC-1α variant mRNAs in primary human and mouse islets (Figure S1C, D), yet only PGC-1α1 and PGC-1α4/NT proteins were detectable by Western blot (Figure 1B, S1E).
Figure 1.

Pgc-1α/β expression in response to acute treatment with high glucose, palmitate, and cAMP treatment in pancreatic islets. (A) qPCR of human islets treated for 1 h with glucose (Glu) and/or palmitate (Palm) (n = 4). (B) PGC-1α protein levels in primary islets from βPgc1 KO mice and wild-type floxed (WT fl/fl) littermates following 4 h of vehicle (DMSO) or forskolin treatment. (C) qPCR of primary islets (n = 10). (D) qPCR of peripheral tissues and brain nuclei (n = 10). Data are means ± SEM, *p < 0.05 and ***p < 0.001 compared to control (white bars). CNS – central nervous system, ns – non-specific band, loading control.
PGC-1β can functionally overlap with PGC-1α in many ways [32], [33], [34] and may compensate for reduced PGC-1α in vivo [12], [24]. Consistent with this, knockdown of either Pgc-1α or Pgc-1β in cultured INS1 β-cells increased mRNA expression of the other (Figure S2). Thus, to fully elucidate the functional role of PGC-1s in adult β-cells, we simultaneously reduced both Pgc-1α and Pgc-1β in mature β-cells of mice to determine their role in mitochondrial metabolism, endocrine function, and whole body glucose homeostasis.
3.2. βPgc1 KO mice have reduced Pgc-1α/β expression specifically in islets
β-cell function and mass in adult mice is negatively impacted by over-expression of Pgc-1α during embryonic development [17]. Thus, to determine effects of reduced PGC-1 activity in adult β-cells while avoiding potentially confounding developmental issues, we used mice with tamoxifen-inducible β-cell-specific Cre-recombinase activity (MIP-CreERT1Lphi) [22]. To confirm that recombination was restricted to the islet and was tamoxifen-dependent, we introduced a double fluorescent reporter (mT/mG) [23] that expresses red fluorescent protein (tdTomato) constitutively until Cre-recombinase-mediated gene excision replaces the signal with green-fluorescent protein (GFP). In islets, no GFP+ cells were detected in mT/mG reporter mice lacking the MIP-CreERT transgene (Figure S3A). A very low level of spontaneous recombination in double-transgenic mT/mG:MIP-CreERT mice was observed in the absence of tamoxifen (Figure S3B); however, following tamoxifen, the majority of islet cells in mT/mG:MIP-CreERT mice were GFP+ (Figure S3C). This is in accordance with recent reports demonstrating an efficiency of more than 90% in β-cells using this MIP-CreERT line [22], [35]. Additionally, no GFP+ cells were detected in liver, spleen, muscle, kidney, testes (Figure S4) or various nuclei within the central nervous system [36]. These results demonstrate the tightly controlled and highly islet-specific activity of Cre-recombinase activity in MIP-CreERT mice.
Five weeks following tamoxifen administration, Pgc-1αfl/fl/βfl/fl:MIP-CreERT (βPgc1 KO) mice had ∼80% reduction in both Pgc-1α and Pgc-1β mRNA (Figure 1C) and almost complete ablation of all PGC-1α proteins in isolated primary islets compared to Pgc-1αfl/fl/βfl/fl (WT fl/fl) controls (Figure 1B). Despite extensive testing, we have yet to find a commercially available antibody that recognizes endogenous PGC-1β protein by Western blotting in tissue including islets, adipose, liver, or muscle (data not shown). We also noted that while total Pgc-1 mRNAs were at appreciable levels in all conditions tested, PGC-1α proteins were difficult to detect in untreated primary islets, yet significantly increased by cAMP (Figure 1B, S1E), similar to what has been shown in mouse liver [9]. This may be due to the low sensitivity of PGC-1α antibodies in general [31], but also demonstrates the highly inducible and regulated nature of this protein. mRNA expression of the coactivators was unchanged in cortical brain, liver, muscle or white adipose tissue (Figure 1D) confirming tissue-specificity of gene ablation. In contrast, we noted modest yet significantly increased Pgc-1α and Pgc1β levels in the ventromedial hypothalamus of βPgc1 KO mice (Figure 1D), highlighting the possibility of pancreas-CNS crosstalk in this model.
During the course of these studies, we identified that MIP-CreERT mice artifactually express human growth hormone (hGH) in islets [36]. While this remains an important caveat for aspects of this study, we report here only PGC-1-dependent effects confirmed in gain/loss of function experiments in cell models and unchanged in MIP-CreERT control islets and mice.
3.3. Loss of β-cell PGC-1 expression decreased glucose-stimulated insulin secretion in vivo
When challenged with oral glucose, mice lacking PGC-1s in β-cells had significantly reduced in vivo insulin secretion after five minutes (first phase), followed by a prolonged plateau of plasma insulin producing an area under the curve similar to controls (Figure 2A–B). Despite lower GSIS, body weights, fasting insulin, and fasting/refed blood glucose levels were not significantly different (Figure 2C–E) and glucose tolerance was identical to controls (Figure 2F). Insulin sensitivity was also similar, but trended upward in βPgc1 KO mice (Figure 2G). In concurrent experiments, we previously demonstrated that chow-fed MIP-CreERT mice on an identical genetic background (Cre-only controls) exhibit no differences in whole body glycemia, glucose-stimulated insulin secretion, glucose tolerance, or insulin sensitivity compared to WT littermates [36].
Figure 2.

βPgc1 KO mice had deficient glucose-stimulated insulin secretion in vivo. (A) Blood insulin following an oral glucose tolerance test (n = 24), and (B) area under the insulin curve for βPgc1 KO mice (black squares) compared to WT fl/fl littermates (white circles). (C) Weights, (D) overnight fasted insulin, and (E) fasted and refed glucose (n = 10–13). (F) Oral glucose and (G) insulin tolerance tests (n = 10–13). (H) Blood glucose, (I) glucose infusion rate (GIR), and (J) insulin levels with (K) area under the insulin curve during a hyperglycemic clamp (n = 8–9). (L) C-peptide levels at 60 min and (M) insulin following arginine bolus given at end of clamp. (N) M/I index and (O) insulin clearance during clamp. Data are means ± SEM. Data are means ± SEM, *p < 0.05 compared to WT fl/fl controls.
During a hyperglycemic clamp to assess β-cell function in vivo, the glucose infusion rate required to maintain glycemia at ∼15 mM was similar in WT fl/fl and βPgc1 KO mice (Figure 2H–I). Similar to results following the oral-glucose challenge, βPgc1 KO mice had significantly decreased insulin secretion throughout the clamp in both first and second phases (Figure 2J–K). Serum C-peptide levels were also decreased (Figure 2L), while insulin release in response to arginine (a non-nutrient-dependent secretagogue) was unaltered (Figure 2M). M/I index (an indirect measure of whole body insulin sensitivity) and insulin clearance were similar (Figure 2N–O). M/I index may be more appropriately calculated during a hyperinsulinemic-euglycemic clamp; however, these data in combination with the normal insulin tolerance and clearance (Figure 2G, 2O) suggest that normoglycemia in βPgc1 KO mice was not the result of large adaptive changes in peripheral insulin sensitivity. Taken together, these data demonstrate that β-cell PGC-1s are essential for efficient glucose-stimulated insulin secretion in vivo.
3.4. βPgc1 KO islets have normal mitochondrial mass and function in response to glucose
No differences were detected in islet morphology, insulin or glucagon staining, or islet area in WT fl/fl versus βPgc1 KO mice (Figure 3A–C). A trend toward increased total islet mass in βPgc1 KOs was noted (Figure 3C, inset); however, we have observed a similar increase in mice carrying only the MIP-CreERT transgene [36]. mRNA expression of multiple factors essential for adult β–cell differentiation and function, including MafA and Pdx1, were unaltered (Figure 3D). As PGC-1s are important for mitochondrial dynamics, biogenesis, and oxidative phosphorylation in many tissues [4], we investigated whether these parameters were altered by concurrent loss of both PGC-1s in adult β-cells. Mitofusin (Mfn) and optic atrophy 1 (Opa1) mRNAs were similarly expressed, and, although there was a significant decrease (28%) in Dynamin-related protein 1 (Drp1) (Figure 4A), there were no differences in total mitochondrial mass in dispersed primary islets (Figure 4B) or cross-sectional area of individual mitochondria measured by electron microscopy (Figure 4C–D). Islets of MIP-CreERT control mice also have normal architecture [36] and no alterations in mitochondrial gene expression, mass or morphology (Figure S5A–C).
Figure 3.

Islet architecture, cell composition, β-cell area and gene expression were unaltered in βPgc1 KO islets. Representative images of (A) insulin and (B) glucagon immunostaining from βPgc1 KO mice and WT fl/fl littermates (n = 6 mice). (C) Islet area and islet mass (inset) (n = 6). (D) qPCR analysis of primary islets (n = 10).
Figure 4.

Minor effects of reduced β-cell PGC-1α/β on mitochondrial gene expression, mass, and function. (A) qPCR analysis of primary islets from βPgc1 KO mice and WT fl/fl littermate controls (n = 10). (B) Mitochondrial mass in dispersed primary islets (n = 4). (C) Electron microscopy images of β-cells in primary islets – mitochondria (black triangles), insulin granules (white triangles), and nucleus (N) and (D) average mitochondrial area (n = 250 mitochondria). (E–F) qPCR of primary islets (n = 10). (G) Mitochondrial oxygen consumption rate (OCR) in whole, live islets (n = 5). (H) Glucose-stimulated increases in ATP from dispersed islets (n = 5 experiments in triplicate, pooled). Data are means ± SEM, *p < 0.05 compared to islets of WT fl/fl controls.
Surprisingly, only a small subset of canonical PGC-1-responsive mitochondrial genes were significantly decreased in βPgc1 KO islets, including succinate dehydrogenase (Sdh – complex II), multiple components of the ATP synthase (Atp5b, Atp5e and Atp5j2 – Complex V), and the nuclear transcription factor Errα (Figure 4E–F). These genes were unaltered in MIP-CreERT control islets (Figure S5C). Consistent with normal mass and only modestly decreased ETC-component gene expression, mitochondrial oxygen consumption rate (OCR) in response to increased glucose (11 mM), oligomycin (ATP-coupled), FCCP (maximal respiratory capacity), and rotenone (non-mitochondrial respiration) were similar between WT fl/fl and βPgc1 KO islets (Figure 4G). In addition, ATP production in dispersed βPgc1 KO islets stimulated with glucose were similar to WT fl/fl controls (Figure 4H). These results illustrate that PGC-1s are largely dispensable for mitochondrial content and glucose-stimulated oxidative capacity in adult β-cells.
3.5. Primary islets with reduced PGC-1s exhibit decreased fatty-acid potentiated insulin secretion and mitochondrial respiration
We next assessed whether decreased GSIS in βPgc1 KO mice was cell-autonomous. Interestingly, we found that primary islets of WT fl/fl and βPgc1 KO mice had similar insulin secretion in vitro in response to 16 mM glucose alone, while the potentiation of insulin secretion that is normally observed during co-stimulation with glucose and palmitate was essentially absent in βPgc1 KO islets (Figure 5A). This difference was not the result of decreased exocytotic capacity or islet insulin content (Figure 5B–C). Importantly, primary islets of MIP-CreERT mice showed no differences in insulin secretion in response to glucose, palmitate, or KCl compared to WT fl/fl littermates (Figure S6A–C). Similarly, mitochondrial OCR potentiated by concurrent high glucose and fatty acid (Figure S7) was significantly blunted in βPgc1 KO islets (Figure 5D), and stimulated ATP levels trended lower (Figure 5E) in this condition. Taken together with in vivo data, we concluded that PGC-1s are required for efficient insulin secretion, possibly due to novel roles in β-cell lipid sensing or metabolism.
Figure 5.

Fatty-acid potentiated insulin secretion is dependent on β-cells PGC-1s expression. (A) Insulin secretion from primary islets (n = 16–20). (B) KCl response (n = 8) and (C) insulin content of primary islets (n = 10). Data are means ± SEM. Statistical comparisons are: *p < 0.05, **p < 0.01, ****p < 0.0001 comparing treatment effect to basal (2.8 mM glucose) of similar genotype; ###p < 0.001 comparing effect of palmitate on glucose response (versus 16 mM glucose) of similar genotype; &&p < 0.01 represents genotype effect (WT fl/fl versus βPgc-1 KO). (D) OCR in primary islets (n = 5). (E) Glucose/palmitate-stimulated ATP increases in dispersed islet cells (n = 5, pooled). Data are means ± SEM, *p < 0.05 compared to islets of WT fl/fl controls.
3.6. Loss of PGC-1 expression in adult β-cells alters lipid metabolism
As palmitate potentiation of GSIS was abolished by loss of PGC-1s, we evaluated pathways of lipid sensing and fatty acid metabolism known to be important for insulin secretion [26], [37], [38], [39]. Fatty acid transporter Cd36 mRNA expression was unaltered and although expression of the cell-surface fatty acid sensor G-protein coupled receptor 40 (Gpr40/FFAR1) was decreased in primary βPgc1 KO islets (Figure S8A), potentiation of GSIS in response to TAK-875, a potent and selective GPR40 agonist, was equal in WT fl/fl and βPgc1 KO primary islets (Figure S8B). This pointed toward inefficient intracellular lipid metabolism as a potential explanation for the reduced fatty acid-induced insulin response.
Upon incubation of primary islets overnight with trace 14C-oleate, we found that βPgc1 KO islets accumulated substantially more radiolabeled lipid (Figure 6A). Although Pgc-1 expression is required for efficient fatty acid β-oxidation in other tissues including liver and heart [9], [40], mRNA levels of many key enzymes involved in mitochondrial and peroxisomal fatty acid oxidation were not altered in βPgc1 KO (or MIP-CreERT) islets (Figure 6B, S5C). Consistent with gene expression, oxidation of palmitate was equivalent between genotypes (Figure 6C), and high glucose efficiently suppressed fatty acid oxidation in islets of both WT fl/fl and βPgc1 KO (Figure 6C), implying glucose utilization and generation of malonyl-CoA were also normal.
Figure 6.

βPgc1 KO islets accumulated intracellular lipids but exhibited normal oxidation of fatty acids. (A) Total lipid levels in primary islets after overnight culture in trace 14C-Oleate (n = 5–8). (B–C) qPCR of primary islets from βPgc1 KO mice compared to WT fl/fl littermate controls (n = 10). (D) Palmitate oxidation in primary islets cultured in low and high glucose (n = 10–14). Data are means ± SEM, *p < 0.05.
As pathways of fatty acid oxidation appeared intact, we next investigated whether enzymes responsible for the production and breakdown of stored acyl-glycerols were dependent on PGC-1s. Loss of β-cell PGC-1s led to a substantial decrease in adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL) protein, rate-limiting enzymes involved in acyl-glycerol catabolism [41] (Figure 7A). Decreases in protein corresponded to a ∼27% decrease in Hsl (Lipe) mRNA in βPgc1 KO islets, but no apparent expression changes in the gene encoding ATGL, patatin-like phospholipase domain containing 2 (Pnpla2/Atgl) and a ∼46% increase in monoglyceride lipase (Mgll) mRNA (Figure 7B). Expression of lysosomal acid lipase A (Lipa), an enzyme important for the breakdown of cholesterol esters and triglyceride, also trended downward. In accordance with dysregulated lipolytic enzyme expression, total acyl-glycerol levels were increased 59% in βPgc-1 KO primary islets compared to WT fl/fl controls (Figure 7C). Differences in lipolytic gene expression and increased lipid content were not observed in MIP-CreERT control islets (Figure S9). Separation of radiolabeled lipid species by TLC revealed that βPgc-1 KO islets and INS1 β-cells transduced with shRNA had significantly increased cholesterol esters (CE) and mono-acylglycerol (MAG) (Figure 7D), and diacylglycerol (DAG) and triglyceride (TAG) (Figure S10), respectively. Overexpression of Pgc-1α1, but not Pgc-1α4, in INS1 β-cells significantly increased gene expression of Atgl, Hsl, and Lipa, but not Mgll (Figure 7E, S11). Increased Pgc-1β induced only Atgl, but to a similar magnitude as Pgc-1α1. Taken together, these data suggest that in β-cells, expression levels of key, rate-limiting lipolytic enzymes are dependent on the PGC-1s transcriptional coactivators.
Figure 7.

PGC-1s regulate expression of lipolytic enzymes important for acyl-glycerol metabolism in response in β-cells. (A) Western blot of lipolytic enzymes in primary islets (representative blot of n = 4). (B) qPCR of primary islets from Pgc1 KO mice and WT fl/fl littermate controls (n = 10). (C) Acyl-glycerol content of primary islets (n = 4). (D) Radio-labeled acyl-glycerol species incorporated by primary islets, separated by thin layer chromatography (MAG-Monoacylglycerol, DAG-Diacylglycerol, TG-Triglycerides, CE-Cholesterol ester) (n = 5–8). Data are means ± SEM, *p < 0.05, **p < 0.01 compared to WT fl/fl controls (E) qPCR analysis of genes following overexpression of Pgc-1α1, Pgc-1α4, or Pgc-1β in INS1 cells (n = 3). Data are means + SD, *p < 0.05 compared to cells infected with viral vector (Ad-control).
3.7. Loss of PGC-1s expression in adult β-cells does not promote glucose intolerance in mice challenged with a high fat/high sucrose diet
Hyperglycemia associated with obesity is often exacerbated by inefficient insulin secretion. We challenged βPgc1 KO mice and controls with a high-fat/high sucrose diet (HF/HS) for 16 weeks to determine whether defects in insulin secretion and lipid metabolism in βPgc1 KO islets would worsen glucose tolerance in a model of insulin resistance. βPgc1 KO mice fed a HF/HS diet for 20 weeks had similar body weights (Figure 8A). Consistent with a defect in β-cell glycerolipid catabolism, primary islets of βPgc1 KO mice on a HF/HS diet had higher acyl-glycerol levels (Figure 8B). As in chow fed-mice, βPgc1 KO mice exhibited significantly decreased in vivo GSIS during an OGTT (Figure 8C), with the area under the curve now significantly lower than controls (Figure 8D). Fasting insulin and glucose were unaltered (Figure 8E–F). Despite the decrease in GSIS and increased β-cell acyl-glycerol levels, we did not observe measurable differences in whole body glucose or insulin tolerance (Figure 8G–H).
Figure 8.

βPgc1 KO mice on a HF/HS have decreased in vivo insulin secretion but no difference in glucose or insulin tolerance. Mice were subject to a HF/HS diet for 20 weeks. (A) Total body weights of mice following HF/HS diet (n = 10–11). (B) Total acyl-glycerol species in primary islets of HF/HS–fed mice. (C) Serum insulin and (D) area under the curve of OGTT of HF/HS-fed mice. Overnight fasted (E) insulin and (F) glucose. Blood glucose levels during (G) OGsTT and (H) ITT (n = 8–9). Data are means ± SEM, *p < 0.05, **p < 0.01 compared to WT fl/fl controls.
4. Discussion
Using in vivo β-cell specific knockout mice and in vitro cell models, we demonstrate that β-cell lipid metabolism is disrupted by loss of PGC-1s and identify a novel role for PGC-1 coactivators in the potentiation of insulin secretion by fatty acids. PGC-1 loss of function did not affect mitochondrial function or insulin secretion in response to glucose in vitro, yet caused a significant accumulation of acyl-glycerols and cholesterol esters and abrogated palmitate-potentiation of GSIS. We also demonstrate that β-cell PGC-1 expression is acutely regulated by extracellular nutrients and that these coactivators are essential to maintain expression of the rate-limiting lipolytic enzymes ATGL and HSL. Interestingly, the increased lipid accumulation and decreased insulin secretion resulting from PGC-1 loss are not sufficient to impair whole body glucose tolerance in lean or obese mice.
In general, Pgc-1α gene expression is highly inducible [1], while fewer stimuli have been found to significantly impact Pgc-1β expression [42]. We demonstrate here that both Pgc-1α and Pgc-1β expression in islets is acutely responsive to high glucose and palmitate, suggesting an important role for these coactivators in linking extracellular nutrient signals to transcriptional control of metabolism and β-cell function. We were surprised to observe that unlike in heart and skeletal muscle [24], [43], loss of both PGC-1α and PGC-1β expression in adult mouse β-cells did not significantly disrupt basal mitochondrial content or function. In vitro, glucose-stimulated oxygen consumption and GSIS were normal, implying that mitochondrial activity in the absence of PGC-1s is sufficient under these conditions. We focused on PGC-1α/β as dysregulation of these genes is observed in obesity and diabetes [13], [44], [45], [46], [47], [48]; however, β-cells may rely on the related PRC co-activator for maintenance of basal oxidative capacity [30]. Prc was expressed at similar levels in islets, yet, unlike Pgc-1α or -1β, was not regulated by glucose or palmitate. Future work to determine over-lapping versus tissue-specific roles for distinct PGC-1 family members may shed light on this question. To our knowledge, this is the first study to induce loss of PGC-1s in adult mice, which could explain the lack of a significant mitochondrial phenotype. PGC-1α/β loss during murine cell development and differentiation [10], [11], [43] could affect early β-cell mitochondrial formation and subsequent metabolism. Thus, it remains to be established whether PGC-1s in β-cells (and/or other tissues) ensure maturation of the mitochondria during development, but are largely dispensable for mitochondrial function and maintenance in differentiated cells.
Our data suggest that decreased nutrient-stimulated insulin secretion associated with low PGC-1 stems from altered intracellular lipid metabolism. In PGC-1 deficient islets, significantly blunted insulin secretion and mitochondrial OCR were only observed following co-treatment with high glucose/palmitate. While this may seem paradoxical in light of intact lipid oxidation, the acute, palmitate-potentiated O2 consumption and GSIS were measured in the presence of high glucose, an environment in which oxidation is effectively repressed by high malonyl-CoA (Figure 6C). The decrease in OCR could be explained by the specific sensitivity of ETC complex II (Sdh) to PGC-1 loss. Thus, CO2 produced during oxidation could be normal, while the succinate and FADH2 generated is inefficiently processed through the ETC, leading to lower ATP production under high glucose/palmitate conditions. This may also contribute to the greater decrease in GSIS observed in HF/HS-fed mice (Figure 8C, D).
In addition to reduced energy production by fatty acids, key lipolytic enzymes linked to insulin secretion were significantly decreased in βPgc1 KO islets. Metabolism of acyl-glycerols is coupled to insulin secretion in β-cells and increased intracellular triglycerides inhibit GSIS [38], [49], [50]. Hsl and Atgl expression was increased by ectopic PGC-1α1 and PGC-1β in β-cells, and protein expression was significantly reduced when PGC-1s were absent, implicating these rate-limiting lipolytic enzymes as important downstream effectors of PGC-1 transcriptional activity in β-cells. ATGL and HSL are essential for efficient insulin secretion in mouse islets [41], [50], [51], [52], [53] and their loss disrupts acyl-glycerol and cholesterol hydrolysis [41], [50], [52], [54], corresponding well with both lipid accumulation and GSIS data in islets lacking PGC-1s (even in the absence of additional exogenous lipid). Interestingly, ATGL was recently shown to promote PGC-1α signaling by increasing SIRT1 deacetylase activity [55], implying the existence of a feedback mechanism to enhance lipid catabolism in times of energy demand. We also found Mgll, an enzyme responsible for the hydrolysis of MAG, was not augmented by increased PGC-1 in INS1 cells, yet its expression was increased in β-cells lacking the coactivators. This seemingly compensatory response in βPgc-1 KO islets may reflect upregulation of other lipolytic enzymes to metabolize accumulating acyl-glycerol pools in vivo following long-term coactivator loss. This is supported by our data in INS1 cells demonstrating that only TAG and DAG were increased following short-term PGC-1s knock-down (Figure S9). Recently, it was shown that certain MAG species stimulate GSIS by promoting insulin granule exocytosis via Munc13-1 [56]. Thus, it would be interesting to determine if loss of PGC-1s alters the identity and/or relative proportion of different acyl-glycerol species in β-cells. Furthermore, since we observed normal GSIS in βPgc-1 KO islets following stimulation with glucose alone in vitro, it is possible that production of MAG is maintained by upregulation of MGLL or other lipases (i.e. ABHD6), yet fatty acid-potentiation of GSIS is dependent on mechanisms not compensated for in our PGC-1s loss-of-function model.
An important aspect of this work was to address whether reduced PGC-1 expression in patients might play a role in the development or pathophysiology of diabetes. Despite disruption of insulin secretion and islet lipid accumulation in chow- and high fat/high sucrose-fed mice, βPgc-1 KO mice had normal glucose tolerance. This may not be unexpected in light of models demonstrating that significantly larger reductions in GSIS (50–75% reduction in insulin gene expression) have no significant effect on glucose tolerance, even in obese mice [57]. However, long-term accumulation of potentially toxic intermediate lipid species (i.e. DAG and CE) in cells with low PGC-1 could have detrimental effects on β-cell survival and/or function [58], [59] that may be revealed under other metabolic challenges (i.e. ageing). This implies that while reduced β-cell PGC-1α observed in islets of diabetic patients [13] may be sufficient to disrupt β-cell function, this likely acts in concert with other metabolic disruptions to drive hyperglycemia associated with type 2 diabetes. For example, decreased GSIS and lipid metabolism resulting from low β-cell PGC-1 could exacerbate effects of reduced PGC-1α expression in other tissues, which include insulin resistance [9], [10], [11], [12] and decreased cardiac function [24]. This is important to note, as alterations of PGC-1 (PPARGC1A) at the genomic level via SNPs [6], [7] and/or epigenetic modifications [13], [60] are associated with metabolic disease.
In conclusion, we demonstrate that the PGC-1s are essential, inducible regulators of fatty acid-stimulated insulin secretion in mature β-cells and that low levels of the coactivators disrupt β-cell lipid catabolism. Thus, restoration of PGC-1 activity may prove beneficial to improve the health and integrity of β-cells in metabolic diseases including diabetes.
Acknowledgments
Authors would like to thank: Dr. Bruce Spiegelman for the floxed Pgc-1α mice and Pgc-1α1, Pgc-1α4, Pgc-1β, and shPgc-1β adenoviral constructs; Dr. Daniel Kelly for the floxed Pgc-1β mice; and Dr. Marc Montminy for the shPgc-1α virus. Special thanks to Rana Gupta, Jessica Wiwczar, Marie-Claude Lavallée, Naveen Khan, Aurèle Besse-Patin, Alexandra Abboud and Adam Albarghouthi for technical assistance. We thank the Rodent Metabolic Phenotyping core of CRCHUM for assistance with hyperglycemic clamps. KB was supported by a fellowship from Diabète Québec and NJ was supported by a post-doctoral fellowship from the Montreal Diabetes Research Centre. JLE is the guarantor of this work and takes responsibility for contents of the article. Supported by operating funds to JLE from CDA (OG3123843JE), J.A. DeSève Foundation, CIHR (INMD, 137495), FRQS, CFI (30343); LHP from NIH/NIDDK P30 DK02059, DK092616; TA from CIHR (MOP115042); and VP from CIHR (MOP86454). CIHR and FRSQ provided salary awards to JLE and TA. VP holds a Canada Research Chair in Diabetes and Pancreatic Beta Cell Function.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molmet.2015.08.001.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- 1.Finck B.N., Kelly D.P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. Journal of Clinical Investigation. 2006;116(3):615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 3.Patti M.E., Butte A.J., Crunkhorn S., Cusi K., Berria R., Kashyap S. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villena J.A. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. The FEBS Journal. 2015;282(4):647–672. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 5.Westerbacka J., Kolak M., Kiviluoto T., Arkkila P., Siren J., Hamsten A. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56(11):2759–2765. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- 6.Barroso I., Luan J., Sandhu M.S., Franks P.W., Crowley V., Schafer A.J. Meta-analysis of the Gly482Ser variant in PPARGC1A in type 2 diabetes and related phenotypes. Diabetologia. 2006;49(3):501–505. doi: 10.1007/s00125-005-0130-2. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y., Mo X., Chen S., Lu X., Gu D. Association of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PPARGC1A) gene polymorphisms and type 2 diabetes mellitus: a meta-analysis. Diabetes/Metabolism Research and Reviews. 2011;27(2):177–184. doi: 10.1002/dmrr.1158. [DOI] [PubMed] [Google Scholar]
- 8.Liu C., Lin J.D. PGC-1 coactivators in the control of energy metabolism. Acta Biochimica et Biophysica Sinica (Shanghai) 2011;43(4):248–257. doi: 10.1093/abbs/gmr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estall J.L., Kahn M., Cooper M.P., Fisher F.M., Wu M.K., Laznik D. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes. 2009;58(7):1499–1508. doi: 10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handschin C., Choi C.S., Chin S., Kim S., Kawamori D., Kurpad A.J. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. Journal of Clinical Investigation. 2007;117(11):3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleiner S., Mepani R.J., Laznik D., Ye L., Jurczak M.J., Jornayvaz F.R. Development of insulin resistance in mice lacking PGC-1alpha in adipose tissues. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9635–9640. doi: 10.1073/pnas.1207287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sczelecki S., Besse-Patin A., Abboud A., Kleiner S., Laznik-Bogoslavski D., Wrann C.D. Loss of Pgc-1alpha expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. American Journal of Physiology. Endocrinology and Metabolism. 2014;306(2):E157–E167. doi: 10.1152/ajpendo.00578.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ling C., Del Guerra S., Lupi R., Ronn T., Granhall C., Luthman H. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 2008;51(4):615–622. doi: 10.1007/s00125-007-0916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P., Liu C., Zhang C., Zhang Y., Shen P., Zhang J. Free fatty acids increase PGC-1alpha expression in isolated rat islets. FEBS Letters. 2005;579(6):1446–1452. doi: 10.1016/j.febslet.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 15.Kim J.W., You Y.H., Ham D.S., Cho J.H., Ko S.H., Song K.H. Suppression of peroxisome proliferator-activated receptor gamma-coactivator-1alpha normalizes the glucolipotoxicity-induced decreased BETA2/NeuroD gene transcription and improved glucose tolerance in diabetic rats. Endocrinology. 2009;150(9):4074–4083. doi: 10.1210/en.2009-0241. [DOI] [PubMed] [Google Scholar]
- 16.Oberkofler H., Hafner M., Felder T., Krempler F., Patsch W. Transcriptional co-activator peroxisome proliferator-activated receptor (PPAR)gamma co-activator-1beta is involved in the regulation of glucose-stimulated insulin secretion in INS-1E cells. Journal of Molecular Medicine (Berl) 2009;87(3):299–306. doi: 10.1007/s00109-008-0425-0. [DOI] [PubMed] [Google Scholar]
- 17.Valtat B., Riveline J.P., Zhang P., Singh-Estivalet A., Armanet M., Venteclef N. Fetal PGC-1alpha overexpression programs adult pancreatic beta-cell dysfunction. Diabetes. 2013;62(4):1206–1216. doi: 10.2337/db12-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon J.C., Xu G., Deeney J.T., Yang S.N., Rhee J., Puigserver P. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Developmental Cell. 2003;5(1):73–83. doi: 10.1016/s1534-5807(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 19.Mulder H., Ling C. Mitochondrial dysfunction in pancreatic beta-cells in type 2 diabetes. Molecular and Cellular Endocrinology. 2009;297(1–2):34–40. doi: 10.1016/j.mce.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Supale S., Li N., Brun T., Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends in Endocrinology and Metabolism. 2012;23(9):477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Anello M., Lupi R., Spampinato D., Piro S., Masini M., Boggi U. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48(2):282–289. doi: 10.1007/s00125-004-1627-9. [DOI] [PubMed] [Google Scholar]
- 22.Tamarina N.A., Roe M.W., Philipson L. Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic beta-cells. Islets. 2014;6(1) doi: 10.4161/isl.27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 24.Lai L., Leone T.C., Zechner C., Schaeffer P.J., Kelly S.M., Flanagan D.P. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes & Development. 2008;22(14):1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fergusson G., Ethier M., Guevremont M., Chretien C., Attane C., Joly E. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Molecular Metabolism. 2014;3(9):848–854. doi: 10.1016/j.molmet.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alquier T., Peyot M.L., Latour M.G., Kebede M., Sorensen C.M., Gesta S. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes. 2009;58(11):2607–2615. doi: 10.2337/db09-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sniderman A.D., Qi Y., Ma C.I., Wang R.H., Naples M., Baker C. Hepatic cholesterol homeostasis: is the low-density lipoprotein pathway a regulatory or a shunt pathway? Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(11):2481–2490. doi: 10.1161/ATVBAHA.113.301517. [DOI] [PubMed] [Google Scholar]
- 28.Roduit R., Masiello P., Wang S.P., Li H., Mitchell G.A., Prentki M. A role for hormone-sensitive lipase in glucose-stimulated insulin secretion: a study in hormone-sensitive lipase-deficient mice. Diabetes. 2001;50(9):1970–1975. doi: 10.2337/diabetes.50.9.1970. [DOI] [PubMed] [Google Scholar]
- 29.Asfari M., Janjic D., Meda P., Li G., Halban P.A., Wollheim C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130(1):167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- 30.Scarpulla R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Annals of the New York Academy of Sciences. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Redondo V., Pettersson A.T., Ruas J.L. The hitchhiker's guide to PGC-1alpha isoform structure and biological functions. Diabetologia. 2015 doi: 10.1007/s00125-015-3671-z. [DOI] [PubMed] [Google Scholar]
- 32.Lin J., Puigserver P., Donovan J., Tarr P., Spiegelman B.M. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. The Journal of Biological Chemistry. 2002;277(3):1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 33.Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 34.St-Pierre J., Lin J., Krauss S., Tarr P.T., Yang R., Newgard C.B. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. The Journal of Biological Chemistry. 2003;278(29):26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 35.Wicksteed B., Brissova M., Yan W., Opland D.M., Plank J.L., Reinert R.B. Conditional gene targeting in mouse pancreatic ss-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59(12):3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oropeza D., Jouvet N., Budry L., Campbell J.E., Bouyakdan K., Lacombe J. Phenotypic characterization of MIP-CreERT1Lphi mice with transgene-driven islet expression of human growth hormone. Diabetes. 2015 doi: 10.2337/db15-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Azzouny M., Evans C.R., Treutelaar M.K., Kennedy R.T., Burant C.F. Increased glucose metabolism and glycerolipid formation by fatty acids and GPR40 receptor signaling underlies the fatty acid potentiation of insulin secretion. The Journal of Biological Chemistry. 2014;289(19):13575–13588. doi: 10.1074/jbc.M113.531970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prentki M., Matschinsky F.M., Madiraju S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metabolism. 2013;18(2):162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 39.Latour M.G., Alquier T., Oseid E., Tremblay C., Jetton T.L., Luo J. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56(4):1087–1094. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chambers K.T., Chen Z., Crawford P.A., Fu X., Burgess S.C., Lai L. Liver-specific PGC-1beta deficiency leads to impaired mitochondrial function and lipogenic response to fasting-refeeding. PloS One. 2012;7(12):e52645. doi: 10.1371/journal.pone.0052645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peyot M.L., Nolan C.J., Soni K., Joly E., Lussier R., Corkey B.E. Hormone-sensitive lipase has a role in lipid signaling for insulin secretion but is nonessential for the incretin action of glucagon-like peptide 1. Diabetes. 2004;53(7):1733–1742. doi: 10.2337/diabetes.53.7.1733. [DOI] [PubMed] [Google Scholar]
- 42.Lin J., Yang R., Tarr P.T., Wu P.H., Handschin C., Li S. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120(2):261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 43.Rowe G.C., Patten I.S., Zsengeller Z.K., El-Khoury R., Okutsu M., Bampoh S. Disconnecting mitochondrial content from respiratory chain capacity in PGC-1-deficient skeletal muscle. Cell Reports. 2013;3(5):1449–1456. doi: 10.1016/j.celrep.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen G., Wegner L., Yanagisawa K., Rose C.S., Lin J., Glumer C. Evidence of an association between genetic variation of the coactivator PGC-1beta and obesity. Journal of Medical Genetics. 2005;42(5):402–407. doi: 10.1136/jmg.2004.026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrulionyte L., Peltola P., Chiasson J.L., Laakso M. Single nucleotide polymorphisms of PPARD in combination with the Gly482Ser substitution of PGC-1A and the Pro12Ala substitution of PPARG2 predict the conversion from impaired glucose tolerance to type 2 diabetes: the STOP-NIDDM trial. Diabetes. 2006;55(7):2148–2152. doi: 10.2337/db05-1629. [DOI] [PubMed] [Google Scholar]
- 46.Bhat A., Koul A., Rai E., Sharma S., Dhar M.K., Bamezai R.N. PGC-1alpha Thr394Thr and Gly482Ser variants are significantly associated with T2DM in two North Indian populations: a replicate case-control study. Human Genetics. 2007;121(5):609–614. doi: 10.1007/s00439-007-0352-0. [DOI] [PubMed] [Google Scholar]
- 47.Ek J., Andersen G., Urhammer S.A., Gaede P.H., Drivsholm T., Borch-Johnsen K. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino acid polymorphisms to type II diabetes mellitus. Diabetologia. 2001;44(12):2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- 48.Kunej T., Globocnik Petrovic M., Dovc P., Peterlin B., Petrovic D. A Gly482Ser polymorphism of the peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) gene is associated with type 2 diabetes in Caucasians. Folia Biologica (Praha) 2004;50(5):157–158. [PubMed] [Google Scholar]
- 49.Kelpe C.L., Johnson L.M., Poitout V. Increasing triglyceride synthesis inhibits glucose-induced insulin secretion in isolated rat islets of langerhans: a study using adenoviral expression of diacylglycerol acyltransferase. Endocrinology. 2002;143(9):3326–3332. doi: 10.1210/en.2002-220402. [DOI] [PubMed] [Google Scholar]
- 50.Tang T., Abbott M.J., Ahmadian M., Lopes A.B., Wang Y., Sul H.S. Desnutrin/ATGL activates PPARdelta to promote mitochondrial function for insulin secretion in islet beta cells. Cell Metabolism. 2013;18(6):883–895. doi: 10.1016/j.cmet.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mulder H., Yang S., Winzell M.S., Holm C., Ahren B. Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes. 2004;53(1):122–128. doi: 10.2337/diabetes.53.1.122. [DOI] [PubMed] [Google Scholar]
- 52.Peyot M.L., Guay C., Latour M.G., Lamontagne J., Lussier R., Pineda M. Adipose triglyceride lipase is implicated in fuel- and non-fuel-stimulated insulin secretion. The Journal of Biological Chemistry. 2009;284(25):16848–16859. doi: 10.1074/jbc.M109.006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fex M., Haemmerle G., Wierup N., Dekker-Nitert M., Rehn M., Ristow M. A beta cell-specific knockout of hormone-sensitive lipase in mice results in hyperglycaemia and disruption of exocytosis. Diabetologia. 2009;52(2):271–280. doi: 10.1007/s00125-008-1191-9. [DOI] [PubMed] [Google Scholar]
- 54.Fex M., Olofsson C.S., Fransson U., Bacos K., Lindvall H., Sorhede-Winzell M. Hormone-sensitive lipase deficiency in mouse islets abolishes neutral cholesterol ester hydrolase activity but leaves lipolysis, acylglycerides, fat oxidation, and insulin secretion intact. Endocrinology. 2004;145(8):3746–3753. doi: 10.1210/en.2003-1673. [DOI] [PubMed] [Google Scholar]
- 55.Khan S.A., Sathyanarayan A., Mashek M.T., Ong K.T., Wollaston-Hayden E.E., Mashek D.G. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1alpha/PPAR-alpha signaling. Diabetes. 2015;64(2):418–426. doi: 10.2337/db14-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao S., Mugabo Y., Iglesias J., Xie L., Delghingaro-Augusto V., Lussier R. Alpha/beta-hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metabolism. 2014;19(6):993–1007. doi: 10.1016/j.cmet.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 57.Mehran A.E., Templeman N.M., Brigidi G.S., Lim G.E., Chu K.Y., Hu X. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metabolism. 2012;16(6):723–737. doi: 10.1016/j.cmet.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 58.Ishikawa M., Iwasaki Y., Yatoh S., Kato T., Kumadaki S., Inoue N. Cholesterol accumulation and diabetes in pancreatic beta-cell-specific SREBP-2 transgenic mice: a new model for lipotoxicity. Journal of Lipid Research. 2008;49(12):2524–2534. doi: 10.1194/jlr.M800238-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Kruit J.K., Wijesekara N., Westwell-Roper C., Vanmierlo T., de Haan W., Bhattacharjee A. Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes. 2012;61(3):659–664. doi: 10.2337/db11-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barres R., Osler M.E., Yan J., Rune A., Fritz T., Caidahl K. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metabolism. 2009;10(3):189–198. doi: 10.1016/j.cmet.2009.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.