

Abstract
Objective
Placenta growth factor (PLGF), a vascular endothelial growth factor-A (VEGF-A) related protein, mediates collateral enlargement via monocytes but plays little role in capillary proliferation. In contrast, VEGF-A mediates both collateral enlargement and capillary proliferation. PLGF has been less thoroughly studied than VEGF-A, and questions remain regarding its regulation and function. Therefore, our goal was to characterize the expression of PLGF by vascular cells. We hypothesized that vascular smooth muscle cells (SMC) would express more PLGF than EC, since VEGF-A is primarily expressed by non-EC.
Methods
We compared PLGF and VEGF-A across 8 EC and SMC lines, then knocked down PLGF and evaluated cell function. We also assessed the effect of hypoxia on PLGF expression and promoter activity.
Results
PLGF was most highly expressed in EC, whereas VEGF-A was most highly expressed in SMC. PLGF knockdown did not affect EC number, migration, or tube formation, but reduced monocyte migration towards EC. Monocyte migration was rescued by exogenous PLGF. Hypoxia increased PLGF protein without activating PLGF gene transcription.
Conclusions
PLGF and VEGF-A have distinct patterns of expression in vascular cells. EC derived PLGF may function primarily in communication between EC and circulating cells. Hypoxia increases EC PLGF expression post-transcriptionally.
Keywords: Growth factors, endothelial cells, vascular smooth muscle cells, arteriogenesis
Introduction
In the setting of ischemic cardiovascular diseases such as coronary artery disease and peripheral artery disease, collateral artery enlargement (arteriogenesis) can rescue distal tissue at risk of ischemia by providing an alternate route for blood flow around vascular occlusions. Placenta growth factor, a vascular endothelial growth factor family protein, is a key mediator of arteriogenesis. Exogenous PLGF induces arteriogenesis in ischemic skeletal muscle [25] and skin [29]. Micro-CT studies have shown that PLGF selectively increases the volume of 96–136 μm-diameter vessels in ischemic skeletal muscle [23]. Studies in PLGF−/− mice have suggested that PLGF is required for arteriogenesis in the setting of hindlimb ischemia, but is not essential for embryonic vasculogenesis [7]. Likewise, PLGF appears to play little role in angiogenesis (capillary proliferation). However, PLGF appears to augment the angiogenic effect of VEGF-A [2, 25]. These findings have led to the suggestion that PLGF may function as a “master switch” for arteriogenesis [11].
The specificity of PLGF signaling for arteriogenesis contrasts with the combined vasculogenic/angiogenic/arteriogenic effect of VEGF-A. The prominent effects of VEGF-A on vascular development and capillary proliferation are well recognized. VEGF-A is also able to induce collateral enlargement, and has been shown to be essential for arteriogenesis in the setting of repetitive coronary occlusion [43]. Interestingly, however, studies in rodent ischemic hindlimb have found that PLGF is more effective than VEGF-A at increasing the number of collateral side branches and the total collateral perfusion area [25], and that PLGF improves angioscore and collateral conductance to a greater extent than VEGF-A [31]. Collateral enlargement and blood flow recovery are significantly delayed in PLGF knockout mice [37], suggesting that PLGF plays a key role in early stages of arteriogenesis. Indeed, PLGF expression is upregulated in rat hindlimb collaterals immediately following femoral artery occlusion [32]. These observations of the relative role of VEGF-A and PLGF in arteriogenesis may reflect differences in the signaling pathways activated in the coronary model (in which remodeling collaterals are located in/near hypoxic tissue) and the hindlimb ischemia model (in which remodeling collaterals are distant from the site of hypoxia). Nevertheless, it is clear that the primarily arteriogenic effect of PLGF differs from the mixed angiogenic/arteriogenic effect of VEGF-A, and that these two factors likely operate in concert during active remodeling. PLGF is therefore a promising target for arteriogenic therapy. However, although PLGF was described soon after VEGF-A [27], it has not been studied to the same extent. Therefore, many questions regarding its regulation and function remain.
Capillary proliferation is driven primarily by tissue hypoxia. Hypoxia may also influence collateral growth. Remodeling collaterals can be located near or within hypoxic tissues. There is also evidence to suggest that hypoxia can influence arteriogenesis even when present in regions distant from the site of collateral growth, although the mechanism remains to be defined [46]. Although hypoxia may contribute to both angiogenesis and arteriogenesis, it is generally well accepted that a key difference between capillary proliferation and arteriogenesis lies in the additional influence of hemodynamic stimuli such as shear stress on arteriogenesis. [15, 30, 34–36]. Thus, although there is a large overlap in signaling between angiogenesis and arteriogenesis, there must necessarily be some distinct mechanisms between the two processes.
Given the only partially overlapping roles of PLGF and VEGF-A in vascular remodeling, it seems likely that the regulatory mechanisms controlling PLGF and VEGF-A expression have at least some unique features. Downstream signaling events induced by PLGF and VEGF-A are also not identical, as PLGF and VEGF-A have differing receptor binding specificities. Whereas VEGF-A binds to VEGFR-1 and VEGFR-2, PLGF binds to VEGFR-1 only [3, 9]. PLGF and VEGF-A are dimers in vivo and the existence of PLGF/VEGF heterodimers has been reported [13]. VEGFR-1 and VEGFR-2 can also heterodimerize upon ligand binding, and their tyrosine phosphorylation patterns and subsequent downstream signaling events can vary depending on the identity of the ligand (PLGF homodimer, VEGF-A homodimer, or PLGF/VEGF heterodimer) [26]. Thus, PLGF is expected to influence VEGF-A signaling and vice versa.
PLGF is non-mitogenic for endothelial cells, in contrast to VEGF-A [7]. Rather, PLGF stimulates arteriogenesis via a monocyte-dependent mechanism. Monocytes express VEGFR-1 but not VEGFR-2 and respond to PLGF with chemotaxis [3, 9, 31, 42]. Migration of monocytes into the arterial wall is a key component of arteriogenesis [1, 4, 20, 21, 38].
The expression of PLGF by adult vascular cells has not been systematically characterized. Thus, the goal of this study was to determine whether the expression pattern of PLGF by endothelial cells and smooth muscle cells is similar to the expression pattern of VEGF-A. Given that the role of PLGF in arteriogenesis appears to be mediated through monocytes, we hypothesized that SMC would be the primary vascular cell type expressing PLGF, which would facilitate monocyte migration into the vascular wall. To test this hypothesis, we compared the expression of PLGF and VEGF-A in eight different EC and SMC lines. We then performed functional studies to determine whether endogenous PLGF has a critical role in vascular cell function. Finally, we assessed whether PLGF expression in EC is influenced by hypoxia. These studies expand our knowledge of PLGF biology and function and suggest important questions for further research.
Methods
Established cell lines
Vascular smooth muscle cells (A10), endothelial cells (EOMA), and monocytes/macrophages (U937) were purchased from American Type Culture Collection (Manassas, VA). A10 and EOMA cells were grown in DMEM (Invitrogen, Carlsbad, CA). U937 cells were cultured in RPMI 1640 and were maintained at 1 × 105–2 × 106 cells/mL. All cells were grown in a humidified incubator (5% CO2) with added penicillin-streptomycin (1%) and FBS (10%, Invitrogen).
Primary human cells
HCASMC, HLMVEC, and HCAEC were purchased from Lonza (Walkersville, MD). HUVEC were purchased from ScienCell (Carlsbad, CA). HCASMC were grown in SMGM-2 (Lonza). HLMVEC and HCAEC were grown in EGM-2MV (Lonza). HUVEC were grown in EGM-2 (Lonza).
Primary porcine cells
Hearts were obtained from a local packing plant (Ralph’s Meats, Perkins, OK) after slaughter and stored in physiological saline solution on ice until use. Coronary arteries were dissected and cleaned of adventitia and surface fat, then dipped briefly in 70% ethanol and rinsed in cold, sterile phosphate-buffered saline (PBS). PCASMC were isolated by enzymatic dissociation. The dissociation solution was prepared in HBSS containing isoproterenol (10 μM), amino acid standard (1.3%), DNase I type IV (60 U/mL), bovine serum albumin (1.5%), trypsin inhibitor (0.1%), Mg-ATP (4 mM), elastase (Calbiochem, 1 U/mL), collagenase (Worthington, 500 U/mL), CaCl2 (0.5 mM), and MgSO4 (1.16 mM). Dissociation solution was syringe-filtered before use. Arteries were cut into ~1 cm segments, opened longitudinally, and pinned lumen side up in glass vials. Dissociation solution was added and the vials placed in a shaking water bath at 37°C for 45–60 min. The EC layer was removed by forcefully rinsing the tissue with a pipettor. This solution was discarded and the vessel was scraped lightly with a sterile instrument to remove any remaining EC, then rinsed with HBSS. Fresh dissociation solution was added and the tissue incubated for 30–45 min at 37°C with shaking. PCASMC were dissociated as described above for EC. The resulting cell suspension was centrifuged at 900 rpm for 3 min to pellet cells. The supernatant was removed and the cells resuspended in HBSS. PCASMC were plated in standard culture vessels and grown in DMEM + 1% penicillin-streptomycin + 5% FBS until ready for use.
RT-PCR
Cell culture medium was aspirated and the cells were rinsed briefly in Dulbecco’s PBS (Invitrogen). Total RNA was extracted using Trizol (Invitrogen) and treated to remove genomic DNA (Turbo DNAFree, Ambion, Austin, TX). Total RNA was analyzed spectrophotometrically to assess quantity and purity. RNA was reverse transcribed to cDNA using qScript cDNA SuperMix (Quanta BioSciences, Gaithersburg, MD). Real-time quantitative RT-PCR was used to determine mRNA expression of the target genes in an ABI 7500 Fast instrument (Applied Biosystems) using PerfeCTa SYBR Green FastMix, Low ROX (Quanta BioSciences). Primers were designed using Primer Express software and custom-synthesized by Invitrogen. Primer sequences were as follows: human PLGF forward 5′-CCTACGTGGAGCTGACGTTCT-3′; human PLGF reverse 5′-TCCTTTCCGGCTTCA TCTTCT-3′; human VEGF-A forward 5′-ACGAGGGCCTGGAGTGTGT-3′; human VEGF-A reverse 5′-GATCCGCATAATCTGCATGGT-3′; mouse and rat PLGF forward 5′-CTGCTGGGAACAACTCAACAGA-3′; mouse PLGF reverse 5′-GCGACCCCACACTTCGTT-3′; rat PLGF reverse 5′-GCGGCCCCACACTTCATT-3′; mouse VEGF-A forward, 5′-CCCTGGCTTTACTGCTGTACCT-3′; mouse VEGF-A reverse, 5′-CTTGATCACTTCATGGGACTTCTG-3′; rat VEGF-A forward, 5′-TTCAAGCCGTCCTGTGTGC-3′; rat VEGF-A reverse, 5′-TCCAGGGCTTCATCATTGC-3′; pig PLGF forward, 5′-GGAGACGGTCAATGTCACCAT-3′; pig PLGF reverse, 5′-GAGAATGTCAGCTCCACGTAG-3′; pig VEGF-A forward, 5′-CATGCAGATTATGCGGATCAA-3′; pig VEGF-A reverse, 5′-TTTGTTGTGCTGTAGGAAGCT-3′; rodent β-actin forward, 5′-AGTTCGCCATGGATGACGAT-3′; rodent β-actin reverse, 5′-TGCCGGAGCCGTTGTC-3′; human β-actin forward, 5′-TGCCGACAGGATGCAGAAG-3′; human β-actin reverse, 5′-CTCAGGAGGAGCAATGATCTTGAT-3′; pig β-actin forward, 5′-CTCTTCCAGCCCTCCTTCCT-3′; pig β-actin reverse, 5′-CGACGTCGCACTTCATGATG-3′. PLGF and VEGF-A mRNA expression was normalized to β-actin and relative gene expression was quantified using the ΔΔCt method.
ELISA
Medium was collected 3 days after cells reached 95–100% confluency for measurement of secreted PLGF or VEGF-A protein. Medium was concentrated using filters (Icon, 7mL/9K, Pierce). Protease inhibitor cocktail (Halt, Pierce) was added (1:100) to protect proteins from degradation. ELISA was performed using the Quantikine Human PLGF, VEGF-A, and VEGF/PLGF heterodimer ELISA kits (R&D Systems, Minneapolis, MN). ELISA results were normalized to total protein in the concentrated medium as determined by BCA assay (Pierce).
siRNA transfection of HCAEC and HCASMC
Predesigned double-stranded 21-mer siRNA corresponding to PLGF mRNA and a negative control siRNA (silencer no.1 siRNA; scRNA) were purchased from Invitrogen. PLGF siRNA had the following sequences: sense, 5′-AGGUGGAAGUGGUACCCUU-3′, overhang dTdT; antisense, 5′-AAGGGUACCACUUCCACCU-3′, overhang, dCdT. HCAEC and HCASMC were plated 24 h before transfection in 6- and 96-well plates. The cultures were incubated for 6 h with 5 nM siRNA precomplexed in Opti-MEM medium (Invitrogen) with lipofectamine™ RNAiMAX transfection reagent (Invitrogen) according to manufacturer’s protocol. After incubation, the medium was replaced by complete medium, and cells were cultivated under standard conditions for another 18 h, followed by RT-PCR or functional assays.
Viability assay
HCAEC and HCASMC were seeded into black 96-well plates with clear bottoms (Corning) at 7000 cells/well and grown under standard conditions for 1 d. On the second day, cells were ~30–50% confluent. Cells were then transfected with siPLGF or scRNA as described above. Alamar Blue (10%; Invitrogen) was added to each well 24 h later, and the plate was returned to the incubator for 3 h.
Fluorescence was read in a Bio-Tek Synergy HT multimode plate reader (Winooski, VT; ex 570 nm, em 585 nm). Background fluorescence was calculated from cell-free wells and subtracted from experimental values. A standard curve was created by plating known amounts of cells (2,000–14,000) and was used to calculate the number of cells in each experimental well.
Migration assay
HCAEC were cultured and transfected with siPLGF or scRNA as described above. Six hours after transfection, cells were serum-starved for 24 h in 2% serum media made by diluting EGM-2MV medium (5% FBS) with serum-free DMEM. Then, 1 × 105 cells were seeded into each insert of a BD Falcon FluoroBlok endothelial migration plate (BD Biosciences). Recombinant human VEGF165 (100 _g/mL in DPBS with 0.1% BSA, R&D Systems) was added to the bottom well to serve as a chemoattractant for EC. The final concentrations of VEGF were 0.8, 10 and 50 ng/mL. Plates were then returned to the incubator for 22 h. The medium was removed, and the inserts were transferred to a second plate containing 5 μg/ml calcein AM (BD Bioscences) in HBSS per well. The plate was incubated for 90 min, and migrated cells were detected by measuring fluorescence at ex 494 nm/em 517 nm.
Tube formation assay
HCAEC were cultured in T-25 flasks till 50% confluent, then transfected with siPLGF or scRNA. After 24 h, cells were starved with 2% serum medium for 24 h. Geltrex reduced growth factor basement membrane matrix (Invitrogen) was added to a 96-well plate at 100 μL per cm2 and allowed to polymerize for 30 min at 37°C. HCAEC were trypsinized and seeded in the matrix-coated plate at 1.6 × 104 cells/well. After 6 h, cells were imaged under a phase-contrast inverted microscope with a digital camera.
Monocyte migration assay
The human histocytic lymphoma cell line U937 was used to test the effect of PLGF knockdown on monocyte migration. This cell line displays characteristics typical of immature monocytes [19, 41] and has been demonstrated to mainly differentiate along the monocyte/macrophage pathway[5]. HCAEC were seeded into 12 well plates (85,000 cells/well). After 24 h, cells were transfected with either PLGF siRNA or negative control siRNA as described above. Following transfection, media was replaced with phenol red free medium containing reduced concentrations of added growth factors (2%) and cells were allowed to recover for 18 h before assessing monocyte migration. Migration of U937 cells towards HCAEC was assayed using Transwell inserts (Corning Costar, 3 μm pore size). U937 cells were suspended in 2% serum media at a density of 1 × 106 cells/mL and labeled by incubation with calcein AM (Invitrogen, 8 μM) for 1 h. After labeling, cells were resuspended in fresh medium at a density of 2 × 106 cells/mL. Transwell inserts were placed in wells containing HCAEC treated with either scRNA or PLGF siRNA, and 200 μl of the labeled U937 cell suspension was added. Recombinant human PLGF (ab174025, Abcam; 500 pg/mL) was added to an additional set of wells containing PLGF siRNA treated EC. Cell migration was determined 2 h after addition of monocytes by collecting 100 μL of medium from the lower chamber of each well and reading the fluorescence intensity at 485nm/528nm (Bio-Tek, Synergy HT).
Hypoxia
To mimic the effects of hypoxia on gene transcription, HCAEC were treated with the HIF-1α inducer cobalt chloride (CoCl2, 100 μM) or were exposed to 1% O2 in a cell culture chamber (Stem Cell Technologies) for up to 24 h.
Plasmids
To assess PLGF promoter activity, we utilized a firefly luciferase pGL3 basic plasmid (Promega) containing the PLGF promoter sequence inserted at the Sst1 restriction site (SRI International, Menlo Park, CA) [18]. A Renilla luciferase plasmid (pRL, Promega) was used as the transfection efficiency control. A 50:1 ratio of firefly luciferase plasmid to Renilla luciferase plasmid was used, as recommended by the manufacturer. HCAEC (passage 5; 5 × 105 cells) were co-transfected with 2 μg of PLGF-luc and 40 ng pRL using the Amaxa Nucleofector System, program S-005 (Lonza) as we previously described [39].
Statistical analyses
All data are presented as mean ± SEM. Experiments were replicated at least three times and the results were averaged. Data which were normally distributed were analyzed by ANOVA, followed by post-hoc testing. Non-normally distributed data were analyzed by Mann-Whitney rank sum test (for two groups) or ANOVA on ranks (for multiple groups) followed by post-hoc testing. Differences were considered to be significant at p<0.05.
Results
Expression of PLGF and VEGF-A varies strikingly between EC and SMC
PLGF mRNA expression varied over a wide range across the EC and SMC lines tested, with the lowest expression found in the A10 SMC line and the highest expression found in the EOMA EC line (Fig. 1A). A clear cell-type-specific pattern of expression was found, with the 4 SMC lines tested expressing less PLGF than the 4 EC lines (p<0.001). PLGF mRNA expression in primary adult human cells was similar to that in the established cell lines and porcine primary cells. Thus, human primary adult EC and SMC were used for all further analyses, as a more physiologically relevant model. ELISA analysis of secreted PLGF protein in conditioned medium from the 5 human primary cell lines tested showed that PLGF protein levels were much lower in SMC-conditioned medium than in EC-conditioned medium (Fig. 1B, p<0.01), consistent with the mRNA results. PLGF mRNA and protein levels were well-correlated across the five human cell lines and were best fit with an exponential curve (Fig. 1C, r2=0.96).
Figure 1. PLGF gene and protein expression in EC and SMC.

Analysis of PLGF mRNA across a total of eight SMC and EC lines demonstrated that PLGF is more highly expressed in EC (black bars) than SMC (gray bars) (A, p<0.001). Measurement of PLGF protein in medium conditioned by the five primary human cell lines studied confirmed high PLGF expression by EC (B, p<0.01). Exogenous PLGF protein was not detected in culture medium in amounts significant enough to interfere with analysis. PLGF mRNA and protein were well-correlated in the human cell lines (C, r2=0.96). All measurements were repeated in at least 3 separate experiments.
VEGF-A mRNA expression also varied across the cell lines tested in a cell-type-specific manner, but over a narrower range than PLGF, and with the opposite expression pattern. The highest VEGF-A expression was found in the SMC lines, and the lowest expression in the EC lines (Fig. 2A; p<0.001). Analysis of VEGF-A protein in EC-conditioned culture medium was confounded both by the presence of exogenous VEGF-A in the medium and by the fact that exogenous VEGF-A was consumed by EC. Nevertheless, these data confirmed the high expression of VEGF-A by SMC (Fig. 2B, p<0.001). Disappearance of VEGF-A protein from EC-conditioned medium was determined to be an active, cell-mediated process by incubating the same media formulation without cells. A similar decrease in VEGF-A levels in medium did not occur in the absence of EC (data not shown).
Figure 2. VEGF-A gene and protein expression in EC and SMC.

Analysis of VEGF-A mRNA across a total of eight SMC and EC lines demonstrated that VEGF-A is more highly expressed in SMC (gray bars) than EC (black bars) (A, p<0.001). Measurement of VEGF-A protein in medium conditioned by the five primary human cell lines studied essentially confirmed the mRNA results, although measurement of VEGF-A protein in EC culture medium was confounded by presence of exogenous VEGF-A in culture medium (open bars) and also by utilization of VEGF-A by EC (B, p<0.001).
PLGF/VEGF heterodimer protein expression varies unpredictably
Since both EC and SMC expressed both PLGF and VEGF-A, although in different proportions, we next analyzed production of the PLGF/VEGF heterodimer protein to determine whether heterodimer production varied between EC and SMC. Somewhat surprisingly, PLGF/VEGF heterodimer levels in culture medium did not show a clear pattern and were not readily predictable based on the mRNA or protein data for PLGF or VEGF-A (Fig. 3, p=NS).
Figure 3. PLGF/VEGF heterodimer protein expression in EC and SMC.

Analysis of PLGF/VEGF heterodimer protein in medium conditioned by the five primary human cell lines did not reveal a clear pattern of expression that was readily predictable from the results for PLGF or VEGF-A alone (P=NS). Minor amounts of heterodimer protein were detected in both EC and SMC culture medium (open bars).
Relative expression of PLGF and VEGF-A differs between EC and SMC
Examination of the relative difference in PLGF and VEGF-A expression within each EC and SMC line demonstrated that VEGF-A mRNA levels were ~1–3 orders of magnitude higher than PLGF mRNA levels in SMC, whereas PLGF mRNA levels were ~1 order of magnitude higher than VEGF-A mRNA levels in EC (Fig. 4, p<0.001). As described above, ELISA analysis of VEGF-A in EC was confounded by exogenous VEGF-A and by EC VEGF-A uptake, and thus the ratio of PLGF and VEGF-A protein was not calculated.
Figure 4. Relative gene expression of PLGF and VEGF-A in EC and SMC.

Comparison of the relative expression levels for PLGF and VEGF-A in the eight cell lines examined demonstrated that VEGF-A mRNA is ~1–3 orders of magnitude higher than PLGF in SMC (gray bars), whereas PLGF mRNA is ~1 order of magnitude higher than VEGF-A mRNA in EC (black bars) (p<0.001).
Functional significance of high PLGF expression in adult EC
PLGF has previously been shown to be non-mitogenic for EC [7]. Thus, we next investigated whether the high PLGF expression in EC had functional significance for EC survival, migration, or tube formation. PLGF mRNA was knocked down in HCAEC using siRNA (Fig. 5A, p<0.05). ELISA analysis confirmed that PLGF protein was reduced to ~10% of control 24 h post-siRNA treatment (Fig. 5B, p<0.001). PLGF gene knockdown did not significantly affect VEGF-A gene expression (Fig. 5C, P=NS).
Figure 5. siRNA knockdown of PLGF gene and protein expression.
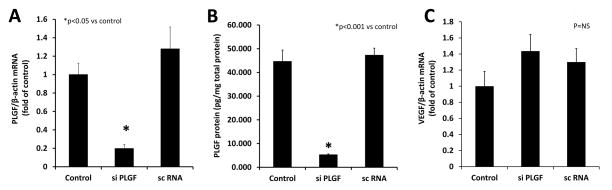
A. Treatment of primary human coronary artery endothelial cells (HCAEC) with siRNA effectively knocked down PLGF gene expression (p<0.05). B. PLGF protein level was also decreased following siRNA treatment (p<0.001). A control siRNA did not affect either PLGF mRNA or protein expression. C. VEGF-A mRNA levels were not significantly affected by knockdown of PLGF mRNA (p=NS).
To determine whether PLGF affects the balance between survival/proliferation and apoptosis/necrosis in EC, we assessed the effect of PLGF knockdown on HCAEC cell number. HCAEC treated with siPLGF showed no change in cell number (Fig. 6A, p=NS). Likewise, knockdown of PLGF expression did not affect HCASMC cell number (Fig. 6B, p=NS). We next assessed migration of HCAEC in response to VEGF-A to determine if endogenous PLGF production by EC modulates the migratory response of EC to VEGF-A. Knockdown of PLGF had no significant effect on migration of HCAEC towards VEGF-A (Fig. 7, p=NS). We also assessed the ability of HCAEC to form tubes in an extracellular matrix material following PLGF knockdown. Tube formation did not appear to be affected by siPLGF treatment (Fig. 8).
Figure 6. Effect of PLGF siRNA treatment on cell number of HCAEC and HCASMC.

Knockdown of PLGF gene expression did not significantly affect cell number of either primary human coronary artery endothelial cells (HCAEC, A) or primary human coronary artery smooth muscle cells (HCASMC, B) (p=NS).
Figure 7. Effect of PLGF siRNA treatment on migration of HCAEC.

Knockdown of PLGF gene expression did not significantly affect migration of HCAEC towards VEGF-A (p=NS). Migration was assessed using the BD Falcon FluoroBlok endothelial migration assay at VEGF-A concentrations of 0.8, 10, and 50 ng/mL.
Figure 8. Effect of PLGF siRNA treatment on tube formation by HCAEC.

Knockdown of PLGF gene expression had no apparent effect on the ability of HCAEC to form tubes in GelTrex extracellular matrix material.
Since high basal PLGF expression did not appear to be critical for EC survival, migration, or tube formation, we next tested the effect of PLGF knockdown on monocyte migration towards HCAEC, to determine whether high EC PLGF expression may be important in paracrine communication. PLGF knockdown significantly reduced migration of U937 monocytes towards HCAEC (Fig. 9). The reduced migration was rescued by exogenous PLGF at concentrations equivalent to those found in HCAEC conditioned medium (500 pg/mL).
Figure 9. Effect of PLGF siRNA treatment on migration of monocytes towards HCAEC.

Knockdown of PLGF gene expression significantly reduced migration of U937 monocytes towards HCAEC (p<0.05). Inhibition of monocyte migration towards HCAEC was rescued by exogenous PLGF (500 pg/mL).
Although shear stress is an important stimulus for arteriogenesis, collateral growth can occur within hypoxic tissue. Thus we sought to determine whether hypoxia can modulate EC PLGF expression. HCAEC were treated with the HIF-1α inducer CoCl2 to mimic the effects of hypoxia. CoCl2 treatment was able to stimulate hypoxia-inducible gene expression, as confirmed by upregulation of VEGF-A mRNA within 4 h of treatment as a positive control (Fig. 10A). PLGF protein was upregulated by CoCl2, although the increase was not evident until 22 h post-treatment (Fig. 10B). Exposure of HCAEC to 1% O2 increased PLGF protein expression similarly to CoCl2 (Fig. 10C). Activation of hypoxia-inducible gene expression by 1% O2 was confirmed by upregulation of VEGF-A mRNA; however, PLGF mRNA was not significantly affected by 1% O2 (relative PLGF mRNA in 1% O2 exposed HCAEC, fold of control: 4 h, 1.11 ± 0.10; 8 h, 1.46 ± 0.08; 24 h, 1.00 ± 0.21). Analysis of PLGF promoter activity showed that the CoCl2-induced increase in PLGF protein was not due to increased transcription of PLGF mRNA (Fig. 10D).
Figure 10. Effect of hypoxia on PLGF expression by HCAEC.

Treatment of HCAEC with CoCl2 (100 μM) upregulated VEGF-A gene expression as a positive control for hypoxia-inducible gene expression (A, p<0.05). PLGF protein was increased by CoCl2 treatment at the 22 h time point (B, p<0.05). PLGF protein was similarly increased by 1% O2 (C, p<0.05). However, PLGF gene transcription was not activated by CoCl2 (D, p=NS).
Discussion
Comparison of PLGF and VEGF-A gene and protein expression across a variety of EC and SMC lines revealed that adult EC express high levels of PLGF. PLGF mRNA and protein levels were generally well correlated. In contrast, VEGF-A was more highly expressed by SMC than EC, in agreement with previously reported results showing that VEGF-A is primarily expressed by non-EC, and is secreted to act on EC in a paracrine manner [28, 40]. Indeed, VEGF-A protein disappeared from EC culture media over time, suggesting active uptake and/or degradation of VEGF-A by EC. This observation is in agreement with previous reports showing that EC take up VEGF-A both by receptor-mediated endocytosis and by an alternate pathway resulting in nuclear accumulation [22, 33, 44].
Interestingly, PLGF expression was lower in human lung microvascular endothelial cells than in the other EC lines examined. PLGF levels in HLMVEC were similar to levels in HCASMC, suggesting that EC PLGF expression levels can overlap with SMC PLGF expression at the lower end of the EC range, depending on the vascular bed from which the cells are derived. Although the number of cell lines examined is not sufficient to draw firm conclusions, these data suggest the possibility that microvascular EC and conduit EC may differ in their PLGF expression. Whether systematic differences in microvascular vs conduit EC PLGF expression exist, and whether such differences are physiologically significant are important questions for further study.
The mechanism underlying differential expression of PLGF and VEGF-A in vascular cells remains to be determined. The VEGF-A and PLGF promoter regions share several common response elements, including binding sites for NF-κB, HIF-1α, and Sp1 [10, 12, 18]. However, despite common elements, these genes are differentially regulated. Although HIF-1α is a well-known regulator of VEGF-A expression, it has been reported to have no effect on transcription at the PLGF promoter in placental cells [17]. This is consistent with our observation that exposure of EC to hypoxia (1% O2) upregulates VEGF-A mRNA, but not PLGF mRNA. Differential PLGF expression may also be due to activation of specific factors which drive PLGF, but not VEGF-A, expression. For example, GCM-1 has been reported to mediate high constitutive expression of PLGF in trophoblast cells, as compared to non-trophoblast cells [8]. BF-2/FoxD1 activates the PLGF promoter during development of the kidney, but is not required for VEGF-A expression in this organ [47]. The transcription factor MTF-1 has also been identified as an important regulator of PLGF expression [10, 18]. Thus, the differential basal expression patterns of VEGF and PLGF in EC versus SMC may reflect both differing responses to common transcription factors and the activation of transcriptional elements promoting differential expression. This complex regulatory pathway warrants further investigation.
PLGF and VEGF-A have the ability to form heterodimers in vivo with altered signaling properties, compared to PLGF or VEGF-A homodimers [6, 13]. Thus, we also assessed the levels of the heterodimer protein in medium from EC and SMC. We expected that heterodimer levels would exhibit a relationship to the levels of PLGF and VEGF-A expressed by the cells. With the exception of HUVEC, however, PLGF/VEGF heterodimer levels were similar between EC and SMC and no consistent pattern could be identified. Further research is needed to determine how PLGF/VEGF heterodimers are formed and regulated.
Primary human SMC and EC were grown in culture media containing supplemental growth factors for studies of the relative expression of PLGF and VEGF-A in SMC and EC, raising the possibility that growth factors (including VEGF-A) present in the media could have affected PLGF and/or VEGF-A expression levels. This was an unavoidable limitation of the study, as in our experience, human primary EC do not survive when cultured without supplemental growth factors. Fujii and coauthors recently reported that exogenous VEGF-A can upregulate PLGF expression in HUVEC and in HPAEC [16]. However, the striking cell-type-specific pattern of PLGF expression observed in the present study cannot be attributed solely to the presence of exogenous VEGF-A in EC culture media. In the study of Fujii et al., treatment of HUVEC and HPAEC with 10 ng/mL VEGF-A resulted in a moderate (~1.5–2 fold) increase in PLGF protein [16]. Culture media used in the present study contained <4 ng/mL VEGF-A according to our analysis (data not shown). If we assume that the effect of VEGF-A on PLGF expression is linear between 4 and 10 ng/mL, then we might reasonably attribute a 0.8-fold increase in PLGF expression to the presence of VEGF-A in EC culture medium. It is evident from Fig. 1 that the striking differences between EC and SMC PLGF expression levels would still be present even if EC expression was decreased by 0.8-fold. Finally, human neonatal dermal microvascular EC have previously been reported to express much higher levels of PLGF mRNA than VEGF mRNA [45], in agreement with our results. In the present study, we have extended this earlier finding from human neonatal EC to adult human EC from multiple vascular beds, and have confirmed that the relative differences in PLGF and VEGF-A mRNA expression in EC are maintained at the protein level.
The high expression of PLGF in the EC lines we examined prompted us to question whether PLGF might have a role in adult EC function that was not apparent in earlier reported studies. Although PLGF was initially reported to induce proliferation of an EC line [27], later studies failed to show a direct mitogenic effect of PLGF on either EC [7] or SMC [25]. Interestingly, however, the mitogenic effect of VEGF-A on both vascular cell types appears to require the presence of PLGF [7, 25]. Comparison of wild-type EC with EC isolated from PLGF−/− mice similarly suggested that PLGF itself does not induce EC migration, but is required for EC migration in response to VEGF-A [7]. We examined EC survival, EC migration towards VEGF-A, and the ability of EC to form tubes in extracellular matrix to determine whether acute knockdown of PLGF expression with siRNA could affect these processes. PLGF knockdown did not significantly affect survival of HCAEC. In contrast to the previous findings discussed above, we also did not note any impairment of migration towards VEGF-A following siRNA treatment. This disparity could be due to residual PLGF expression in the siRNA-treated cells, as PLGF protein levels were ~10% of control in the siRNA-treated cells. The ability of EC to form tubes in extracellular matrix material also appeared to be normal following siRNA knockdown of PLGF. These findings suggest that high basal expression of PLGF is not critical for EC function, and that low levels of PLGF are sufficient to maintain normal EC responses to VEGF-A. We also examined survival of HCASMC following PLGF knockdown to determine if PLGF had a pro-survival action in this cell type. Treatment of HCASMC with PLGF siRNA did not affect SMC survival.
Although we cannot entirely rule out a role for PLGF in supporting EC functions such as survival, migration, and tube formation, we conclude that the high level of PLGF expression and secretion by EC most likely mediates paracrine signaling between EC and other cell types. This conclusion is supported by our findings that PLGF knockdown reduces migration of monocytes towards EC, and that this reduction is rescued by exogenous PLGF. Circulating monocytes and other bone-marrow derived cells are known to express VEGFR-1 [9] and have been shown to be essential for PLGF-induced arteriogenesis [31, 37]. Our results suggest that altered PLGF expression by EC in disease may contribute to abnormal vascular growth and/or function by affecting monocyte migration into the vessel wall.
Lastly, to determine whether the high baseline expression of PLGF by EC is subject to regulation by stimuli associated with vascular remodeling, we exposed HCAEC to the hypoxia mimetic CoCl2 or to 1% O2. Previous reports of the effects of low O2 levels on PLGF expression by ECs are inconsistent. Du and coauthors reported increased PLGF expression in rat brain microvascular EC under glucose-free, anoxic conditions [14]. In contrast, Loboda et al reported a decrease in PLGF mRNA in human microvascular EC exposed to 1% O2 [24], while Yonekura et al reported no effect of 0–20% O2 on PLGF mRNA expression in human neonatal dermal microvascular EC [45]. VEGF-A mRNA expression was increased by CoCl2 and 1% O2, as a positive control for hypoxia-inducible gene expression. PLGF protein was also increased by CoCl2 and 1% O2. Hypoxia has previously been reported to increase PLGF promoter activity in fibroblasts via NF-κB and MTF-1 [10, 18]. However, we did not detect an increase in endothelial cell PLGF promoter activity in response to CoCl2 treatment, suggesting that the hypoxia-mediated increase in PLGF protein occurred via a post-transcriptional mechanism. Our results could thus be considered to agree with both Du (increased protein) and Yonekura (unchanged mRNA) and suggest that distinct mechanisms regulate PLGF expression in different cell types. Importantly, we previously reported that regulation of PLGF by reactive oxygen species appears to have an important post-transcriptional component [39]. Our results suggest that results of studies in which only PLGF mRNA is measured should be interpreted with caution.
Although the arteriogenic activity of PLGF has been well documented, there have been relatively few studies of the cell biology of this important growth factor, compared to VEGF-A. Thus, many gaps in our knowledge of the biology of PLGF remain to be filled. In these studies we described a cell-specific, differential pattern of expression of PLGF and VEGF-A in adult vascular cells. Although PLGF was originally described in placenta and its levels are increased during pregnancy, the consistently high level of PLGF expression that we found in adult EC demonstrates that PLGF expression is not restricted to the setting of pregnancy or neonatal life and likely has an important role in adult vascular biology. Our studies of the effects of acute PLGF knockdown on EC function and EC/monocyte interaction suggest that EC expression of PLGF is probably more important in paracrine signaling between EC and circulating cells than in autocrine support of EC function. PLGF levels and signaling may be abnormal in conditions in which the endothelium is damaged or dysfunctional. This work therefore provide a basis for further studies examining how EC PLGF expression and signaling is altered in disease states such as diabetes. Lastly, we demonstrate that EC PLGF expression is subject to post-transcriptional regulation by hypoxia, a key stimulus associated with vascular remodeling.
These studies lay the groundwork for future studies to characterize the distribution of PLGF and VEGF-A in healthy and diseased intact vessels and to determine the functional significance of vascular cell type specific PLGF expression. Further investigation into the basic biology of PLGF may suggest new possibilities for the treatment of ischemic cardiovascular disease and other conditions in which vascular growth and function is abnormal.
Perspectives.
Adult human endothelial cells and vascular smooth muscle cells display cell-type-specific patterns of PLGF and VEGF-A expression, with PLGF primarily expressed by EC and VEGF-A primarily expressed by SMC. Differential expression of these growth factors in the vessel wall may play an important role in regulation of vascular remodeling. Endothelial dysfunction may alter the balance of PLGF and VEGF-A in vessels and contribute to abnormal vascular function in disease.
Acknowledgments
This work was supported by a grant from the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH R01 HL-084494, PL).
List of Abbreviations
- BCA
Bicinchoninic acid
- DMEM
Dulbecco’s modified Eagle’s medium
- EC
Endothelial cells
- EGM-2
Endothelial cell growth medium
- EGM-2MV
Microvascular endothelial cell growth medium
- ELISA
Enzyme linked immunosorbent assay
- FBS
Fetal bovine serum
- HBSS
Hank’s balanced salt solution
- HCAEC
Human coronary artery endothelial cells
- HCASMC
Human coronary artery smooth muscle cells
- HLMVEC
Human lung microvascular endothelial cells
- HPAEC
Human pulmonary artery endothelial cells
- HUVEC
Human umbilical vein endothelial cells
- HUVSMC
Human umbilical vein smooth muscle cells
- PBS
Phosphate-buffered saline
- PCASMC
Pig coronary artery smooth muscle cells
- PLGF
Placenta growth factor
- scRNA
Scrambled RNA
- siPLGF
Small interfering RNA to PLGF
- siRNA
Small interfering RNA
- SMC
Smooth muscle cells
- SMGM-2
Smooth muscle growth medium–2
- VEGF-A
Vascular endothelial growth factor A
- VEGFR-1
Vascular endothelial growth factor receptor 1/Flt-1
- VEGFR-2
Vascular endothelial growth factor receptor 2/KDR
References
- 1.Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. Journal of Clinical Investigation. 1998;101:40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Autiero M, Luttun A, Tjwa M, Carmeliet P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost. 2003;1:1356–1370. doi: 10.1046/j.1538-7836.2003.00263.x. [DOI] [PubMed] [Google Scholar]
- 3.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 4.Bergmann CE, Hoefer IE, Meder B, Roth H, van Royen N, Breit SM, Jost MM, Aharinejad S, Hartmann S, Buschmann IR. Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J Leukoc Biol. 2006;80:59–65. doi: 10.1189/jlb.0206087. [DOI] [PubMed] [Google Scholar]
- 5.Caligo Maria Adelaide, Cipollini Giovanna, Petrini Mario, Valentini Paola, Bevilacqua Generoso. Down regulation of NM23.H1, NM23.H2 and c-myc genes during differentiation induced by 1,25 dihydroxyvitamin D3. Leukemia Research. Leukemia Research. 1996;20:161–167. doi: 10.1016/0145-2126(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 6.Cao Y, Chen H, Zhou L, Chiang MK, nand-Apte B, Weatherbee JA, Wang Y, Fang F, Flanagan JG, Tsang ML. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. Journal of Biological Chemistry. 1996;271:3154–3162. doi: 10.1074/jbc.271.6.3154. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T, Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vandendriessche T, Ponten A, Eriksson U, Plate KH, Foidart JM, Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, Collen D, Persico MG. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nature Medicine. 2001;7:575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 8.Chang M, Mukherjea D, Gobble R, Groesch K, Torry RJ, Torry DS. Glial Cell Missing 1 Regulates Placenta Growth Factor (PGF) Gene Transcription in Human Trophoblast. Biol Reprod. 2007 doi: 10.1095/biolreprod.107.065599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clauss M, Weich H, Breier G, Knies U, Rockl W, Waltenberger J, Risau W. The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. Journal of Biological Chemistry. 1996;271:17629–34. doi: 10.1074/jbc.271.30.17629. [DOI] [PubMed] [Google Scholar]
- 10.Cramer M, Nagy I, Murphy BJ, Gassmann M, Hottiger MO, Georgiev O, Schaffner W. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol Chem. 2005;386:865–872. doi: 10.1515/BC.2005.101. [DOI] [PubMed] [Google Scholar]
- 11.De Muinck ED, Simons M. Re-evaluating therapeutic neovascularization. J Mol Cell Cardiol. 2004;36:25–32. doi: 10.1016/j.yjmcc.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Depoix C, Tee MK, Taylor RN. Molecular regulation of human placental growth factor (PlGF) gene expression in placental villi and trophoblast cells is mediated via the protein kinase a pathway. Reprod Sci. 2011;18:219–228. doi: 10.1177/1933719110389337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiSalvo J, Bayne ML, Conn G, Kwok PW, Trivedi PG, Soderman DD, Palisi TM, Sullivan KA, Thomas KA. Purification and characterization of a naturally occurring vascular endothelial growth factor. placenta growth factor heterodimer. Journal of Biological Chemistry. 1995;270:7717–7723. doi: 10.1074/jbc.270.13.7717. [DOI] [PubMed] [Google Scholar]
- 14.Du H, Li P, Pan Y, Li W, Hou J, Chen H, Wang J, Tang H. Vascular endothelial growth factor signaling implicated in neuroprotective effects of placental growth factor in an in vitro ischemic model. Brain Res. 2010;1357:1–8. doi: 10.1016/j.brainres.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 15.Eitenmuller I, Volger O, Kluge A, Troidl K, Barancik M, Cai WJ, Heil M, Pipp F, Fischer S, Horrevoets AJ, Schmitz-Rixen T, Schaper W. The range of adaptation by collateral vessels after femoral artery occlusion. Circ Res. 2006;99:656–662. doi: 10.1161/01.RES.0000242560.77512.dd. [DOI] [PubMed] [Google Scholar]
- 16.Fujii T, Yonemitsu Y, Onimaru M, Inoue M, Hasegawa M, Kuwano H, Sueishi K. VEGF function for upregulation of endogenous PlGF expression during FGF-2-mediated therapeutic angiogenesis. Atherosclerosis. 2008;200:51–57. doi: 10.1016/j.atherosclerosis.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Gobble RM, Groesch KA, Chang M, Torry RJ, Torry DS. Differential regulation of human PlGF gene expression in trophoblast and nontrophoblast cells by oxygen tension. Placenta. 2009;30:869–875. doi: 10.1016/j.placenta.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green CJ, Lichtlen P, Huynh NT, Yanovsky M, Laderoute KR, Schaffner W, Murphy BJ. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res. 2001;61:2696–2703. [PubMed] [Google Scholar]
- 19.Harris P, Ralph P. Human leukemic models of myelomonocytic development: a review of the HL-60 and U937 cell lines. Journal of Leukocyte Biology. 1985;37:407–422. doi: 10.1002/jlb.37.4.407. [DOI] [PubMed] [Google Scholar]
- 20.Heil M, Ziegelhoeffer T, Pipp F, Kostin S, Martin S, Clauss M, Schaper W. Blood monocyte concentration is critical for enhancement of collateral artery growth. American Journal of Physiology Heart and Circulatory Physiology. 2002;283:2411–9. doi: 10.1152/ajpheart.01098.2001. [DOI] [PubMed] [Google Scholar]
- 21.Hoefer IE, Grundmann S, van Royen N, Voskuil M, Schirmer SH, Ulusans S, Bode C, Buschmann IR, Piek JJ. Leukocyte subpopulations and arteriogenesis: specific role of monocytes, lymphocytes and granulocytes. Atherosclerosis. 2005;181:285–293. doi: 10.1016/j.atherosclerosis.2005.01.047. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Keller G. VEGF nuclear accumulation correlates with phenotypical changes in endothelial cells. J Cell Sci. 2000;113(Pt 9):1525–1534. doi: 10.1242/jcs.113.9.1525. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Shen W, Gill R, Corbly A, Jones B, Belagaje R, Zhang Y, Tang S, Chen Y, Zhai Y, Wang G, Wagle A, Hui K, Westmore M, Hanson J, Chen YF, Simons M, Singh J. High-resolution quantitative computed tomography demonstrating selective enhancement of medium-size collaterals by placental growth factor-1 in the mouse ischemic hindlimb. Circulation. 2006;113:2445–2453. doi: 10.1161/CIRCULATIONAHA.105.586818. [DOI] [PubMed] [Google Scholar]
- 24.Loboda A, Jazwa A, Jozkowicz A, Molema G, Dulak J. Angiogenic transcriptome of human microvascular endothelial cells: Effect of hypoxia, modulation by atorvastatin. Vascul Pharmacol. 2006;44:206–214. doi: 10.1016/j.vph.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, Hicklin DJ, Carmeliet P. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nature Medicine. 2002;8:831–40. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- 26.Mac Gabhann F, Popel AS. Systems biology of vascular endothelial growth factors. Microcirculation. 2008;15:715–738. doi: 10.1080/10739680802095964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maglione D, Guerriero V, Viglietto G, li-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9267–9271. doi: 10.1073/pnas.88.20.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maharaj AS, Saint-Geniez M, Maldonado AE, D’Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168:639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odorisio T, Schietroma C, Zaccaria ML, Cianfarani F, Tiveron C, Tatangelo L, Failla CM, Zambruno G. Mice overexpressing placenta growth factor exhibit increased vascularization and vessel permeability. Journal of Cell Science. 2002;115:2559–2567. doi: 10.1242/jcs.115.12.2559. [DOI] [PubMed] [Google Scholar]
- 30.Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G, Ritter R, Balzer J, Scheler C, Schaper W, Schmitz-Rixen T. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004;24:1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4. [DOI] [PubMed] [Google Scholar]
- 31.Pipp F, Heil M, Issbrucker K, Ziegelhoeffer T, Martin S, van den Heuvel J, Weich H, Fernandez B, Golomb G, Carmeliet P, Schaper W, Clauss M. VEGFR-1-selective VEGF homologue PlGF is arteriogenic: evidence for a monocyte-mediated mechanism. Circulation Research. 2003;92:378–85. doi: 10.1161/01.RES.0000057997.77714.72. [DOI] [PubMed] [Google Scholar]
- 32.Prior BM, Lloyd PG, Ren J, Li H, Yang HT, Laughlin MH, Terjung RL. Time course of changes in collateral blood flow and isolated vessel size and gene expression after femoral artery occlusion in rats. American Journal of Physiology Heart and Circulatory Physiology. 2004;287:H2434–H2447. doi: 10.1152/ajpheart.00398.2004. [DOI] [PubMed] [Google Scholar]
- 33.Santos SC, Miguel C, Domingues I, Calado A, Zhu Z, Wu Y, Dias S. VEGF and VEGFR-2 (KDR) internalization is required for endothelial recovery during wound healing. Exp Cell Res. 2007;313:1561–1574. doi: 10.1016/j.yexcr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 34.Schaper W. New paradigms for collateral vessel growth. Basic Res Cardiol. 1993;88:193–198. doi: 10.1007/BF00794992. [DOI] [PubMed] [Google Scholar]
- 35.Schierling W, Troidl K, Mueller C, Troidl C, Wustrack H, Bachmann G, Kasprzak PM, Schaper W, Schmitz-Rixen T. Increased intravascular flow rate triggers cerebral arteriogenesis. J Cereb Blood Flow Metab. 2009;29:726–737. doi: 10.1038/jcbfm.2008.165. [DOI] [PubMed] [Google Scholar]
- 36.Schierling W, Troidl K, Troidl C, Schmitz-Rixen T, Schaper W, Eitenmuller IK. The role of angiogenic growth factors in arteriogenesis. J Vasc Res. 2009;46:365–374. doi: 10.1159/000189797. [DOI] [PubMed] [Google Scholar]
- 37.Scholz D, Elsaesser H, Sauer A, Friedrich C, Luttun A, Carmeliet P, Schaper W. Bone marrow transplantation abolishes inhibition of arteriogenesis in placenta growth factor (PlGF) −/− mice. Journal of Molecular and Cellular Cardiology. 2003;35:177–184. doi: 10.1016/s0022-2828(02)00304-8. [DOI] [PubMed] [Google Scholar]
- 38.Scholz D, Ito W, Fleming I, Deindl E, Sauer A, Wiesnet M, Busse R, Schaper J, Schaper W. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis) Virchows Arch. 2000;436:257–70. doi: 10.1007/s004280050039. [DOI] [PubMed] [Google Scholar]
- 39.Shaw JH, Lloyd PG. Post-transcriptional regulation of placenta growth factor mRNA by hydrogen peroxide. Microvasc Res. 2012;84:155–160. doi: 10.1016/j.mvr.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 41.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 42.Tchaikovski V, Fellbrich G, Waltenberger J. The molecular basis of VEGFR-1 signal transduction pathways in primary human monocytes. Arterioscler Thromb Vasc Biol. 2008;28:322–328. doi: 10.1161/ATVBAHA.107.158022. [DOI] [PubMed] [Google Scholar]
- 43.Toyota E, Warltier DC, Brock T, Ritman E, Kolz C, O’Malley P, Rocic P, Focardi M, Chilian WM. Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation. 2005;112:2108–2113. doi: 10.1161/CIRCULATIONAHA.104.526954. [DOI] [PubMed] [Google Scholar]
- 44.Wang D, Lehman RE, Donner DB, Matli MR, Warren RS, Welton ML. Expression and endocytosis of VEGF and its receptors in human colonic vascular endothelial cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1088–G1096. doi: 10.1152/ajpgi.00250.2001. [DOI] [PubMed] [Google Scholar]
- 45.Yonekura H, Sakurai S, Liu X, Migita H, Wang H, Yamagishi S, Nomura M, Abedin MJ, Unoki H, Yamamoto Y, Yamamoto H. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J Biol Chem. 1999;274:35172–35178. doi: 10.1074/jbc.274.49.35172. [DOI] [PubMed] [Google Scholar]
- 46.Yun J, Rocic P, Pung YF, Belmadani S, Carrao AC, Ohanyan V, Chilian WM. Redox-dependent mechanisms in coronary collateral growth: the “redox window” hypothesis. Antioxid Redox Signal. 2009;11:1961–1974. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Palmer R, Gao X, Kreidberg J, Gerald W, Hsiao L, Jensen RV, Gullans SR, Haber DA. Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol. 2003;13:1625–1629. doi: 10.1016/j.cub.2003.08.054. [DOI] [PubMed] [Google Scholar]