

Abstract
Background/Aims:
MiR-26a has been identified as a tumor suppressor in various tumors, but the relationship between miR-26a and the sensitivity of gastric cancer to chemotherapies has not been established. The present study was performed to investigate the effect of miR-26a on drug sensitivity in gastric cancer (GC).
Materials and Methods:
The expression level of miRNA-26a in cisplatin-resistant SGC-7901/DDP cells and parent SGC-7901 cells was evaluated by qRT-PCR. The effect of miR-26a on sensitivity of GC cells to cisplatin was assayed using MTS method. The effect of miR-26a on cisplatin-induced apoptosis were determined by Annexin V/propidium iodide (PI) double staining method and flow cytometry. The targets of miR-26a were identified using a luciferase activity assay and miR-26a-mediated target genes expression analysis. Furthermore, the role of the targets neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) and E2F2 on sensitivity of chemotherapy in GC by MTS and apoptotic cell analysis was assessed.
Results:
We found that miR-26a was downregulated in cisplatin-resistant SGC-7901/DDP cells compared with SGC-7901 cells. Using both gain- and loss-of-function analyses, we further revealed that miR-26a could improve the sensitivity of GC cells to cisplatin. Furthermore, miR-26a has target sites in the 3′-UTR of NRAS and E2F2 by luciferase reporter assay and reduces the expression levels of NRAS and E2F2. In addition, knockdown of NRAS or E2F2 sensitize GC cells to cisplatin.
Conclusion:
Our results suggest that miR-26a can improve the sensitivity of GC cells to cisplatin-based chemotherapies through targeting NRAS and E2F2, and provide the first evidence of the potential utility of miR-26a as a sensitizer in chemotherapy for GC.
Keywords: Cisplatin sensitivity, E2F2, gastric cancer, MiR-26a, neuroblastomaRAS viral (v-ras) oncogene homolog
Gastric cancer (GC) is the second most common cause of death from cancer in the world.[1] The incidence is higher in eastern countries (such as Japan, Korea, and China) compared with the West.[1] Because the disease is often diagnosed at an advanced stages, the available therapeutic methods to treat the disease in most patients are limited. Meanwhile, chemotherapy is an important option. Platinum-based drugs and cisplatin in particular, have been the major clinical agents for the chemotherapy of GC.[2] However, despite tremendous efforts, cisplatin treatment often results in the development of drug resistance, which has been a major obstacle for successful treatment of GC.[3] The prognosis with a reported 5-year survival rate still remains poor.[3] The challenge for improving chemotherapeutic efficacy is in the development of strategies that enhance cancer cell sensitivity to treatment.
Recently, microRNAs (miRNAs) have been proposed to play essential roles in the development of drug resistance.[4,5,6,7] microRNAs are a group of endogenously expressed, non-coding small RNAs (20–25 nucleotides in length). MicroRNAs negatively regulate the expression of target mRNAs by suppressing translation or decreasing the stability of mRNAs.[8] It has been found that miRNAs play crucial roles in various biological processes, including development, differentiation, apoptosis, and cell proliferation.[9] An increasing number of studies have demonstrated that miRNAs can function as oncogenes or tumor suppressors, and they are often dysregulated in tumors.[10,11,12,13] MiR-26a belongs to the miR-26 family, which contains another member miR-26b, both of which house identical sequence with the exception of two different nucleotides in mature miRNAs. MiR-26a has been independently reported to be tumor suppressor gene in various cancers.[14,15,16,17] MiR-26a is strongly downregulated in GC and its expression levels were associated with overall survival and replase-free survival of GC.[17] Ectopic expression of miR-26a inhibited GC cell proliferation and GC metastasis in vitro and in vivo.[17] As reported in recent studies, miR-26a is involved in the radiosensitivity of glioblastoma multiforme cells.[18] However, it remains unknown whether miR-26a can modulate the sensitivity of chemotherapy in GC, and the target gene of miR-26a in regulating drug sensitivity remains unacquainted.
MATERIALS AND METHODS
Cell lines and culture
The SGC-7901 human gastric adenocarcinoma cell line was purchased from Academy of Military Medical Science (Beijing, China). Its drug-resistant SGC7901/DDP cell line was purchased from Keygen Biotech (Nanjing, China). The cell lines were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco, Carlsbad, CA, USA). Human Embryonic Kidney 293T (HEK293T) cells were maintained in DMEM medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA).
Quantitative RT-PCR analysis
The total RNAs were extracted from cells with TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). For the detection of miR-26a, RT and PCR reactions were performed by means of qSYBR-green-containing PCR kit (Genecopoie, Guangzhou, China), and U6 snRNA was used as an endogenous control for miRNA detection. For the detection of neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) or E2F2, cDNA was synthesized from 1 μg of total RNA by means of Reverse Reaction kit according to the manufacturer's instructions (Promega, Madison, WI, USA). Human GAPDH was amplified in parallel as an internal control. The expression of each gene was quantified by measuring Ct values, and normalized using the 2-ddCt method relative to U6-snRNA or GAPDH.
Cell viability assay
The transfected cells were seeded into 96-well plates (5 × 103 cells/well) and maintained overnight. Then, cells were exposed to various concentrations of cisplatin for 72 h. Cell viability was measured by MTS–formazan reduction (Promega, USA) by absorbance at 490 nm and 50% inhibition of growth (IC50) of the drug was estimated by the relative survival curve. Three independent experiments were performed in quadruplicate.
Apoptosis analysis
For assessment of apoptosis, the Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA) was used. After treating the transfected cells with 5 mg/L cisplatin for 48 h, the cells were collected and stained with 2 μL Annexin V-FITC and 2 μL PI for 15 min at room temperature in the dark for 30 min. The samples were analyzed with an FACS Calibur instrument (BD, San Diego, CA, USA) and the collected data were analyzed using FlowJosoftware.
Luciferase reporter assay
The 3′-UTR of NRAS or E2F2 was amplified from human blood genomic DNA and then was cloned into pMir-Report vector (Ambion). Yielding mutant constructs, mutations were introduced in potential miR-26a binding sites using the QuikChange site-directed mutagenesis Kit (Stratagene). One microgram of the wild-type or mutant UTR plasimds were cotransfected either with 50 nM of miR-26a mimic or mimic control into HEK293T cells using Lipofectamine™ 2000 (Invitrogen). Cells were harvested 48 h after transfection and assayed using the Dual Luciferase Reporter Assay System (Promega).
Western blot analysis
Proteins were separated by 10% SDS-PAGE and then transferred to PVDF membranes (Amersham, Buckinghamshire, UK). The membranes were incubated overnight at 4°C with anti-NRAS antibody (CST), E2F2 (Abcam), or anti-GAPDH (Sigma) antibody followed by HRP-linked secondary antibodies.
Statistical analysis
Statistical analyses were performed using SPSS 17.0. Data are presented as the mean ± standard deviation. The difference between groups was analyzed using a Student's t test when comparing only two groups or one-way analysis of variance when comparing more than two groups. P < 0.05 was considered statistically significant.
RESULTS
MiR-26a modulated the sensitivity of GC cells to cisplatin
To investigate the potential role of miRNA-26a on drug resistance in GC, the expression of miRNA-26a in cisplatin-resistant SGC-7901/DDP cells and parent SGC-7901 cells was evaluated by qRT-PCR. We found that miR-26a was reduced by 60% in SGC-7901/DDP cells compared with SGC-7901 cells [Figure 1a].
Figure 1.
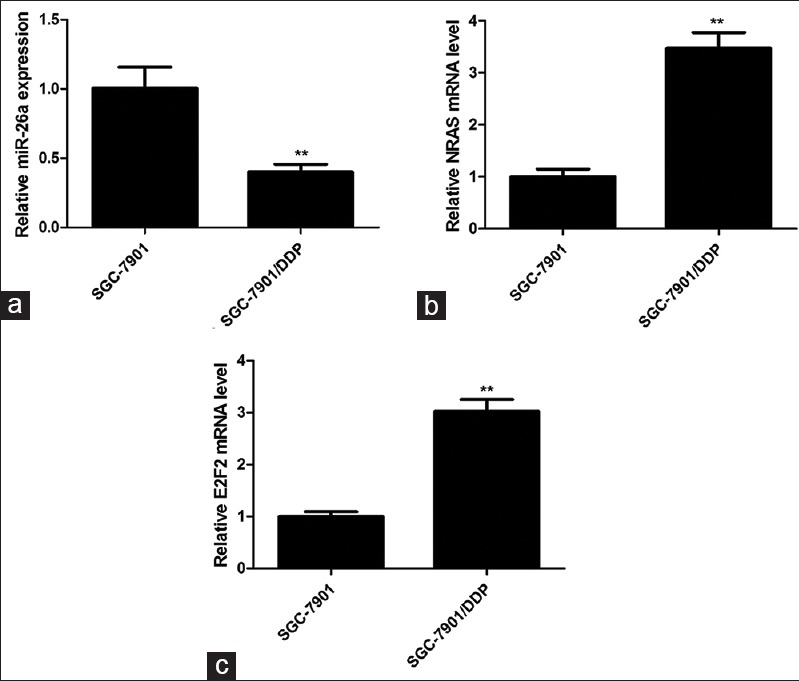
The expression profiles of miR-26a, NRAS, and E2F2 in SGC-7901/DDP and SGC-7901 cells. The expression of miR-26a (a), NRAS (b), and E2F2 (c) was analyzed by qRT-PCR. Data are presented as mean ± SD from at least three independent experiments. **P < 0.01
To further investigate the effects of miR-26a on the sensitivity of GC cells to cisplatin, SGC-7901/DDP or SGC-7901 cells transfected with miR-26a mimic or inhibitor. The effect of miR-26a mimic was determined in SGC-7901/DDP cells, which significantly increased miR-26a expression [Figure 2a]. The effect of miR-26a inhibitor was found to suppress miR-26a expression remarkably in SGC-7901 cells [Figure 2a]. The MTS assay revealed that SGC-7901/DDP cells transfected with miR-26a mimic exhibited greatly increased sensitivity to DDP compared with cells transfected with mimic control [Figure 2b]. In contrast, suppression of the miR-26a level in SGC-7901 cells resulted in a reduced sensitivity to DDP [Figure 2b]. In addition, apoptotic cell analysis by flow cytometry showed the apoptotic rate of SGC-7901/DDP cells transfected with miR-26a mimic and incubated with 5 mg/L DDP for 48 h was significantly higher than that of the control mimic (73.8% ± 4.1% vs. 32.7% ± 4.0%, P = 0.002), whereas the apoptotic rate of SGC-7901 cells transfected with miR-26a inhibitor and incubated with 5 mg/L DDP for 48 h was significantly lower than that of the inhibitor control (29.9% ± 3.6% vs. 48.9% ± 3.3%, P = 0.018; Figure 2c). These results suggested that miR-26a contributed to increase the sensitivity of GC cells to cisplatin.
Figure 2.
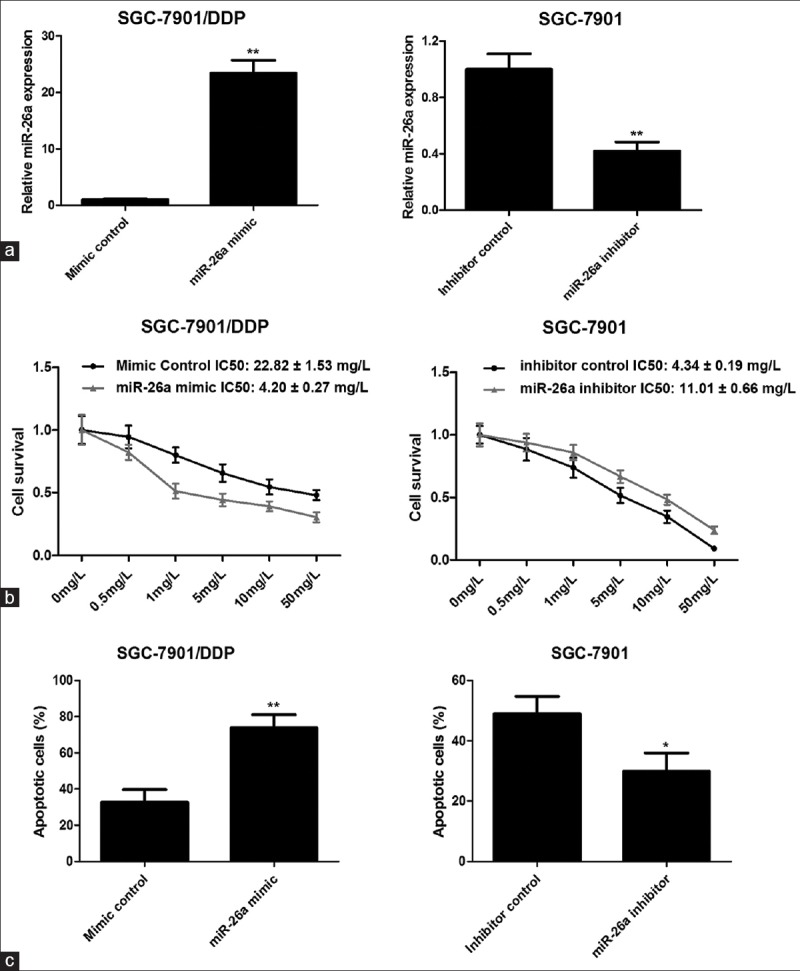
miR-26a increases sensitivity of GC cells to cisplatin. (a) Examination of miR-26a expression in SGC-7901/DDP and SGC-7901 cells transfected with miR-26a mimic or inhibitor by qRT-PCR. (b) The cell survival was examined by MTS assay. (c) The effect of miR-26a on cisplatin-induced cell apoptosis. The transfected cells incubated with 5 mg/L DDP for 48 h and then apoptotic cells were evaluated by Annexin V-FITC and PI staining and analyzed with FACS. *P < 0.05; **P < 0.01
NRAS and E2F2 are the direct targets of miR-26a
We next explored the possible targets of miR-26a in regulating drug sensitivity through different computational algorithms. Silicon analysis revealed NRAS and E2F2 as candidate targets of miR-26a. There was perfect base pairing between the “seed sequence” of mature miR-26a and the 3′-UTRs of NRAS and E2F2 [Figure 3a]. Indeed, in contrast to miR-26a expression mode, the expression levels of NRAS and E2F2 were increased (approximately 3.5 times and 3 times, respectively) in cisplatin-resistant SGC-7901/DDP cells compared with SGC7901 cells [Figure 1b and c]. To verify whether NRAS and E2F2 are the direct targets of miR-26a, the wild-type 3′-UTRs or the mutant (lacking seed sequence) was cloned into a luciferase reporter vector, and a luciferase reporter assay was done. We observed that no reduction of luciferase activity was observed in HEK293T cells transfected with miR-26a mimics and the mutated 3′-UTR of NRAS or E2F2. But greater than 50% reduction of luciferase activity was observed with wild-type NRAS 3′-UTR and 40% reduction of luciferase activity was determined with wild-type E2F2 3′-UTR [Figure 3b]. These data support that miR-26a directly targets 3′-UTRs of NRAS and E2F2.
Figure 3.
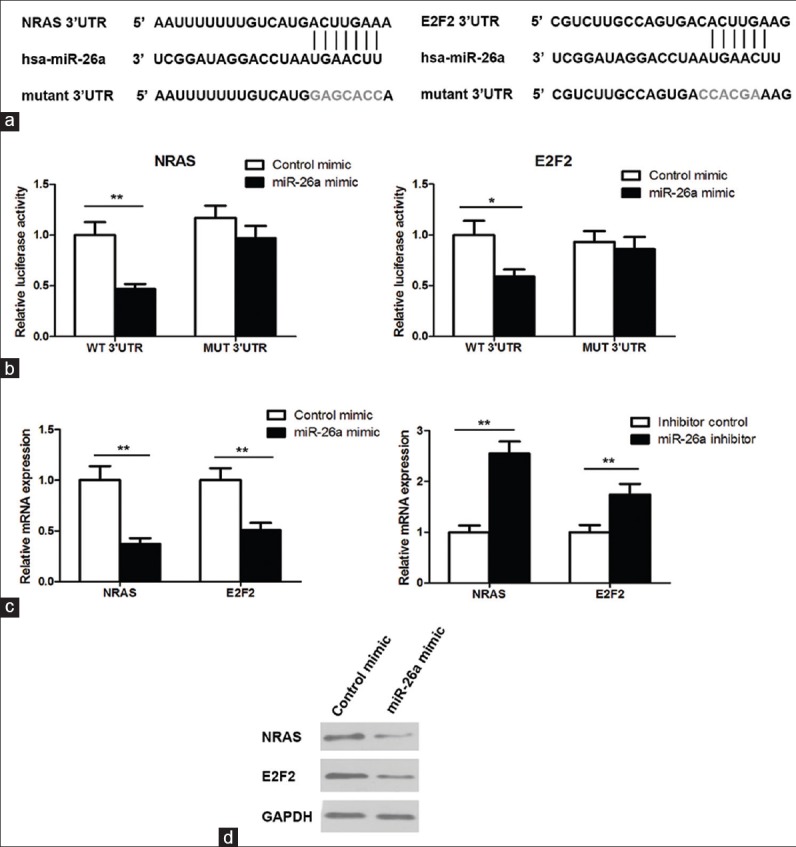
NRAS and E2F2 are direct targets of miR-26a. (a) The 3′-UTR of NRAS or E2F2 contains one predicted miR-26a binding site. The mutagenesis nucleotides are indicated in gray. (b) HEK293T cells were cotransfected with 3′UTR-reporter constructs with miR-26a mimic or control mimic and dual luciferase reporter assay was performed. (c) qRT-PCR to measure the mRNA levels of NRAS and E2F2 in transfected cells. (d) Western blot to measure the protein levels of NRAS and E2F2 in SGC-7901/DDP cells transfected with miR-26a mimic. *P < 0.05; **P < 0.01
Next, we examined the effect of miR-26a on the expression levels of NRAS and E2F2. In agreement, miR-26a overexpression significantly reduced both mRNA and protein expression for NRAS and E2F2 in SGC-7901/DDP cells [Figure 3c and d]. In contrast, knockdown of miR-26a increased their expression levels in SGC-7901 cells [Figure 3c], further indicating that NRAS and E2F2 are the targets of miR-26a in osteosarcoma cells.
Knockdown of NRAS or E2F2 sensitize SGC-7901/DDP cells to cisplatin
Next we sought to find out whether downregulation of NRAS or E2F2 could mimic the promotion of cell sensitivity to cisplatin by miR-26a. We first depleted endogenous NRAS or E2F2 in SGC-7901/DDP cells by transfecting siRNA. The efficiency was confirmed through qRT-PCR and Western blot [Figure 4a]. The MTS assay revealed that the sensitivity of cisplatin was significantly increased in siNRAS or siE2F2 transfected cells compared with cells transfected with control siRNA [Figure 4b]. Furthermore, a marked increase in apoptosis after DDP treatment was observed in SGC-7901/DDP cells transfected with siNRAS or siE2F2 compared with cells transfected with control siRNA in which the apoptotic rates were 67.0 ± 3.9%, 52.0 ± 3.5%, and 32.3 ± 2.8%, respectively [Figure 4c]. These data suggested that downregulation of NRAS or E2F2 could mimic the effect of miR-26a.
Figure 4.
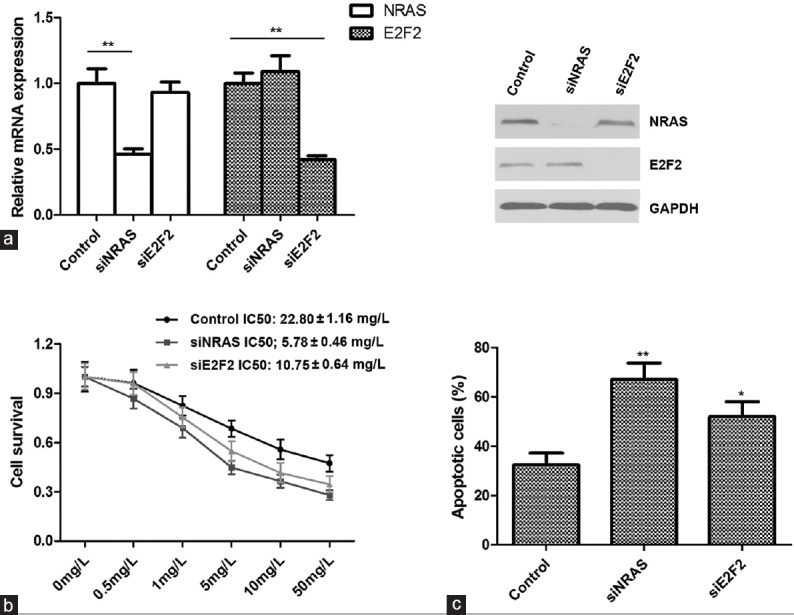
Knockdown of NRAS or E2F2 enhances the sensitivity of SGC-7901/DDP cells to cisplatin. (a) The mRNA and protein levels of NRAS and E2F2 were all reduced after transfection with siRNAs in SGC-7901/DDP cells. (b) The cell survival was examined by MTS assay after siRNA transfection. (c) Knockdown of NRAS or E2F2 enhances cisplatin-induced cell apoptosis. The transfected cells incubated with 5 mg/L DDP for 48 h and the apoptotic rates were evaluated by Annexin V-FITC and PI staining and analyzed with FACS. *P < 0.05; **P < 0.01
DISCUSSION
MiR-26a is a functional miRNA that has merited previous investigation. It is known that miR-26a plays a significant role in the growth, development, and cell differentiation of different tissues.[19] The previous data indicated that miR-26a may play as a tumor-suppressor gene in a number of cancer types. For example, miR-26a is decreased in hepatocellular carcinoma (HCC) and could suppress tumor angiogenesis of HCC through HGF-cMet signaling.[14] Breast cancer also exhibits decreased expression of miR-26a and overexpression of this miRNA results in inhibition of tumor growth and metastasis.[20] It is underexpressed in GC tissues, while overexpression of miR-26a in GC cells inhibits cell proliferation and invasion, and promotes cell apoptosis.[17] However, the relationship between miR-26a and the sensitivity of GC to chemotherapies has not been established.
Accumulating evidence suggests that aberrant miRNA expression is strongly implicated in the development of drug resistance.[4,5,6,7,21] miRNAs might modulate the expression of target proteins, which could include drug transporters, drug targets, or apoptosis and cell cycle-related components, resulting in variations in sensitivity of cells to chemotherapeutic agents.[21]
In this study, we found that miR-26a was decreased in cisplatin-resistant SGC7901/DDP cells compared with that in SGC7901 cells. Furthermore, modulation of miR-26a expression by miR-26a mimic or inhibitor could alter the sensitivity of GC cells to cisplatin. The overexpression of miR-26a contributed to the increased sensitivity of SGC-7901/DDP cells to cisplatin, whereas inhibition of miR-26a expression resulted in decreased sensitivity of SGC-7901 cells to cisplatin. These data confirm that miR-26a is related to the sensitivity of GC to chemotherapies.
We next explored the possible targets of miR-26a in regulating drug sensitivity through different computational algorithms. Silicon analysis revealed NRAS and E2F2 as candidate targets of miR-26a. NRAS and E2F2 were selected out for their role in carcinogenesis and chemotherapy sensitivity.[22,23,24] NRAS, a well-known oncogene, belongs to the guanosine 5’-triphosphate-binding proteins family.[25] Ras proteins are crucial crossroads of signaling pathways that link the activation of cell surface receptors with a wide variety of cellular processes leading to the control of proliferation, apoptosis, invasion, and differentiation.[26] Regarding chemotherapy response, activated mutation or elevated levels of NRAS have previously been implicated in drug resistance.[27] E2F2, a member of the E2F family of transcription factors plays a central role in regulating G1/S transition and progression through S phase, promoting cellular transformation.[28]
Here, NRAS and E2F2 as the direct targets of miR-26a were further confirmed in luciferase activity assays and miR-26a-mediated these two genes expression analysis. Our results also found that knockdown of NRAS or E2F2 sensitize GC cells to cisplatin. miR-26a overexpression has been demonstrated to improve the sensitivity of GC cells to cisplatin and this effect was considered to be mediated via its targets NRAS and E2F2.
CONCLUSION
miR-26a expression was downregulated in cisplatin-resistant GC cells. Upregulation of miR-26a could improve the sensitivity of GC cells to cisplatin-based chemotherapies through the direct downregulation of NRAS and E2F2, suggesting that miR-26a may be a therapeutic target for overcoming cisplatin resistance in GC and the appropriate combination of DDP application with miR-26a upregulation might be a potential strategy for the treatment of GC.
Footnotes
Source of Support: This work was supported by grant from the Nature Scientific Foundation of China (No. 81101526)
Conflict of Interest: None declared.
REFERENCES
- 1.Lee JH, Kim KM, Cheong JH, Noh SH. Current management and future strategies of gastric cancer. Yonsei Med J. 2012;53:248–57. doi: 10.3349/ymj.2012.53.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cervantes A, Roda D, Tarazona N, Rosello S, Perez-Fidalgo JA. Current questions for the treatment of advanced gastric cancer. Cancer Treat Rev. 2013;39:60–7. doi: 10.1016/j.ctrv.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Orditura M, Galizia G, Sforza V, Gambardella V, Fabozzi A, Laterza MM, et al. Treatment of gastric cancer. World J Gastroenterol. 2014;20:1635–49. doi: 10.3748/wjg.v20.i7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Chen X, Xiu YL, Sun KX, Zong ZH, Zhao Y. microRNA 490-3P enhances the drug-resistance of human ovarian cancer cells. J Ovarian Res. 2014;7:84. doi: 10.1186/s13048-014-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu R, Liu X, Zheng Y, Gu J, Xiong S, Jiang P, et al. MicroRNA-7 sensitizes non-small cell lung cancer cells to paclitaxel. Oncol Lett. 2014;8:2193–200. doi: 10.3892/ol.2014.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolfo C, Fanale D, Hong DS, Tsimberidou AM, Piha-Paul SA, Pauwels P, et al. Impact of microRNAs in resistance to chemotherapy and novel targeted agents in non-small cell lung cancer. Curr Pharm Biotechnol. 2014;15:475–85. doi: 10.2174/1389201015666140519123219. [DOI] [PubMed] [Google Scholar]
- 7.Hong L, Han Y, Zhang Y, Zhang H, Zhao Q, Wu K, et al. MicroRNA-21: A therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets. 2013;17:1073–80. doi: 10.1517/14728222.2013.819853. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.He L, Hannon GJ. MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 10.Sharma G, Dua P, Agarwal SM. A comprehensive review of dysregulated miRNAs involved in cervical cancer. Curr Genomics. 2014;15:310–23. doi: 10.2174/1389202915666140528003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XJ, Ren ZJ, Tang JH. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014;5:e1327. doi: 10.1038/cddis.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng M, Liu JF, Gu YX, Zheng GP, He ZM. [miR-216b suppresses cell proliferation and invasion by targeting PKCalpha in nasopharyngeal carcinoma cells] Zhonghua Zhong Liu Za Zhi. 2013;35:645–50. [PubMed] [Google Scholar]
- 13.Deng M, Ye Q, Qin Z, Zheng Y, He W, Tang H, et al. miR-214 promotes tumorigenesis by targeting lactotransferrin in nasopharyngeal carcinoma. Tumour Biol. 2013;34:1793–800. doi: 10.1007/s13277-013-0718-y. [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Zhang XF, Lu X, Jia HL, Liang L, Dong QZ, et al. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology. 2014;59:1874–85. doi: 10.1002/hep.26941. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, He ML, Wang L, Chen Y, Liu X, Dong Q, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71:225–33. doi: 10.1158/0008-5472.CAN-10-1850. [DOI] [PubMed] [Google Scholar]
- 16.Yang X, Liang L, Zhang XF, Jia HL, Qin Y, Zhu XC, et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58:158–70. doi: 10.1002/hep.26305. [DOI] [PubMed] [Google Scholar]
- 17.Deng M, Tang HL, Lu XH, Liu MY, Lu XM, Gu YX, et al. miR-26a suppresses tumor growth and metastasis by targeting FGF9 in gastric cancer. PLoS One. 2013;8:e72662. doi: 10.1371/journal.pone.0072662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo P, Lan J, Ge J, Nie Q, Guo L, Qiu Y, et al. MiR-26a enhances the radiosensitivity of glioblastoma multiforme cells through targeting of ataxia-telangiectasia mutated. Exp Cell Res. 2014;320:200–8. doi: 10.1016/j.yexcr.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Gao J, Liu QG. The role of miR-26 in tumors and normal tissues (Review) Oncol Lett. 2011;2:1019–23. doi: 10.3892/ol.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Li L, Wu M, Liu M, Xie X, Guo J, et al. MiR-26a inhibits proliferation and migration of breast cancer through repression of MCL-1. PLoS One. 2013;8:e65138. doi: 10.1371/journal.pone.0065138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie L, Jing R, Qi J, Lin Z, Ju S. Drug resistance-related microRNAs in hematological malignancies: Translating basic evidence into therapeutic strategies. Blood Rev. 2015;29:33–44. doi: 10.1016/j.blre.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Wong DJ, Robert L, Atefi MS, Lassen A, Avarappatt G, Cerniglia M, et al. Antitumor activity of the ERK inhibitor SCH722984 against BRAF mutant, NRAS mutant and wild-type melanoma. Mol Cancer. 2014;13:194. doi: 10.1186/1476-4598-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedersen M, Viros A, Cook M, Marais R. (G12D) NRAS and kinase-dead BRAF cooperate to drive naevogenesis and melanomagenesis. Pigment Cell Melanoma Res. 2014;27:1162–6. doi: 10.1111/pcmr.12293. [DOI] [PubMed] [Google Scholar]
- 24.Dong Q, Meng P, Wang T, Qin W, Qin W, Wang F, et al. MicroRNA let-7a inhibits proliferation of human prostate cancer cells in vitro and in vivo by targeting E2F2 and CCND2. PLoS One. 2010;5:e10147. doi: 10.1371/journal.pone.0010147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunz M. Oncogenes in melanoma: An update. Eur J Cell Biol. 2014;93:1–10. doi: 10.1016/j.ejcb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120:3397–406. doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milosevic Z, Pesic M, Stankovic T, Dinic J, Milovanovic Z, Stojsic J, et al. Targeting RAS-MAPK-ERK and PI3K-AKT-mTOR signal transduction pathways to chemosensitize anaplastic thyroid carcinoma. Transl Res. 2014;164:411–23. doi: 10.1016/j.trsl.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Timmers C, Sharma N, Opavsky R, Maiti B, Wu L, Wu J, et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27:65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]