

TRAM interaction with TRAF6 regulates the inflammatory response to TLR4 activation, and adds further intricacy to TLR signaling.
Keywords: Toll-like receptors, innate immunity, inflammation, signal transduction
Abstract
TLRs act as sentinels in professional immune cells to detect and initiate the innate immune response to pathogen challenge. TLR4 is a widely expressed TLR, responsible for initiating potent immune responses to LPS. TRAM acts to bridge TLR4 with TRIF, orchestrating the inflammatory response to pathogen challenge. We have identified a putative TRAF6-binding motif in TRAM that could mediate a novel signaling function for TRAM in TLR4 signaling. TRAM and TRAF6 association was confirmed by immunoprecipitation of endogenous, ectopically expressed and recombinant proteins, which was ablated upon mutation of a key Glu residue in TRAM (TRAM E183A). TRAF6 and TRAM were observed colocalizing using confocal microscopy following ectopic expression in cells and the ability of TRAM and TRAM E183A to activate luciferase-linked reporter assays was determined in HEK293 and TRAF6-deficient cells. Importantly, TRAM-deficient macrophages reconstituted with TRAM E183A display significantly reduced inflammatory TNF-α, IL-6, and RANTES protein production compared with WT TRAM. These results demonstrate a novel role for TRAM in TLR4-mediated signaling in regulating inflammatory responses via its interaction with TRAF6, distinct from its role as a bridging adaptor between TLR4 and TRIF.
Introduction
Recognition of microbial pathogens is an essential element for the initiation of innate immune responses such as inflammation. Detection is mediated by germ line-encoded PRRs that are widely expressed on professional immune cells, such as macrophages and dendritic cells. PRRs recognize molecular patterns broadly shared by pathogens, known as PAMPs [1]. The TLRs are a critical family of innate immune PRRs responsible for sensing components of pathogens and the initiation of the proinflammatory response [2, 3]. Recognition of PAMPs by TLRs leads to activation of inflammatory transcription factors, such as NF-κB and IRF3, which promote robust transcription of inflammatory cytokines and IFNs [2, 3].
The cytosolic segment of TLRs is distinguished by a conserved protein motif, termed the TIR domain, which mediates recruitment of cytosolic TIR-containing adaptor proteins following ligand-induced receptor dimerization [4, 5]. There are five TIR-containing adaptors: MyD88, Mal/TIR domain-containing adaptor protein, TRIF/TICAM-1, TRAM/TICAM-2, and SARM [6].
All TLRs, with the exception of TLR3, recruit MyD88 to the receptor complex, initiating recruitment of IRAK-1 and IRAK-4, followed by TRAF6, to the complex [2, 7]. This signaling complex activates the so-called canonical pathway, resulting in the nuclear translocation of NF-κB and the initiation of the proinflammatory response. Conversely, TLR3 recruits TRIF directly to its receptor complex and initiates activation of NF-κB and IRF3 via an alternative pathway to induce inflammatory cytokine production and IFN-β expression.
Whereas all TLRs activate NF-κB, the TLRs demonstrate specificity in the recruitment of TIR-containing adaptor proteins that provide a molecular basis for the variation of gene-expression profiles induced by distinct TLRs. TLR2 and TLR4 specifically interact with Mal upstream of MyD88 recruitment [8–10], whereas TLR4 uses TRAM upstream [11] of TRIF recruitment [12, 13]. Although not required for positive TLR signaling, SARM has been described to regulate negatively TRIF-dependent signaling [14].
TRAM is unique among the TIR-containing adaptors, as it is only required for TLR4 signaling and is specific in its recruitment to endosomes following TLR4 ligation [15]. TLR4, which is responsible for the sensing of LPS or endotoxin, one of the most powerful immunostimulatory factors known and perhaps the most widely studied TLR displays the most complex signaling of all TLRs. Whereas TRAM is constitutively found at the plasma membrane via an N-terminal myristoylation site [16], following TLR4 activation, TRAM localizes to the endosome, allowing recruitment of TRIF to the signaling complex and initiation of the alternative signaling pathway to NF-κB and nuclear translocation of IRF3 to induce IFN-β [16].
Mal and TRAM have been described as “bridging adaptors”, responsible for recruiting MyD88 and TRIF to the activated TLR2 and TLR4 complexes. We have previously demonstrated an interaction between Mal and TRAF6 via a TRAF6-binding motif identified in Mal, which characterized a unique role for Mal in mediating activation of NF-κB via a noncanonical route [17, 18]. Furthermore, TRIF was also demonstrated to interact with TRAF6 via a TRAF6-binding motif [19]. However, it is unclear whether other TIR adaptors engage TRAF6 directly.
In this study, we have identified a TRAF6-binding motif within TRAM, commensurate with those observed in Mal and TRIF, and a direct functional link between TRAM and TRAF6. We have demonstrated direct interaction between TRAM and TRAF6 that is dependent on this binding motif. TRAM colocalization with TRAF6 was dependent on this motif, and TRAM-induced NF-κB, IFN-β, and p38 kinase reporter activity is dependent on an intact TRAF6-binding motif. The protein variant with a mutation of a critical Glu residue with the TRAF6-binding motif (termed TRAM E183A) acted as a dominant-negative to inhibit TLR4-mediated reporter activation, and TRAM-induced reporter activity is inhibited in TRAF6-deficient cells. Critically, TRAM-deficient macrophages reconstituted with TRAM E183A display significantly decreased protein expression of TNF-α, IL-6, and RANTES compared with reconstitution with WT TRAM. These studies therefore identify that similar to Mal and TRIF, TRAM contains a TRAF6-binding motif that is important in TLR4-induced signal transduction, providing a distinguishing signaling feature for TRAM, distinct of its role in bridging TLR4 and TRIF.
MATERIALS AND METHODS
Cell lines and reagents
HEK293, HEK293 T, HEK293 stably transfected cells expressing TLR4 and MD2, and TRAF6-deficient MEFs were incubated in DMEM, supplemented with 10% FCS and 2 mM glutamine and maintained in a 37°C humidified atmosphere. Immortalized TRAM-deficient macrophages were generated from TRAM-deficient mice, as described previously [20], and cultured in DMEM/10% FCS plus glutamine. THP-1 monocytes were maintained in RPMI -1640 medium, 2 mM glutamine, 10% FCS at 37°C, and 5% CO2. LPS K235 (Sigma, St. Louis, MO, USA) was repurified as described previously [21], and ANTI-FLAG M2 agarose beads were from Sigma.
Plasmids
TRAM-HA and IFN-β-luciferease reporter were kindly provided by Katherine Fitzgerald (University of Massachusetts, North Worcester, MA, USA). TRAM E183A was generated using the QuikChange site-directed mutagenesis kit with PfuTurbo (Stratagene, La Jolla, CA, USA), using the TRAM-HA template. Cells were lysed with passive lysis buffer (Promega, Madison, WI, USA) and assayed for luciferase and β-galactosidase activity using luciferase assay reagent (Promega).
Transient transfections and gene reporter assays
HEK293 cells (2×104) were seeded in 96-well plates, 24 h before transfection. Transfections were performed with FuGENE 6 (Roche Diagnostics, Indianapolis, IN, USA). NF-κB-dependent gene expression was determined using the 5× κB-luciferase reporter construct (Stratagene) concomitantly with indicated vectors. The p125-luc luciferase plasmid contains the full-length IFN-β promoter upstream of the firefly luciferase gene [22] and is referred to as the IFN-β-luciferase reporter. With the use of the PathDetect transient transfection kit (Stratagene), cotransfection of pFR-luciferase in combination with Gal4 fusions p65, pFA-Jun, pFA-CHOP, or pFA-Elk-1, respectively, was used to analyze activation of p65 transactivation and MAPK, respectively. The Renilla luciferase-TK (pRL-TK)-encoding plasmid (Promega) was used to normalize for transfection efficiency, and pEF-BOS empty vector was used to maintain constant DNA. Transfected cells were lysed using passive lysis buffer (Promega) and assayed for luciferase and Renilla activity using luciferase assay reagent (Promega). Luminescence readings were corrected for Renilla activity and expressed as fold increases over nonstimulated control values.
TRAF6-deficient MEFs were seeded at 2 × 105 cells/ml in 96-well format, 24 h before transfection with indicated plasmids: TK-Renilla and FuGENE 6. Cells were harvested 24 h later with passive lysis buffer and assayed as described above.
Protein production and purification
DNA-encoding residues 346–504 of TRAF6 (TRAF6346–504) were amplified by PCR and inserted into the pET28b vector using the NcoI and XhoI restriction enzyme cleavage sites. The resulting construct encodes C-terminal Myc and His6 tags. TRAM1–235, TRAM70–235, and CD40216–245 constructs were inserted into the pMCSG10 vector using ligation-independent cloning [23]. The resulting constructs encode N- terminal His6 and GST tags. All of the constructs were verified by sequencing. The proteins were expressed in Escherichia coli BL21 (DE3) cells using autoinduction media [24]. Cells were grown at 37°C until the midexponential phase (optical density of 600 nm of 0.6–0.8) was reached. The temperature was then reduced to 20°C, and the cultures were grown for ∼16 h before harvesting.
The cells were lysed using sonication, and the resulting supernatant was applied onto a 5-ml HisTrap or a 5-ml GSTrap FF column (GE Healthcare, Pittsburgh, PA, USA). Bound protein was eluted using a linear gradient of imidazole (30–250 mM) or 5 mM reduced glutathione. The fractions containing the protein of interest were pooled and loaded onto a Superdex 75 or 200 HiLoad 26/60 gel-filtration column (GE Healthcare), pre-equilibrated with 10 mM HEPES, pH 7.4, 150 mM NaCl, and 1 mM DTT. The peak fractions were pooled, concentrated to 0.75–1.5 mg/ml, and stored in aliquots at −80°C.
Immunoprecipitation and immunoblot analysis
HEK293 T cells (2×106 cells/10 cm dish) were transfected using FuGENE 6 (Promega) with the indicated plasmids, where the total amount of DNA (2.5 μg/dish) was kept constant. Twenty-four hours later, the cells were lysed in KalB buffer, as described [25]. The indicated antibodies (2 μg) or ANTI-FLAG-Sepharose beads (20 μl, 50% slurry) were incubated with the cell lysates for 2 h, followed by the addition of 40 μl 50% protein G slurry for 1 h. The immune complexes were precipitated, washed, eluted by the addition of sample buffer, followed by SDS-PAGE, and immunoblotted using the indicated antibodies. For GST-pulldown experiments, the lysates prepared from HEK293 T cells transfected with indicated vectors were used in a GST-pulldown assay, whereby cell lysates were incubated for 2 h at 4°C with recombinant GST fusion protein coupled to glutathione-Sepharose. The complexes were washed three times in lysis buffer, separated by SDS-PAGE, and immunoblotted as indicated in the figure legend. Endogenous immunoprecipitations of TRAM and TRAF6 were performed as described previously [18]. Anti-TRAF6 and anti-TRAM antibodies were sourced from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
To assay the interactions between purified proteins, a bait/prey mixture, consisting of 10 μg-purified TRAF6346–504-Myc and 100 μg-purified GST fusion protein in 1.5 ml binding/wash buffer (50 mM HEPES, pH 7.4, 250 mM NaCl, 0.5 mM EDTA, 1 mM DTT, and 0.5% Triton X-100) with 1% BSA, was incubated with glutathione-Sepharose resin for 4 h. After incubation, the resin was washed three times with 1 ml binding/wash buffer, and bound protein was eluted by adding 50 μl SDS-PAGE sample buffer. Eluted complexes were separated by SDS-PAGE and stained with Coomassie brilliant blue or immunoblotted, as indicated in the figure legend.
siRNA knockdown of TRIF
The 25-bp duplex of targeting stealth siRNA or NT siRNA (Invitrogen, Carlsbad, CA, USA) was transfected into subconfluent HEK293 T cells, using siRNA jetPRIME (Polyplus-Transfection, Illkirch, France). Double-stranded siRNAs, containing equal parts of the following antisense sequences, were used to knock down TRIF: siRNA2, 5′-CCCAUUGACGGUGUUUCGGACUGGA-3′; siRNA3, 5′-CCAUCACUUCCUAGCGCCUUCGACA-3′. The NT siRNA were low GC and medium GC stealth RNA interference-negative control duplexes (Invitrogen). Forty-eight hours after transfection, the cells were transfected with the relevant constructs or analyzed by real-time quantitative PCR or Western blot using anti-TRIF (Cell Signaling Technology, Danvers, MA, USA) and β-actin (Ambion, Austin, TX, USA).
Cell imaging
HEK293 cells (4×104 cells/well) were plated onto eight-well plates overnight and transfected with 115 ng TRAM-Cherry and TRAF6-FLAG, respectively, for 24 h. Cells were washed three times PBS, permeabilized with 0.1% saponin, 2.5% FCS, followed by a 2-h incubation with anti-M2 (FLAG) antibody (1:1000; Sigma). Cells were washed three times with PBS, supplemented with 0.1% saponin and 2.5% FCS, and three times with PBS. Imaging was performed on a Leica SP5 multichannel Acousto-Optical Beam Splitter confocal laser-scanning microscope and imaged as deconvoluted z-stacks. Images were analyzed using Imaris Scientific software (Bitplane, Zurich, Switzerland).
Generation of TRAM WT/E183A macrophages and LPS challenge
HEK293 T cells were plated at 0.5 × 106 cells/well in a six-well plate and left to adhere overnight at 37°C. Cells were then tranfected with 2 μg pRH-TRAM WT-citrine or pRH-TRAM E183A-citrine, 1 μg gag-pol, and 100 ng VSV-G, mixed with transduction reagent FuGENE 6 (Promega) at a ratio of 3:1. This mixture was then added to cells, and the cells incubated for 24 h at 37°C. Media were then removed and replaced with DMEM containing 30% heat-inactivated FCS, and the cells incubated for 24 h at 37°C. Media were then harvested and filtered through a 0.45-μm filter. TRAM-deficient macrophages were transduced with retrovirus (VSV-G coat protein) containing TRAM WT or TRAM E183A mutant. Macrophages were seeded at 1 × 105 cells/ml in a 24-well plate and incubated overnight at 37°C. Media were then removed and replaced with media containing retrovirus for 24 h at 37°C. Media containing virus were then removed and replaced with DMEM, and the cells incubated for a further 48 h at 37°C. Cells were then sorted using FACS and the positive cells collected and cultured for further experiments. Cells were seeded at 4 × 104/well in a 96-well plate, 24 h before stimulation with LPS (0.312–10 ng/ml) for a further 8 h. Cultured supernatants were collected and assayed for TNF-α, IL-6 (BD Biosciences, San Jose, CA, USA), and RANTES (R&D Systems, Minneapolis, MN, USA), according to manufacturers' instructions.
Statistical analysis
For statistical analysis, the data obtained from triplicate experiments are presented as the means ± sem as indicated. All statistical analysis was performed using GraphPad PRISM software (GraphPad Software, La Jolla, CA, USA). Mean differences were calculated using Student's t-test. The levels of significance are: *P < 0.05, **P < 0.01, and ***P < 0.001.
RESULTS
TRAM interacts with TRAF6 via a TRAF6-binding motif
Structural analysis of TRAF6-binding partners [26] identified the consensus sequence Pro-X-Glu-X-X-(aromatic/acidic residue) as the TRAF6-binding motif. We have previously identified such a motif in the TIR domain of TRAM: PRERTP {residues 181–186 (TRAM181–186) [17]}. To test if TRAM could interact with TRAF6, we performed coimmunoprecipitation experiments in HEK293 T cells that were transiently transfected with FLAG-tagged TRAF6 and HA-tagged TRAM. As shown in Fig. 1, we were able to detect complexes containing TRAM- and TRAF6-tagged proteins by immunoprecipitation (Fig. 1C, lane 2), demonstrating an association between TRAM and TRAF6.
Figure 1. TRAF6 associates with TRAM.

(A) Schematic representation of TRAM, indicating the position of the TIR domain (aa 73–235) and the location of the putative TRAF6-binding motif (PRERTP: aa 181–186). (B) HEK293 T cells (2×106) were transiently cotransfected with FLAG-tagged TRAF6 and HA-tagged TRAM where indicated. Cells were probed [immunoprecipitated (IP)] with anti (α)-FLAG M2-agarose beads to precipitate TRAF6 and interacting complexes. Protein complexes were separated by SDS-PAGE, transferred to nitrocellulose, and immunoblotted (IB) with anti-HA antibody to identify interacting proteins. Cell extracts were also immunoblotted to check expression of HA- and FLAG-tagged constructs and FLAG immunoprecipitation assessed by immunoblotting with anti-FLAG antibody. (C) Human monocytic THP1 cells (2×107) were stimulated with LPS (100 ng/ml) for indicated times. The cells were lysed, and cellular debris was removed by centrifugation (14,000 rpm, 10 min, 4°C). The lysates were precleared with protein A-Sepharose beads, and the lysates were probed with α-TRAF6 mAb (1 μg) bound to α-mouse IgG beads. Detection of immunoprecipitated endogenous TRAM was analyzed by immunoblotting with α-TRAM antibody. Results presented represent three independent experiments.
Having established an ectopic interaction between TRAM and TRAF6, we next wished to demonstrate if TLR stimulation could induce association between the two proteins. As can be observed in Fig. 1C, TRAF6 immunoprecipitated TRAM in a time-dependent manner within 10–20 min of LPS stimulation of human THP-1 monocytic cells, decreasing after 30 min. Together, these results clearly demonstrate a TLR4-induced, direct interaction between TRAM and TRAF6.
To evaluate further TRAM interaction with TRAF6, we demonstrated that recombinant GST-tagged TRAM (i.e., TRAM73–232), consisting of the TIR domain and TRAF6-binding motif, was able to recognize and precipitate ectopically expressed FLAG-TRAF6 from cellular lysates (Fig. 2A, lane 2). As TRIF associates with TRAF6 via three TRAF6-binding motifs [19] located distal to its TIR domain (i.e., TRIF84–91, −248–255, and −299–309), we also determined whether the recombinant GST-TRIF TIR domain (TRIF390–460) could precipitate ectopically expressed FLAG-TRAF6. As can be observed in Fig. 2A, lane 3), TRIF was able to recognize TRAF6, albeit with a significantly lower affinity than that observed with TRAM (compare Fig. 2A, lanes 2 and 3). Importantly, recombinant GST alone was not able to bind TRAF6. Therefore, to determine if the putative TRAF6-binding motif within TRAM was responsible for this association, we probed cellular lysates expressing HA-TRAM or HA-TRAM E183A, which encodes a point mutation of the critical Glu residue, described previously as crucial for target protein binding of TRAF6 [26]. As can be observed in Fig. 2B, recombinant GST-TRAF6 was able to immunoprecipitate HA-TRAM (Fig. 2B, lane 1) but did not associate with HA-TRAM E183A (Fig. 2B, lane 2), indicating that mutation of the critical Glu residue in TRAM ablated association between TRAM and TRAF6, consistent with previous TIR-containing adaptor protein association studies with TRAF6 [17–19]. Taken together, these results demonstrate that TRAM interacts with TRAF6 via its TRAF6-binding motif, which is contained within the C-terminal region of TRAM.
Figure 2. TRAM interacts directly with TRAF6 via a TRAF6-binding motif.
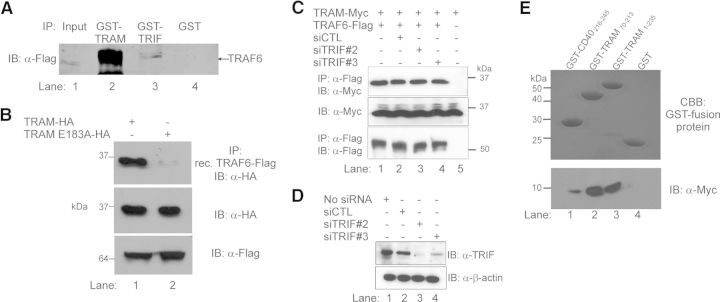
(A) HEK293 T cells were transiently transfected with TRAF6-FLAG for 24 h. Cells were lysed and the cellular lysates probed with equal amounts of recombinant GST-TRAM-TIR (lane 2), GST-TRIF-TIR (lane 3), and GST alone (lane 4) for 2 h. Complexes were immunoprecipitated with glutathione-Sepharose beads and interacting complexes separated by SDS-PAGE and immunoblotted with anti-FLAG antibody to identify TRAF6. Expression of TRAF6-FLAG in transiently transfected cells was confirmed by immunoblot of cell lysates with anti-FLAG antibody (lane 1). (B) HEK293 T cells were transiently transfected with WT TRAM-HA or TRAM with mutated TRAF6-binding motif (TRAM E183A-HA) where indicated. Cellular lysates were then probed with recombinant (rec.) FLAG-tagged GST-TRAF6 protein. TRAF6 was immunoprecipitated with glutathione-sepharose beads and interacting complexes separated by SDS-PAGE and immunoblotted with anti-HA antibody to detect TRAM. Protein expression of TRAM constructs and TRAF6 was confirmed by immunoblotting cell lysates with anti-HA and anti-FLAG, respectively. (C) TRIF-targeted siRNA-treated HEK293 T cells were transfected with full-length Myc-tagged TRAM and Flag-TRAF6 (lanes 1–5). Lane 2 was stimulated with LPS (100 ng/ml) for 30 min. Interacting complexes were immunoprecipitated and imaged as described above. (D) Two different TRIF-targeted siRNAs (siTRIF#2 and siTRIF#3) or NT (CTL) siRNA at 10 nM were transfected into HEK293 T cells and left for 48 h. Cell lysates were immunoblotted with anti-TRIF antibody for endogenous TRIF detection and anti-β-actin for loading control. (E) Pull-down analysis of recombinant GST-TRAM and TRAF6-Myc, expressed and purified in E. coli. The TRAF6-binding region of CD40 was used as a positive control. Protein complexes were coincubated in PBS and interacting complexes immunoprecipitated with glutathione-agarose beads, which were washed, boiled in Laemmli buffer, separated by SDS-PAGE, and then stained with Coomassie brilliant blue (CBB), or transferred to nitrocellulose membranes, which were probed with anti-Myc antibody to identify interacting proteins. All results shown are representative of three independent experiments.
As TRIF has also been documented to associate with TRAF6 [19], we next examined if TRAM could interact with TRAF6 independent of TRIF. As can be observed in Fig. 2C, Flag-tagged TRAF6 was able to immunoprecipitate coexpressed, Myc-tagged TRAM in the presence (Fig. 2C, lanes 1 and 2) or absence (Fig. 2C, lanes 3 and 4) of TRIF depleted by siRNA. Successful depletion of TRIF by siRNA was confirmed by immunoblot of HEK293 T cells (Fig. 2D). To exclude further the possibility that TRIF may be bridging TRAM and TRAF6, we next demonstrated direct interaction by immunoprecipitating TRAM and TRAF6 in a cell-free environment. As can be seen in Fig. 2E, recombinant full-length GST-fusion TRAM1–235 (Fig. 2E, lane 3) and truncated GST-TRAM comprising the TIR domain only (TRAM70–235; Fig. 2E, lane 2) immunoprecipitated recombinant Myc-tagged TRAF6346–504 (Fig. 2E, lower) produced in E. coli. These results clearly demonstrate that TRAM interacts directly with TRAF6, independent of TRIF.
TRAM colocalization with TRAF6 requires an intact TRAF6-binding motif
TRAM localizes to the plasma membrane in resting cells via an N-terminal myristoylation site [16] but relocates to endosomes following TLR4 activation to initiate TRAM-TRIF signaling [15]. As ectopic expression of TRAM can activate NF-κB and IRF3 signaling pathways, mimicking function without the need for TLR4 activation [11, 15, 16, 27, 28], we sought to determine whether localization of TRAM and TRAF6 occurs at the plasma membrane or cytosol in HEK293 cells by fluorescence microscopy. We found that, consistent with earlier reports, Cherry-tagged TRAM (red) was visualized at the plasma membrane (Fig. 3A) and also at structures that appear consistent with previous reports of TRAM-induced endosomal formation in the cytosol (Fig 3, arrows) [11, 16]. Interestingly, although FLAG-TRAF6 (green) was also observed in the cytosol, it also appears to be colocalizing with TRAM along the edge of these endosomes, evident in the fluorescence overlay (yellow). This suggests that TRAF6 is recruited to the endosomes following ectopically expressed, TRAM-induced signaling activation. Conversely, we noted that whereas Cherry-TRAM E183A (red) was also observed located at the plasma membrane (Fig. 3B), we did not observe obvious formation of the cytosolic endosomal structures seen with WT TRAM. Furthermore, we failed to observe colocalization of TRAM E183A and TRAF6, as evidenced by overlay (yellow), and TRAF6 appeared diffuse throughout the cytosol, consistent with an inability of the binding motif mutant of TRAM to recruit TRAF6 to the plasma and endosomal membrane, supporting our immunoprecipitation results.
Figure 3. TRAM colocalization with TRAF6 at the cell membrane requires an intact TRAF6-binding motif.
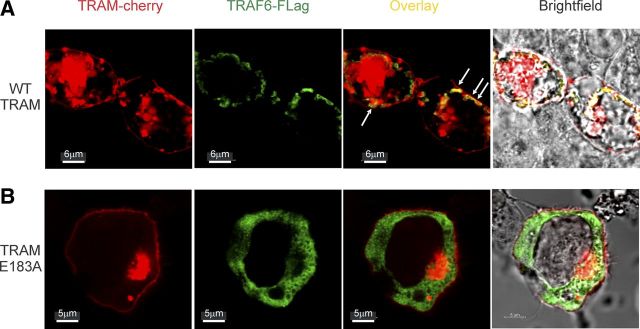
HEK293 cells were transiently transfected with fluorescent monomeric Cherry (mCherry) tagged-TRAM or TRAM E183A fluorescently tagged monomeric Cherry and TRAF6-FLAG, where indicated for 24 h. Immunofluorescence microscopy was performed on individual cells. (A) Colocalization (overlay) illustrates TRAF6-FLAG colocalizing with TRAM-mCherry at the plasma membrane and what appears to be early endosomes (indicated with arrows), consistent with TRAM-mediated activation of the signaling pathway (bars, 6 μm). (B) Expression of TRAM E183A-mCherry demonstrates a lack of colocalization with TRAF6, which appears diffuse throughout the cytosol. Representative images shown are flattened maximum intensity projections of three-dimensional, deconvolved z-stacks using Imaris Scientific software (bars, 5 μm). All images are representative of multiple cells observed in three individual experiments.
TRAM-induced activation of NF-κB and IFN-β reporter activity is dependent on interaction with TRAF6
Having established that TRAM interacts with and localizes with TRAF6 via its TRAF6-binding motif, we next investigated if this interaction affected downstream signaling. As noted above, ectopic expression of TRAM can activate TRAM-TRIF-dependent signaling pathways to induce inflammation [11, 16]. Therefore, we next assessed the ability of TRAM and TRAM E183A to drive NF-κB- and IFN-β-luciferase reporter activity. As expected, WT TRAM was able to induce an eight to 21-fold increase in NF-κB-luciferase reporter activity (Fig. 4A) in a dose-dependent manner. Additionally, WT TRAM also induced an 11- to 12-fold increase in IFN-β-luciferase reporter activity (Fig. 4B). Consistent with our immunoprecipitation and localization data, TRAM E183A, however, displayed a decreased ability to drive NF-κB-luciferase activation (three- to ninefold increase) and ablated IFN-β-luciferase activation compared with WT.
Figure 4. TRAM interaction with TRAF6 mediates NF-κB and IFN-β activation.
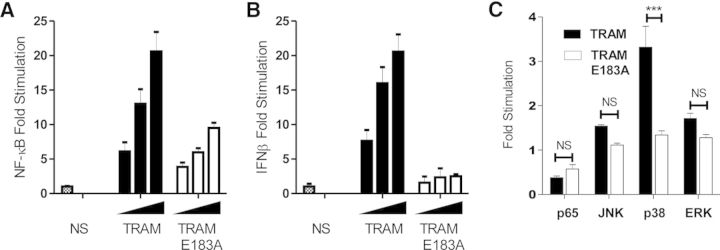
HEK293 cells (2×104/ml) were transiently transfected with TK-Renilla and (A) κB-luciferase and (B) IFN-β-luciferase reporter constructs in conjunction with TRAM or TRAM E183A (10-–40 ng). Readings are normalized as expressed luciferase reporter activity over constitutively expressed TK-Renilla luminescence. Results are mean ± sem of triplicate determinations performed in three independent experiments. (C) HEK293 cells (2×104) were cotransfected with the components of the pFR-luciferase (80 ng) and p65[1–551]-Gal4, c-jun (JNK pathway; 2 ng), CHOP (p38 pathway; 5 ng), and Elk-1 (ERK pathway; 5 ng) Gal4 fusion vectors, respectively, together with TRAM and TRAM E183A plasmids (40 ng). Results are mean ± sem for triplicate determinations (***P<0.001; n=3).
Previously, we have demonstrated that Mal interaction with TRAF6 mediated Mal-induced p65 transactivation and JNK and ERK activation but not p38 kinase activity [17, 18]. Contrary to these results, we observed that TRAM drove p38-dependent luciferase activity (3.3-fold; P<0.001), whereas TRAM E183A failed to induce p38-dependent luciferase activity (Fig. 4C). Furthermore, TRAM failed to induce p65-mediated transactivation-, JNK-, or ERK-dependent luciferase activity, different from what was observed with Mal and TRAF6. Together, these results suggest that TRAM interaction with TRAF6 is required specifically for p38 MAPK, NF-κB, and IRF3 activation.
We next addressed the role of TRAM/TRAF6 interaction in TLR4-mediated activation of NF-κB and IRF3. HEK293 cells stably expressing TLR4/MD2 were transiently transfected with the NF-κB- and IFN-β/IRF3-dependent luciferase reporter plasmid and stimulated with 10 ng/ml LPS. As can be seen in Fig. 5A, TRAM E183A was able to inhibit TLR4-mediated activation of NF-κB- and IFN-β-luciferase reporter activity (Fig. 5B) in a dose-dependent manner.
Figure 5. Expression of TRAM E183A inhibits TLR4-mediated activation of NF-κB- and IFN-β-luciferase activity.

HEK293 cells (2×104), stably expressing TLR4 and MD2, were transiently transfected with TK-Renilla and (A) NF-κB-luciferase and (B) IFN-β-luciferase reporter constructs in conjunction with indicated amounts of TRAM E183A (1–80 ng) for 24 h. Cells were stimulated for 6 h with 10 ng/ml LPS and assessed for luciferase activity. Results are normalized to each individual cell line and represented as means ± sem of three independent experiments.
Next, we wished to examine if the absence of TRAF6 impaired the ability of TRAM to induce NF-κB- or IFN-β-luciferase reporter activity. Whereas ectopic expression of TRAM was able to induce a 2.0- to 3.2-fold, dose-dependent increase in NF-κB-luciferase activity (Fig. 6A) in WT MEFs, expression of TRAM in TRAF6-deficient MEFs displayed ablated responses (1- to 1.5-fold increases). Consistent with what was observed for NF-κB activation, TRAM induced a similar two- to threefold dose-dependent increase in IFN-β-luciferase activation (Fig. 6B), which was similarly ablated in TRAF6-deficient MEFs. Importantly, constitutively active IRF3, which has had five critical Ser residues mutated to phosphomimetic Asp, generated a constitutively active form of IRF3 [29] (termed IRF3 5D) and was able to activate the IFN-β-luciferase reporter in the presence or absence of TRAF6.
Figure 6. TRAF6 is required to mediate TRAM-induced activation of NF-κB and IFN-β.

TRAF6-deficient MEFs (2×105) were transfected with TRAM or TRAM E183A (1–25 ng) in conjunction with TK-Renilla and (A) NF-κB- or (B) IFN-β-luciferase. Cells were also transfected with constitutively active IRF3 5D to assess activation of IFN-β-luciferase promoter activity. Shown is the mean relative luciferase activity ± sem for a representative experiment from three separate experiments. The readings are normalized for reporter luciferase expression over TK Renilla expression.
TRAM association with TRAF6 is critical for TLR4-mediated inflammatory responses
Finally, to determine whether TRAM/TRAF6-mediated signaling plays a role in the inflammatory response to TLR4 stimulation, we investigated TLR4-induced cytokine expression in reconstituted TRAM-deficient macrophages, which were reconstituted with WT TRAM or TRAM E183A tagged with the fluorescent protein citrine. Reconstituted cells expressing commensurate amounts of citrine-tagged WT TRAM and TRAM E183A were sorted by FACS (Fig. 7A) and stimulated with LPS to induce TLR4-dependent signaling. As can be seen in Fig. 7, B and C, whereas LPS induced a robust, dose-dependent induction of NF-κB-dependent cytokines TNF-α and IL-6, respectively, macrophages expressing TRAM E183A display significantly lower expression of both cytokines, particularly at the lower concentrations of LPS. Importantly, TRAM association with TRAF6 was also required for protein expression of the chemokine RANTES (Fig. 7D), which requires NF-κB and IRF3 transcription factors [30], further demonstrating the requirement of TRAF6 association with TRAM for the NF-κB and IRF3 signaling pathways.
Figure 7. TRAM association with TRAF6 is required for TLR4-induced proinflammatory responses.

TRAM-deficient macrophages were reconstituted by retroviral transduction with citrine-tagged WT TRAM or TRAM E183A and (A) selected for comparable protein expression by FACS sorting based on citrine expression. Reconstituted macrophages were seeded at 4 × 104/well, 24 h before stimulation with a dose range of LPS (0.312–10 ng/ml) for 8 h. Cultured supernatants were assayed for (B) TNF-α, (C) IL-6, and (D) RANTES by ELISA, according to the manufacturer's instructions. Results are the means ± sem representing the pooled results of three independent experiments performed in triplicate. *P < 0.05, **P < 0.01, and ***P < 0.001.
DISCUSSION
In this study, we have found a novel feature in TRAM that shows it has activities beyond its function as a bridging adaptor to recruit TRIF to the TLR4 complex. TRAM contains a TRAF6-binding motif that is required for TRAM to signal. The presence of this TRAF6-binding motif is important for TRAM function, as mutation of a critical amino acid within the motif, TRAM E183A, inhibited the ability of TRAM to activate p38 MAPK and NF-κB- and IFN-β-luciferase promoters, while also inhibiting TLR4-induced inflammatory responses. Additionally, whereas TRAM interacts directly with TRAF6, TRAM E183A could no longer recruit TRAF6. Importantly, the mutated TRAM E183A acted as a dominant-negative toward TLR4-mediated signal transduction, and TRAF6 was demonstrated to be important for TRAM-mediated signaling, as TRAF6-deficient MEFs were not responsive to TRAM-induced activation of NF-κB and IFN-β. Based on the new data presented here, we propose that TRAM directly recruits TRAF6 to the TLR4 signaling complex and that this recruitment is important for NF-κB activation and IFN-β induction, promoting a new role for TRAM in addition to its role as a bridging adaptor for TRIF.
Previous studies have demonstrated that Mal and TRIF bind TRAF6 via their respective TRAF6-binding motifs [17–19]. However, it should be noted that the role of TRAF6 in TRIF signaling is controversial. Whereas some studies found that TRAF6 functions exclusively in MyD88-directed signaling, others suggest a secondary role in TRIF-mediated activation of NF-κB [31, 32]. Our study demonstrates that TRAM also interacts with TRAF6 and regulates signaling. Together, these studies suggest that adaptor interaction with TRAF6 may mediate “alternate” signals emanating from the membrane-bound signaling complexes that allow specificity and fine-tuning of TLR signal transduction. It is important to note that MyD88 does not contain a TRAF6-binding motif nor does it interact directly with TRAF6. It would appear that the role of MyD88 is to direct the canonical signaling pathway that recruits TRAF6 to the signaling complex via IRAK1 and IRAK4 to induce NF-κB nuclear translocation in response to multiple TLRs. Thus, the ability of the other adaptors—Mal (in response to TLR2 and TLR4), TRIF (in response to TLR3 and TLR4) and TRAM (in response to TLR4)—to associate independently with TRAF6 allows modulation of the inflammatory response by a subset of TLRs as a means of adding specificity and regulatory control to the wide range of inflammatory responses to different TLRs. Our findings also suggest that TRAF6 plays a crucial role in mediating TRAM-dependent signaling in TLR4 responses.
At present, we have a limited understanding of the molecular mechanisms by which postreceptor complexes form during TLR signaling. Analysis of the crystal structure of Mal [33, 34] and homology model of the TRAM TIR domain shows that the critical glutamate residue in the P0 position of the TRAF6-binding motif (the nomenclature follows) [26] is surface-exposed and can mediate direct interaction with TRAF6 (Fig. 8). It should be noted that the P3 position in the TRAM TRAF6-binding motif is occupied by proline, not the prototypic aromatic/acidic group assigned to a classic TRAF6-binding motif. This may suggest that alternate residues may occupy the P3 position; however, the effect on binding avidity for TRAF6 is unknown. We have also noted that the side-chain of the proline residue at the P−2 position is partially buried in Mal and TRAM, and the overall configuration of the TRAF6 motifs is not optimal compared with the structure of the CD40-TRAF6 complex [26]. Furthermore, neither TRAM nor Mal has acidic residues in the P1 and P2 positions, which have previously been shown to increase the binding affinity to TRAF6 [26] (Fig. 8C). Overall, this suggests that Mal and TRAM may undergo a conformational change upon binding to TRAF6 or that apart for the critical glutamate residue in the P0 position, the binding mode of Mal and TRAM to TRAF6 is distinct from CD40.
Figure 8. The critical glutamate residue in the TRAF6-binding motif is surface-exposed in MAL and TRAM.

(A and B) Transparent surface representations of a crystal structure of the Mal TIR domain (Protein Data Bank ID: 2Y92; A) and a model of the TRAM TIR domains (B) with the TRAF6-binding motif residues displayed in wireframe. The model of the TRAM TIR domain was prepared using the I-TASSER webserver [35] using the crystal structure of Mal as a template. (C) Sequence comparison of the TRAF6-binding motifs in CD40, IRAK2, Mal, TRAM, and TRIF.
An interesting finding from these studies was the requirement of TRAF6 for TRAM-induced activation of the IFN-β-luciferase promoter. These observations may be a result of the previous finding that NF-κB is required to bind the PRDII that forms a component of the IFN-β enhanceosome [36, 37]. NF-κB and IRF3 (via binding of the PRDI/III domain) are therefore both required for IFN-β promoter activation and expression. Therefore, the absence of TRAF6 recruitment to the signaling complex, either through an inability to interact with TRAM (Fig. 2) or the absence of TRAF6 (Fig. 6), would therefore not only affect NF-κB activation, as we observed, but also induction of IFN-β, as a result of a lack of NF-κB in the IFN-β enhanceosome. These results exemplify further the requirement of an orchestrated signaling pathway that converges on multiple transcription factors and promoter binding sites to initiate the optimal inflammatory response. Disruption of this coordinated process can therefore lead to dysregulated inflammation.
We have reported previously that Mal/TRAF6 association regulates the activation of MAPKs, ERK and JNK, but not p38, and that disruption of the Mal/TRAF6 interaction did not ablate TNF-α production, despite inhibiting NF-κB transcriptional activation and IL-6 production [18]. Intriguingly, in this study, we found that TRAM drove p38 activation, which was inhibited upon disruption of the ability of TRAM to recruit TRAF6 (i.e., in TRAM E183A). Importantly, p38 has been shown to play a role in regulating TNF-α message stability and protein expression [38, 39]. TLR4 induction of TNF-α has been described previously as requiring the MyD88- and TRAM/TRIF-dependent pathways to induce optimal expression [40, 41], and TRAM has been implicated further in TLR4-induced translation of TNF-α [42]. As such, it may be that TRAM association with TRAF6 acts to enhance or sensitize responses, as evidenced by TRAM E183, predominately affecting lower concentrations of LPS (Fig. 7), which was not as apparent at higher concentrations, where sensitivity may not be such an issue. Our finding that TRAM association with TRAF6 regulates p38 activation complements our earlier studies on Mal-dependent signaling via TRAF6 and may provide an understanding of the convergence of signaling pathways that promote optimal expression of this prototypic inflammatory cytokine.
In conclusion, we demonstrate TRAM as a novel interacting partner of TRAF6, completing our understanding of how TIR-containing adaptor proteins interact directly, in the case of TRAM, Mal, and TRIF with TRAF6, or indirectly, in the case of MyD88. Interestingly, a recent study found that TRAM was required for IL-18 signaling and acted as a sorting adaptor for MyD88, further suggesting that there are alternate roles for TRAM other than as an obligate bridging adaptor between TLR4 and TRIF. Whereas future studies would concentrate on understanding the molecular mechanisms underpinning how TRAF6/TRAM association regulates signaling pathways, these studies have identified a critical role for TRAM interacting with TRAF6 to modulate downstream inflammation from TLR4, adding further complexity to regulation of TLR signaling. Importantly, TRAM is only used in TLR4 signaling. Therefore, the ability to manipulate the interaction between TRAM and TRAF6 may provide therapeutic opportunities to moderate TLR4-mediated inflammation, which plays such a critical role in many chronic inflammatory diseases. Whereas many current therapies target MyD88, a limiting factor is the generic dependency of nearly all inflammatory pathways on MyD88. Our studies presented here suggest that modulation of TRAM and TRAF6 association may specifically target only TLR4-mediated inflammation, targeting potentially NF-κB and IFN pathways—something that targeting MyD88 alone would not achieve.
ACKNOWLEDGMENTS
This work was funded by the Australian NHMRC (to A.M. and B.K.), the Victorian Government's Operational Infrastructure Support Program and a United Kingdom Medical Research Council Programme Grant (G1000133-E01) to N.J.G. J.S. and A.G.B. were funded by a Science Foundation Ireland Strategic Research Cluster grant (07/SRC/B1144). B.K. is a NHMRC Research Fellow.
We thank Katherine Fitzgerald for the IFN-β-luciferase reporter and FLAG-TRAM constructs.
Footnotes
- HA
- hemagglutinin
- HEK
- human embryonic kidney
- IRAK
- IL-1R-associated kinase
- IRF3
- IFN regulatory factor 3
- Mal
- MyD88-adaptor like
- MD2
- myeloid differentiation protein 2
- MEF
- murine embryonic fibroblast
- NHMRC
- National Health and Medical Research Council
- NT
- nontargeting
- PAMP
- pathogen-associated molecular pattern
- PRD
- positive regulatory domain
- PRR
- pattern recognition receptor
- SARM
- sterile α and hydroxyphenyl-ethyl-aminomethyl-tetralone-armadillo motif
- siRNA
- small interfering RNA
- TICAM
- Toll/IL-1R-containing adaptor molecule
- TIR
- Toll/IL-1R
- TK
- thymidine kinase
- TRAF6
- TNFR-associated factor 6
- TRAM
- TRIF-related adaptor molecule
- TRIF
- Toll/IL-1R domain-containing adaptor-inducing IFN-β
- VSV-G
- vesicular stomatitis virus G
- WT
- wild-type
AUTHORSHIP
B.V., J.S., J.J., T.V., and M.M. performed immunoprecipitations, reporter assays, and cellular reconstitutions. T.V. and B.K. performed the molecular modeling and assisted in the preparation of the manuscript. E.L. assisted in the microscopy. K.H. and R.S. carried out the molecular biology. A.G.B. and N.G. designed experiments, interpreted data, and assisted in the preparation of the manuscript. A.M. conceived of the project, designed experiments, interpreted data, and drafted the manuscript. All authors read and approved the final manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Takeuchi O., Akira S. (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. [DOI] [PubMed] [Google Scholar]
- 3. O'Neill L. A. (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18. [DOI] [PubMed] [Google Scholar]
- 4. Jenkins K. A., Mansell A. (2010) TIR-containing adaptors in Toll-like receptor signalling. Cytokine 49, 237–244. [DOI] [PubMed] [Google Scholar]
- 5. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364. [DOI] [PubMed] [Google Scholar]
- 6. Ve T., Gay N. J., Mansell A., Kobe B., Kellie S. (2012) Adaptors in Toll-like receptor signaling and their potential as therapeutic targets. Curr. Drug Targets 13, 1360–1374. [DOI] [PubMed] [Google Scholar]
- 7. Kawai T., Akira S. (2007) TLR signaling. Semin. Immunol. 19, 24–32. [DOI] [PubMed] [Google Scholar]
- 8. Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., McMurray D., Smith D. E., Sims J. E., Bird T. A., O'Neill L. A. (2001) Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83. [DOI] [PubMed] [Google Scholar]
- 9. Horng T., Barton G. M., Flavell R. A., Medzhitov R. (2002) The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420, 329–333. [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto M., Sato S., Hemmi H., Sanjo H., Uematsu S., Kaisho T., Hoshino K., Takeuchi O., Kobayashi M., Fujita T., Takeda K., Akira S. (2002) Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420, 324–329. [DOI] [PubMed] [Google Scholar]
- 11. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto M., Sato S., Mori K., Hoshino K., Takeuchi O., Takeda K., Akira S. (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J. Immunol. 169, 6668–6672. [DOI] [PubMed] [Google Scholar]
- 13. Beutler B., Hoebe K., Du X., Janssen E., Georgel P., Tabeta K. (2003) Lps2 and signal transduction in sepsis: at the intersection of host responses to bacteria and viruses. Scand. J. Infect. Dis. 35, 563–567. [DOI] [PubMed] [Google Scholar]
- 14. Carty M., Goodbody R., Schroder M., Stack J., Moynagh P. N., Bowie A. G. (2006) The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 7, 1074–1081. [DOI] [PubMed] [Google Scholar]
- 15. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rowe D. C., McGettrick A. F., Latz E., Monks B. G., Gay N. J., Yamamoto M., Akira S., O'Neill L. A., Fitzgerald K. A., Golenbock D. T. (2006) The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc. Natl. Acad. Sci. USA 103, 6299–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mansell A., Brint E., Gould J. A., O'Neill L. A., Hertzog P. J. (2004) Mal interacts with tumor necrosis factor receptor-associated factor (TRAF)-6 to mediate NF-κB activation by Toll-like receptor (TLR)-2 and TLR4. J. Biol. Chem. 279, 37227–37230. [DOI] [PubMed] [Google Scholar]
- 18. Verstak B., Nagpal K., Bottomley S. P., Golenbock D. T., Hertzog P. J., Mansell A. (2009) MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-κB proinflammatory responses. J. Biol. Chem. 284, 24192–24203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato S., Sugiyama M., Yamamoto M., Watanabe Y., Kawai T., Takeda K., Akira S. (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κ B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171, 4304–4310. [DOI] [PubMed] [Google Scholar]
- 20. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirschfeld M., Ma Y., Weis J. H., Vogel S. N., Weis J. J. (2000) Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J. Immunol. 165, 618–622. [DOI] [PubMed] [Google Scholar]
- 22. Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., Fujita T. (1998) Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15. [DOI] [PubMed] [Google Scholar]
- 24. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 25. Mansell A., Smith R., Doyle S. L., Gray P., Fenner J. E., Crack P. J., Nicholson S. E., Hilton D. J., O'Neill L. A., Hertzog P. J. (2006) Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol. 7, 148–155. [DOI] [PubMed] [Google Scholar]
- 26. Ye H., Arron J. R., Lamothe B., Cirilli M., Kobayashi T., Shevde N. K., Segal D., Dzivenu O. K., Vologodskaia M., Yim M., Du K., Singh S., Pike J. W., Darnay B. G., Choi Y., Wu H. (2002) Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447. [DOI] [PubMed] [Google Scholar]
- 27. McGettrick A. F., Brint E. K., Palsson-McDermott E. M., Rowe D. C., Golenbock D. T., Gay N. J., Fitzgerald K. A., O'Neill L. A. (2006) TRIF-related adapter molecule is phosphorylated by PKC{epsilon} during Toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. USA 103, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palsson-McDermott E. M., Doyle S. L., McGettrick A. F., Hardy M., Husebye H., Banahan K., Gong M., Golenbock D., Espevik T., O'Neill L. A. (2009) TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat. Immunol. 10, 579–586. [DOI] [PubMed] [Google Scholar]
- 29. Lin R., Heylbroeck C., Pitha P. M., Hiscott J. (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wietek C., Miggin S. M., Jefferies C. A., O'Neill L. A. (2003) Interferon regulatory factor-3-mediated activation of the interferon-sensitive response element by Toll-like receptor (TLR) 4 but not TLR3 requires the p65 subunit of NF-κ. J. Biol. Chem. 278, 50923–50931. [DOI] [PubMed] [Google Scholar]
- 31. Gohda J., Matsumura T., Inoue J. (2004) Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not Toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signaling. J. Immunol. 173, 2913–2917. [DOI] [PubMed] [Google Scholar]
- 32. Hacker H., Redecke V., Blagoev B., Kratchmarova I., Hsu L. C., Wang G. G., Kamps M. P., Raz E., Wagner H., Hacker G., Mann M., Karin M. (2006) Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207. [DOI] [PubMed] [Google Scholar]
- 33. Lin Z., Lu J., Zhou W., Shen Y. (2012) Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS One 7, e34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valkov E., Stamp A., Dimaio F., Baker D., Verstak B., Roversi P., Kellie S., Sweet M. J., Mansell A., Gay N. J., Martin J. L., Kobe B. (2011) Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. USA 108, 14879–14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., Thanos D. (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell 103, 667–678. [DOI] [PubMed] [Google Scholar]
- 37. Thanos D., Maniatis T. (1995) Virus induction of human IFN β gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- 38. Clark A. R., Dean J. L., Saklatvala J. (2003) Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 546, 37–44. [DOI] [PubMed] [Google Scholar]
- 39. Saklatvala J., Dean J., Clark A. (2003) Control of the expression of inflammatory response genes. Biochem. Soc. Symp. 70, 95–106. [DOI] [PubMed] [Google Scholar]
- 40. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301, 640–643. [DOI] [PubMed] [Google Scholar]
- 41. Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. (2003) TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4, 1144–1150. [DOI] [PubMed] [Google Scholar]
- 42. Wang L., Trebicka E., Fu Y., Waggoner L., Akira S., Fitzgerald K. A., Kagan J. C., Cherayil B. J. (2011) Regulation of lipopolysaccharide-induced translation of tumor necrosis factor-α by the Toll-like receptor 4 adaptor protein TRAM. J. Innate Immun. 3, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]